
Uncovering Forensic Evidence: A Path to Age Estimation through DNA Methylation




Abstract
1. Introduction
1.1. Epigenetics
1.2. DNA Methylation for Forensic Science
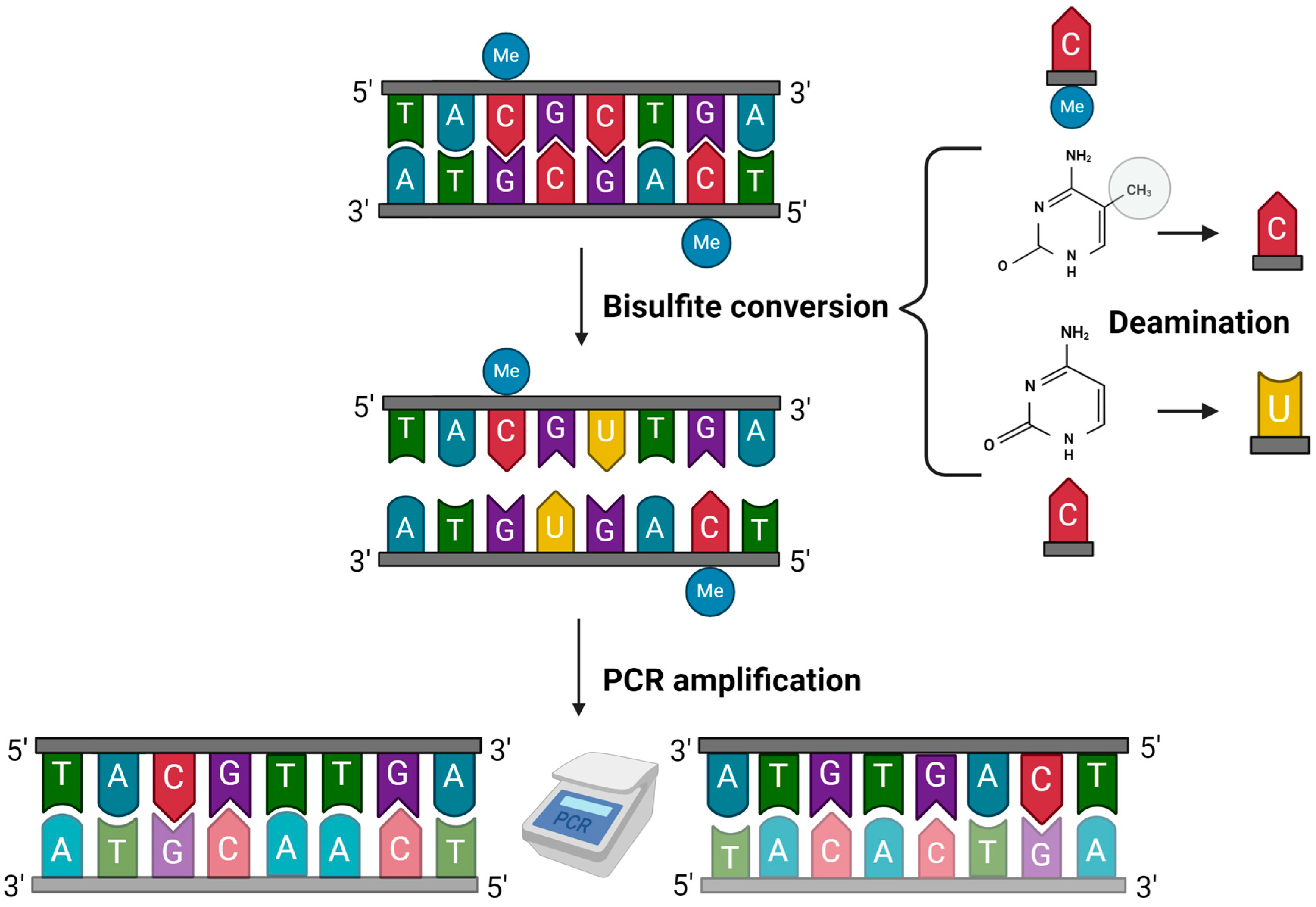
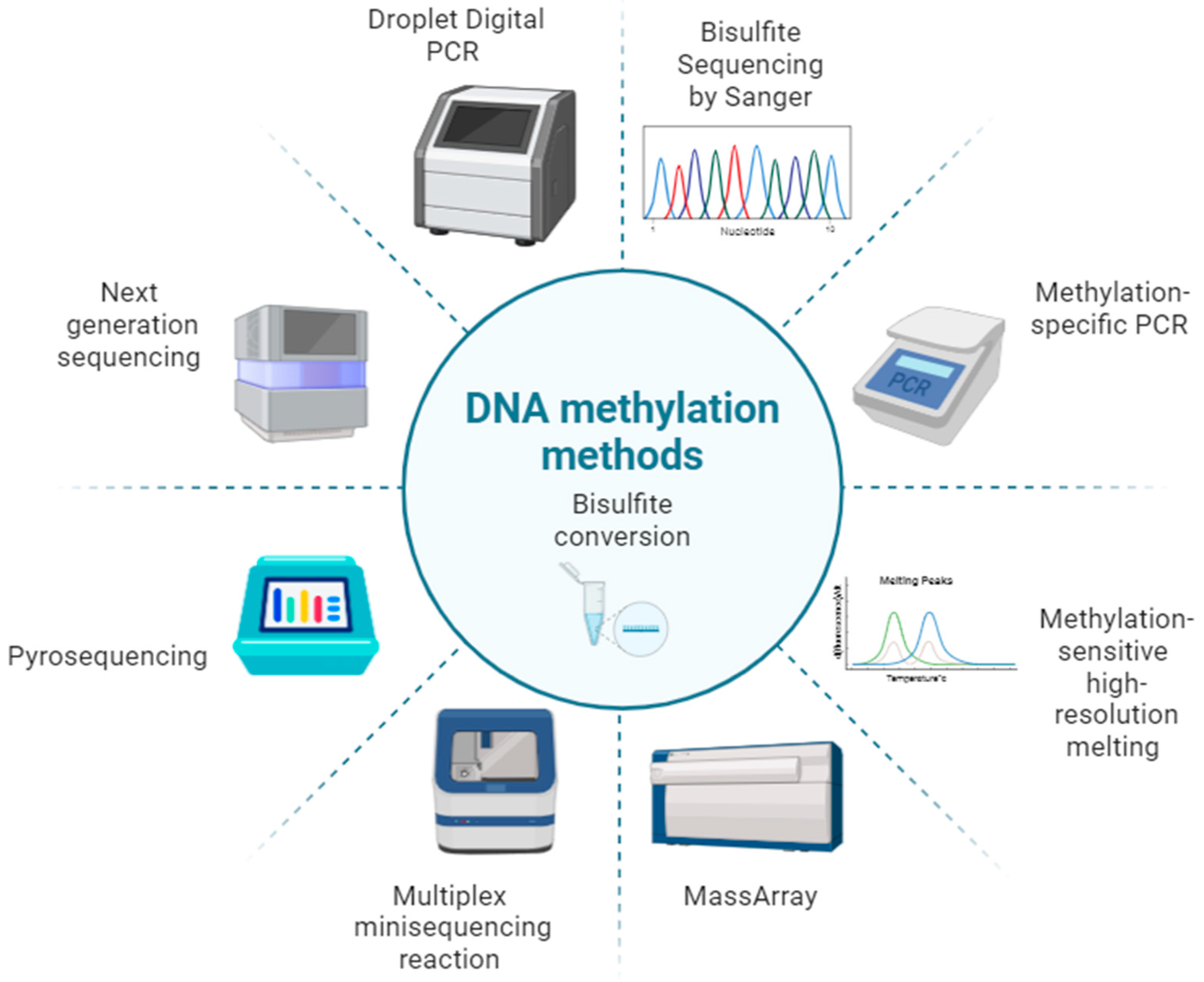
2. Methods for Age Estimation by DNA Methylation Analysis
2.1. Bisulfite Sequencing by Sanger
2.2. Methylation-Specific PCR (MSP)
2.3. Methylation-Sensitive High-Resolution Melting (MS-HRM)
2.4. MassArray (EpiTYPER)
2.5. Multiplex Minisequencing Reaction (SNaPshot)
2.6. Pyrosequencing
2.7. Next Generation Sequencing (NGS)
2.8. Exploring New Approaches in DNA Methylation Analysis
3. Epigenetic Clocks
4. DNA Methylation Analysis for Anthropology
Age Estimation in Children
5. DNA Methylation Analysis for Criminalistics
5.1. Blood
5.1.1. Postmortem Blood Samples
5.1.2. Y-Chromosome in Blood Samples
5.2. Semen
5.3. Saliva and Buccal Swabs
5.4. Multi-Tissue Age Prediction Models
6. Other Factors Impacting DNA Methylation
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Espada, J.; Esteller, M. DNA methylation and the functional organization of the nuclear compartment. Semin. Cell Dev. Biol. 2010, 21, 238–246. [Google Scholar] [CrossRef]
- Zhang, F.F.; Cardarelli, R.; Carroll, J.; Fulda, K.G.; Kaur, M.; Gonzalez, K.; Vishwanatha, J.K.; Santella, R.M.; Morabia, A. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011, 6, 623–629. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Vaiserman, A.; Krasnienkov, D. Telomere Length as a Marker of Biological Age: State-of-the-Art, Open Issues, and Future Perspectives. Front. Genet. 2020, 11, 630186. [Google Scholar] [CrossRef]
- Lujan, S.A.; Longley, M.J.; Humble, M.H.; Lavender, C.A.; Burkholder, A.; Blakely, E.L.; Alston, C.L.; Gorman, G.S.; Turnbull, D.M.; McFarland, R.; et al. Ultrasensitive deletion detection links mitochondrial DNA replication, disease, and aging. Genome Biol. 2020, 21, 248. [Google Scholar] [CrossRef] [PubMed]
- Zapico, S.C.; Ubelaker, D.H. Application of Aspartic Acid Racemization for Age Estimation in a Spanish Sample. Biology 2022, 11, 856. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 2021, 22, 687–698. [Google Scholar] [CrossRef]
- Zgutka, K.; Tkacz, M.; Tomasiak, P.; Tarnowski, M. A Role for Advanced Glycation End Products in Molecular Ageing. Int. J. Mol. Sci. 2023, 24, 9881. [Google Scholar] [CrossRef]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Jin, X.; Ren, Z.; Zhang, H.; Wang, Q.; Liu, Y.; Ji, J.; Huang, J. Systematic Selection of Age-Associated mRNA Markers and the Development of Predicted Models for Forensic Age Inference by Three Machine Learning Methods. Front. Genet. 2022, 13, 924408. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Ż.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef]
- Fiorito, G.; McCrory, C.; Robinson, O.; Carmeli, C.; Ochoa-Rosales, C.; Zhang, Y.; Colicino, E.; Dugué, P.A.; Artaud, F.; McKay, G.J.; et al. Socioeconomic position, lifestyle habits and biomarkers of epigenetic aging: A multi-cohort analysis. Aging 2019, 11, 2045–2070. [Google Scholar] [CrossRef]
- Jovanovic, T.; Vance, L.A.; Cross, D.; Knight, A.K.; Kilaru, V.; Michopoulos, V.; Klengel, T.; Smith, A.K. Exposure to Violence Accelerates Epigenetic Aging in Children. Sci. Rep. 2017, 7, 8962. [Google Scholar] [CrossRef]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef]
- McCartney, D.L.; Zhang, F.; Hillary, R.F.; Zhang, Q.; Stevenson, A.J.; Walker, R.M.; Bermingham, M.L.; Boutin, T.; Morris, S.W.; Campbell, A.; et al. An epigenome-wide association study of sex-specific chronological ageing. Genome Med. 2019, 12, 1. [Google Scholar] [CrossRef]
- Pavanello, S.; Campisi, M.; Tona, F.; Lin, C.D.; Iliceto, S. Exploring Epigenetic Age in Response to Intensive Relaxing Training: A Pilot Study to Slow Down Biological Age. Int. J. Environ. Res. Public. Health 2019, 16, 3074. [Google Scholar] [CrossRef]
- Ambatipudi, S.; Horvath, S.; Perrier, F.; Cuenin, C.; Hernandez-Vargas, H.; Le Calvez-Kelm, F.; Durand, G.; Byrnes, G.; Ferrari, P.; Bouaoun, L.; et al. DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. Eur. J. Cancer 2017, 75, 299–307. [Google Scholar] [CrossRef]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging 2016, 8, 1485–1512. [Google Scholar] [CrossRef]
- Spólnicka, M.; Zbieć-Piekarska, R.; Karp, M.; Machnicki, M.M.; Własiuk, P.; Makowska, Ż.; Pięta, A.; Gambin, T.; Gasperowicz, P.; Branicki, W.; et al. DNA methylation signature in blood does not predict calendar age in patients with chronic lymphocytic leukemia but may alert to the presence of disease. Forensic Sci. Int. Genet. 2018, 34, e15–e17. [Google Scholar] [CrossRef]
- Becker, J.; Böhme, P.; Reckert, A.; Eickhoff, S.B.; Koop, B.E.; Blum, J.; Gündüz, T.; Takayama, M.; Wagner, W.; Ritz-Timme, S. Evidence for differences in DNA methylation between Germans and Japanese. Int. J. Leg. Med. 2022, 136, 405–413. [Google Scholar] [CrossRef]
- Fleckhaus, J.; Freire-Aradas, A.; Rothschild, M.A.; Schneider, P.M. Impact of genetic ancestry on chronological age prediction using DNA methylation analysis. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e399–e400. [Google Scholar] [CrossRef]
- Tajuddin, S.M.; Hernandez, D.G.; Chen, B.H.; Noren Hooten, N.; Mode, N.A.; Nalls, M.A.; Singleton, A.B.; Ejiogu, N.O.; Chitrala, K.N.; Zonderman, A.B.; et al. Novel age-associated DNA methylation changes and epigenetic age acceleration in middle-aged African Americans and whites. Clin. Epigenet. 2019, 11, 119. [Google Scholar] [CrossRef]
- Horvath, S.; Gurven, M.; Levine, M.E.; Trumble, B.C.; Kaplan, H.; Allayee, H.; Ritz, B.R.; Chen, B.; Lu, A.T.; Rickabaugh, T.M.; et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016, 17, 171. [Google Scholar] [CrossRef]
- Spólnicka, M.; Pośpiech, E.; Pepłońska, B.; Zbieć-Piekarska, R.; Makowska, Ż.; Pięta, A.; Karłowska-Pik, J.; Ziemkiewicz, B.; Wężyk, M.; Gasperowicz, P.; et al. DNA methylation in ELOVL2 and C1orf132 correctly predicted chronological age of individuals from three disease groups. Int. J. Leg. Med. 2018, 132, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vidaki, A.; Kayser, M. From forensic epigenetics to forensic epigenomics: Broadening DNA investigative intelligence. Genome Biol. 2017, 18, 238. [Google Scholar] [CrossRef] [PubMed]
- Correia Dias, H.; Cunha, E.; Corte Real, F.; Manco, L. Challenges and (Un)Certainties for DNAm Age Estimation in Future. Forensic Sci. 2022, 2, 601–614. [Google Scholar] [CrossRef]
- Naue, J.; Hoefsloot, H.C.J.; Kloosterman, A.D.; Verschure, P.J. Forensic DNA methylation profiling from minimal traces: How low can we go? Forensic Sci. Int. Genet. 2018, 33, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, L.; Breslin, K.; Zuñiga, S.; Wirken, L.; Pośpiech, E.; Kukla-Bartoszek, M.; Sijen, T.; Knijff, P.; Liu, F.; Branicki, W.; et al. The HIrisPlex-S system for eye, hair and skin colour prediction from DNA: Introduction and forensic developmental validation. Forensic Sci. Int. Genet. 2018, 35, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.M.; Prainsack, B.; Kayser, M. The Use of Forensic DNA Phenotyping in Predicting Appearance and Biogeographic Ancestry. Dtsch. Arztebl. Int. 2019, 51–52, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.R.; Jung, S.E.; Lee, E.H.; Shin, K.J.; Yang, W.I.; Lee, H.Y. DNA methylation-based age prediction from saliva: High age predictability by combination of 7 CpG markers. Forensic Sci. Int. Genet. 2017, 29, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Naue, J.; Sänger, T.; Hoefsloot, H.C.J.; Lutz-Bonengel, S.; Kloosterman, A.D.; Verschure, P.J. Proof of concept study of age-dependent DNA methylation markers across different tissues by massive parallel sequencing. Forensic Sci. Int. Genet. 2018, 36, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Vidaki, A.; Ballard, D.; Aliferi, A.; Miller, T.H.; Barron, L.P.; Syndercombe Court, D. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci. Int. Genet. 2017, 28, 225–236. [Google Scholar] [CrossRef]
- Vidaki, A.; Díez López, C.; Carnero-Montoro, E.; Ralf, A.; Ward, K.; Spector, T.; Bell, J.T.; Kayser, M. Epigenetic discrimination of identical twins from blood under the forensic scenario. Forensic Sci. Int. Genet. 2017, 31, 67–80. [Google Scholar] [CrossRef]
- Vidaki, A.; Kalamara, V.; Carnero-Montoro, E.; Spector, T.D.; Bell, J.T.; Kayser, M. Investigating the Epigenetic Discrimination of Identical Twins Using Buccal Swabs, Saliva, and Cigarette Butts in the Forensic Setting. Genes 2018, 9, 252. [Google Scholar] [CrossRef]
- Lee, H.Y.; Jung, S.-E.; Lee, E.H.; Yang, W.I.; Shin, K.-J. DNA methylation profiling for a confirmatory test for blood, saliva, semen, vaginal fluid and menstrual blood. Forensic Sci. Int. Genet. 2016, 24, 75–82. [Google Scholar] [CrossRef]
- Vidaki, A.; Giangasparo, F.; Syndercombe Court, D. Discovery of potential DNA methylation markers for forensic tissue identification using bisulphite pyrosequencing. Electrophoresis 2016, 37, 2767–2779. [Google Scholar] [CrossRef]
- Planterose Jiménez, B.; Liu, F.; Caliebe, A.; Montiel González, D.; Bell, J.T.; Kayser, M.; Vidaki, A. Equivalent DNA methylation variation between monozygotic co-twins and unrelated individuals reveals universal epigenetic inter-individual dissimilarity. Genome Biol. 2021, 22, 18. [Google Scholar] [CrossRef]
- Park, J.-L.; Woo, K.-M.; Kim, S.-Y.; Kim, Y.S. Potential forensic application of DNA methylation to identify individuals in a pair of monozygotic twins. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e456–e457. [Google Scholar] [CrossRef]
- Marqueta-Gracia, J.J.; Álvarez-Álvarez, M.; Baeta, M.; Palencia-Madrid, L.; Prieto-Fernández, E.; Ordoñana, J.R.; de Pancorbo, M.M. Differentially methylated CpG regions analyzed by PCR-high resolution melting for monozygotic twin pair discrimination. Forensic Sci. Int. Genet. 2018, 37, e1–e5. [Google Scholar] [CrossRef]
- Antunes, J.; Gauthier, Q.; Aguiar-Pulido, V.; Duncan, G.; McCord, B. A data-driven, high-throughput methodology to determine tissue-specific differentially methylated regions able to discriminate body fluids. Electrophoresis 2021, 42, 1168–1176. [Google Scholar] [CrossRef]
- Wang, H.X.; Liu, X.Z.; He, X.M.; Xiao, C.; Huang, D.X.; Yi, S.H. Identification of Mixtures of Two Types of Body Fluids Using the Multiplex Methylation System and Random Forest Models. Curr. Med. Sci. 2023, 43, 908–918. [Google Scholar] [CrossRef]
- Kader, F.; Ghai, M.; Olaniran, A.O. Characterization of DNA methylation-based markers for human body fluid identification in forensics: A critical review. Int. J. Leg. Med. 2020, 134, 1–20. [Google Scholar] [CrossRef]
- Choung, C.M.; Lee, J.W.; Park, J.H.; Kim, C.H.; Park, H.C.; Lim, S.K. A forensic case study for body fluid identification using DNA methylation analysis. Leg. Med. 2021, 51, 101872. [Google Scholar] [CrossRef]
- Silva, D.; Antunes, J.; Balamurugan, K.; Duncan, G.; Alho, C.S.; McCord, B. Developmental validation studies of epigenetic DNA methylation markers for the detection of blood, semen and saliva samples. Forensic Sci. Int. Genet. 2016, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Aliferi, A.; Sundaram, S.; Ballard, D.; Freire-Aradas, A.; Phillips, C.; Lareu, M.V.; Court, D.S. Combining current knowledge on DNA methylation-based age estimation towards the development of a superior forensic DNA intelligence tool. Forensic Sci. Int. Genet. 2022, 57, 102637. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Jung, S.-E.; Hong, S.R.; Lee, E.H.; Lee, J.H.; Lee, S.D.; Lee, H.Y. Independent validation of DNA-based approaches for age prediction in blood. Forensic Sci. Int. Genet. 2017, 29, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Thong, Z.; Tan, J.Y.Y.; Loo, E.S.; Phua, Y.W.; Chan, X.L.S.; Syn, C.K. Artificial neural network, predictor variables and sensitivity threshold for DNA methylation-based age prediction using blood samples. Sci. Rep. 2021, 11, 1744. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Qian, J.; Qu, H.; Ji, Z.; Li, J.; Hu, W.; Cheng, F.; Fang, X.; Yan, J. DNA methylation-based age prediction with bloodstains using pyrosequencing and random forest regression. Electrophoresis 2023, 44, 835–844. [Google Scholar] [CrossRef]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; Van de Voorde, W.; Decorte, R. Improved age determination of blood and teeth samples using a selected set of DNA methylation markers. Epigenetics 2015, 10, 922–930. [Google Scholar] [CrossRef]
- Giuliani, C.; Cilli, E.; Bacalini, M.G.; Pirazzini, C.; Sazzini, M.; Gruppioni, G.; Franceschi, C.; Garagnani, P.; Luiselli, D. Inferring chronological age from DNA methylation patterns of human teeth. Am. J. Phys. Anthropol. 2016, 159, 585–595. [Google Scholar] [CrossRef]
- Zapico, S.C.; Gauthier, Q.; Antevska, A.; McCord, B.R. Identifying Methylation Patterns in Dental Pulp Aging: Application to Age-at-Death Estimation in Forensic Anthropology. Int. J. Mol. Sci. 2021, 22, 3717. [Google Scholar] [CrossRef]
- Correia Dias, H.; Manco, L.; Corte Real, F.; Cunha, E. A Blood-Bone-Tooth Model for Age Prediction in Forensic Contexts. Biology 2021, 10, 1312. [Google Scholar] [CrossRef]
- Lee, H.Y.; Hong, S.R.; Lee, J.E.; Hwang, I.K.; Kim, N.Y.; Lee, J.M.; Fleckhaus, J.; Jung, S.E.; Lee, Y.H. Epigenetic age signatures in bones. Forensic Sci. Int. Genet. 2020, 46, 102261. [Google Scholar] [CrossRef]
- Woźniak, A.; Heidegger, A.; Piniewska-Róg, D.; Pośpiech, E.; Xavier, C.; Pisarek, A.; Kartasińska, E.; Boroń, M.; Freire-Aradas, A.; Wojtas, M.; et al. Development of the VISAGE enhanced tool and statistical models for epigenetic age estimation in blood, buccal cells and bones. Aging 2021, 13, 6459–6484. [Google Scholar] [CrossRef]
- Ambroa-Conde, A.; Girón-Santamaría, L.; Mosquera-Miguel, A.; Phillips, C.; Casares de Cal, M.A.; Gómez-Tato, A.; Álvarez-Dios, J.; de la Puente, M.; Ruiz-Ramírez, J.; Lareu, M.V.; et al. Epigenetic age estimation in saliva and in buccal cells. Forensic Sci. Int. Genet. 2022, 61, 102770. [Google Scholar] [CrossRef]
- Jung, S.E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef]
- Schwender, K.; Hollander, O.; Klopfleisch, S.; Eveslage, M.; Danzer, M.F.; Pfeiffer, H.; Vennemann, M. Development of two age estimation models for buccal swab samples based on 3 CpG sites analyzed with pyrosequencing and minisequencing. Forensic Sci. Int. Genet. 2021, 53, 102521. [Google Scholar] [CrossRef]
- Sukawutthiya, P.; Sathirapatya, T.; Vongpaisarnsin, K. A minimal number CpGs of ELOVL2 gene for a chronological age estimation using pyrosequencing. Forensic Sci. Int. 2021, 318, 110631. [Google Scholar] [CrossRef]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Makowska, Ż.; Spas, A.; Parys-Proszek, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Examination of DNA methylation status of the ELOVL2 marker may be useful for human age prediction in forensic science. Forensic Sci. Int. Genet. 2015, 14, 161–167. [Google Scholar] [CrossRef]
- Montesanto, A.; D’Aquila, P.; Lagani, V.; Paparazzo, E.; Geracitano, S.; Formentini, L.; Giacconi, R.; Cardelli, M.; Provinciali, M.; Bellizzi, D.; et al. A New Robust Epigenetic Model for Forensic Age Prediction. J. Forensic Sci. 2020, 65, 1424–1431. [Google Scholar] [CrossRef]
- Leontiou, C.A.; Hadjidaniel, M.D.; Mina, P.; Antoniou, P.; Ioannides, M.; Patsalis, P.C. Bisulfite Conversion of DNA: Performance Comparison of Different Kits and Methylation Quantitation of Epigenetic Biomarkers that Have the Potential to Be Used in Non-Invasive Prenatal Testing. PLoS ONE 2015, 10, e0135058. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Pajares, M.J.; Palanca-Ballester, C.; Urtasun, R.; Alemany-Cosme, E.; Lahoz, A.; Sandoval, J. Methods for analysis of specific DNA methylation status. Methods 2021, 187, 3–12. [Google Scholar] [CrossRef]
- Holmes, E.E.; Jung, M.; Meller, S.; Leisse, A.; Sailer, V.; Zech, J.; Mengdehl, M.; Garbe, L.A.; Uhl, B.; Kristiansen, G.; et al. Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS ONE 2014, 9, e93933. [Google Scholar] [CrossRef]
- Tierling, S.; Schmitt, B.; Walter, J. Comprehensive Evaluation of Commercial Bisulfite-Based DNA Methylation Kits and Development of an Alternative Protocol With Improved Conversion Performance. Genet. Epigenet. 2018, 10, 1179237X18766097. [Google Scholar] [CrossRef]
- Kint, S.; De Spiegelaere, W.; De Kesel, J.; Vandekerckhove, L.; Van Criekinge, W. Evaluation of bisulfite kits for DNA methylation profiling in terms of DNA fragmentation and DNA recovery using digital PCR. PLoS ONE 2018, 13, e0199091. [Google Scholar] [CrossRef]
- Correia Dias, H.; Corte-Real, F.; Cunha, E.; Manco, L. DNA methylation age estimation from human bone and teeth. Aust. J. Forensic Sci. 2022, 54, 163–176. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9. [Google Scholar] [CrossRef]
- Kondo, M.; Aboshi, H.; Yoshikawa, M.; Ogata, A.; Murayama, R.; Takei, M.; Aizawa, S. A newly developed age estimation method based on CpG methylation of teeth-derived DNA using real-time methylation-specific PCR. J. Oral. Sci. 2020, 63, 54–58. [Google Scholar] [CrossRef]
- Ogata, A.; Kondo, M.; Yoshikawa, M.; Okano, M.; Tsutsumi, T.; Aboshi, H. Dental age estimation based on DNA methylation using real-time methylation-specific PCR. Forensic Sci. Int. 2022, 340, 111445. [Google Scholar] [CrossRef]
- Hernández, H.G.; Tse, M.Y.; Pang, S.C.; Arboleda, H.; Forero, D.A. Optimizing methodologies for PCR-based DNA methylation analysis. Biotechniques 2013, 55, 181–197. [Google Scholar] [CrossRef]
- Wojdacz, T.K.; Dobrovic, A.; Hansen, L.L. Methylation-sensitive high-resolution melting. Nat. Protoc. 2008, 3, 1903–1908. [Google Scholar] [CrossRef]
- Wojdacz, T.K.; Hansen, L.L.; Dobrovic, A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res. Notes 2008, 1, 54. [Google Scholar] [CrossRef]
- Candiloro, I.L.M.; Mikeska, T.; Dobrovic, A. Assessing alternative base substitutions at primer CpG sites to optimise unbiased PCR amplification of methylated sequences. Clin. Epigenet. 2017, 9, 31. [Google Scholar] [CrossRef]
- Hamano, Y.; Manabe, S.; Morimoto, C.; Fujimoto, S.; Ozeki, M.; Tamaki, K. Forensic age prediction for dead or living samples by use of methylation-sensitive high resolution melting. Leg. Med. 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Hamano, Y.; Manabe, S.; Morimoto, C.; Fujimoto, S.; Tamaki, K. Forensic age prediction for saliva samples using methylation-sensitive high resolution melting: Exploratory application for cigarette butts. Sci. Rep. 2017, 7, 10444. [Google Scholar] [CrossRef]
- Alghanim, H.; Balamurugan, K.; McCord, B. Development of DNA methylation markers for sperm, saliva and blood identification using pyrosequencing and qPCR/HRM. Anal. Biochem. 2020, 611, 113933. [Google Scholar] [CrossRef]
- Oka, H.; Dwi Ariani, M.; Akazaki, T.; Miyauchi, M.; Kitagawa, M. Some tips on age estimation using DNA methylation in saliva samples as an index across the Japanese and Indonesian ethnicities. Leg. Med. 2022, 56, 102042. [Google Scholar] [CrossRef]
- Suchiman, H.E.; Slieker, R.C.; Kremer, D.; Slagboom, P.E.; Heijmans, B.T.; Tobi, E.W. Design, measurement and processing of region-specific DNA methylation assays: The mass spectrometry-based method EpiTYPER. Front. Genet. 2015, 6, 287. [Google Scholar] [CrossRef]
- Feng, L.; Peng, F.; Li, S.; Jiang, L.; Sun, H.; Ji, A.; Zeng, C.; Li, C.; Liu, F. Systematic feature selection improves accuracy of methylation-based forensic age estimation in Han Chinese males. Forensic Sci. Int. Genet. 2018, 35, 38–45. [Google Scholar] [CrossRef]
- Freire-Aradas, A.; Giron-Santamaria, L.; Mosquera-Miguel, A.; Ambroa-Conde, A.; Phillips, C.; Casares de Cal, M.; Gomez-Tato, A.; Alvarez-Dios, J.; Pospiech, E.; Aliferi, A.; et al. A common epigenetic clock from childhood to old age. Forensic Sci. Int. Genet. 2022, 60, 102743. [Google Scholar] [CrossRef]
- Freire-Aradas, A.; Phillips, C.; Girón-Santamaría, L.; Mosquera-Miguel, A.; Gómez-Tato, A.; Casares de Cal, M.Á.; Álvarez-Dios, J.; Lareu, M.V. Tracking age-correlated DNA methylation markers in the young. Forensic Sci. Int. Genet. 2018, 36, 50–59. [Google Scholar] [CrossRef]
- Harrison, A.; Parle-McDermott, A. DNA methylation: A timeline of methods and applications. Front. Genet. 2011, 2, 74. [Google Scholar] [CrossRef]
- So, M.H.; Lee, H.Y. Genetic analyzer-dependent DNA methylation detection and its application to existing age prediction models. Electrophoresis 2021, 42, 1497–1506. [Google Scholar] [CrossRef]
- Pan, C.; Yi, S.; Xiao, C.; Huang, Y.; Chen, X.; Huang, D. The evaluation of seven age-related CpGs for forensic purpose in blood from Chinese Han population. Forensic Sci. Int. Genet. 2020, 46, 102251. [Google Scholar] [CrossRef]
- Onofri, M.; Delicati, A.; Marcante, B.; Carlini, L.; Alessandrini, F.; Tozzo, P.; Carnevali, E. Forensic Age Estimation through a DNA Methylation-Based Age Prediction Model in the Italian Population: A Pilot Study. Int. J. Mol. Sci. 2023, 24, 5381. [Google Scholar] [CrossRef]
- Dias, H.C.; Cordeiro, C.; Pereira, J.; Pinto, C.; Real, F.C.; Cunha, E.; Manco, L. DNA methylation age estimation in blood samples of living and deceased individuals using a multiplex SNaPshot assay. Forensic Sci. Int. 2020, 311, 110267. [Google Scholar] [CrossRef]
- Hong, S.R.; Shin, K.-J.; Jung, S.-E.; Lee, E.H.; Lee, H.Y. Platform-independent models for age prediction using DNA methylation data. Forensic Sci. Int. Genet. 2019, 38, 39–47. [Google Scholar] [CrossRef]
- Lee, H.Y.; Jung, S.-E.; Oh, Y.N.; Choi, A.; Yang, W.I.; Shin, K.-J. Epigenetic age signatures in the forensically relevant body fluid of semen: A preliminary study. Forensic Sci. Int. Genet. 2015, 19, 28–34. [Google Scholar] [CrossRef]
- Reed, K.; Poulin, M.L.; Yan, L.; Parissenti, A.M. Comparison of bisulfite sequencing PCR with pyrosequencing for measuring differences in DNA methylation. Anal. Biochem. 2010, 397, 96–106. [Google Scholar] [CrossRef]
- Ghemrawi, M.; Tejero, N.F.; Duncan, G.; McCord, B. Pyrosequencing: Current forensic methodology and future applications-a review. Electrophoresis 2023, 44, 298–312. [Google Scholar] [CrossRef]
- Harrington, C.T.; Lin, E.I.; Olson, M.T.; Eshleman, J.R. Fundamentals of pyrosequencing. Arch. Pathol. Lab. Med. 2013, 137, 1296–1303. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Kayser, M.; Branicki, W.; Parson, W.; Phillips, C. Recent advances in Forensic DNA Phenotyping of appearance, ancestry and age. Forensic Sci. Int. Genet. 2023, 65, 102870. [Google Scholar] [CrossRef]
- Anaya, Y.; Yew, P.; Roberts, K.A.; Hardy, W.R. DNA methylation of decedent blood samples to estimate the chronological age of human remains. Int. J. Leg. Med. 2021, 135, 2163–2173. [Google Scholar] [CrossRef]
- Koop, B.E.; Mayer, F.; Gunduz, T.; Blum, J.; Becker, J.; Schaffrath, J.; Wagner, W.; Han, Y.; Boehme, P.; Ritz-Timme, S. Postmortem age estimation via DNA methylation analysis in buccal swabs from corpses in different stages of decomposition-a “proof of principle” study. Int. J. Leg. Med. 2021, 135, 167–173. [Google Scholar] [CrossRef]
- Li, L.; Song, F.; Lang, M.; Hou, J.; Wang, Z.; Prinz, M.; Hou, Y. Methylation-Based Age Prediction Using Pyrosequencing Platform from Seminal Stains in Han Chinese Males. J. Forensic Sci. 2020, 65, 610–619. [Google Scholar] [CrossRef]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; Van de Voorde, W.; Decorte, R. A selective set of DNA-methylation markers for age determination of blood, teeth and buccal samples. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e144–e145. [Google Scholar] [CrossRef]
- Márquez-Ruiz, A.B.; González-Herrera, L.; Luna, J.D.; Valenzuela, A. DNA methylation levels and telomere length in human teeth: Usefulness for age estimation. Int. J. Leg. Med. 2020, 134, 451–459. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Lau, P.Y.; Fung, W.K. Evaluation of marker selection methods and statistical models for chronological age prediction based on DNA methylation. Leg. Med. 2020, 47, 101744. [Google Scholar] [CrossRef]
- Lee, H.Y.; An, J.H.; Jung, S.E.; Oh, Y.N.; Lee, E.Y.; Choi, A.; Yang, W.I.; Shin, K.J. Genome-wide methylation profiling and a multiplex construction for the identification of body fluids using epigenetic markers. Forensic Sci. Int. Genet. 2015, 17, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Heidegger, A.; Pisarek, A.; de la Puente, M.; Niederstatter, H.; Pospiech, E.; Wozniak, A.; Schury, N.; Unterlander, M.; Sidstedt, M.; Junker, K.; et al. Development and inter-laboratory validation of the VISAGE enhanced tool for age estimation from semen using quantitative DNA methylation analysis. Forensic Sci. Int. Genet. 2022, 56, 102596. [Google Scholar] [CrossRef] [PubMed]
- Piniewska-Rog, D.; Heidegger, A.; Pospiech, E.; Xavier, C.; Pisarek, A.; Jarosz, A.; Wozniak, A.; Wojtas, M.; Phillips, C.; Kayser, M.; et al. Impact of excessive alcohol abuse on age prediction using the VISAGE enhanced tool for epigenetic age estimation in blood. Int. J. Leg. Med. 2021, 135, 2209–2219. [Google Scholar] [CrossRef]
- Pisarek, A.; Pośpiech, E.; Heidegger, A.; Xavier, C.; Papież, A.; Piniewska-Róg, D.; Kalamara, V.; Potabattula, R.; Bochenek, M.; Sikora-Polaczek, M.; et al. Epigenetic age prediction in semen–marker selection and model development. Aging 2021, 13, 19145–19164. [Google Scholar] [CrossRef] [PubMed]
- Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jerónimo, C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes 2018, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Manco, L.; Dias, H.C. DNA methylation analysis of ELOVL2 gene using droplet digital PCR for age estimation purposes. Forensic Sci. Int. 2022, 333, 111206. [Google Scholar] [CrossRef] [PubMed]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Ho Lee, M.; Hee Hwang, J.; Min Seong, K.; Jin Ahn, J.; Jun Kim, S.; Yong Hwang, S.; Lim, S.K. Application of droplet digital PCR method for DNA methylation-based age prediction from saliva. Leg. Med. 2022, 54, 101992. [Google Scholar] [CrossRef] [PubMed]
- Dias, H.C.; Manco, L. Predicting age from blood by droplet digital PCR using a set of three DNA methylation markers. Forensic Sci. Int. 2024, 356, 111950. [Google Scholar] [CrossRef]
- Shi, L.; Jiang, F.; Ouyang, F.; Zhang, J.; Wang, Z.; Shen, X. DNA methylation markers in combination with skeletal and dental ages to improve age estimation in children. Forensic Sci. Int. Genet. 2018, 33, 1–9. [Google Scholar] [CrossRef]
- Yu, M.; Heinzerling, T.J.; Grady, W.M. DNA Methylation Analysis Using Droplet Digital PCR. In Digital PCR: Methods and Protocols; Karlin-Neumann, G., Bizouarn, F., Eds.; Springer: New York, NY, USA, 2018; pp. 363–383. [Google Scholar]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Naue, J.; Lee, H.Y. Considerations for the need of recommendations for the research and publication of DNA methylation results. Forensic Sci. Int. Genet. 2018, 37, e12–e14. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Lee, J.M.; Naue, J.; Fleckhaus, J.; Freire-Aradas, A.; Neubauer, J.; Pośpiech, E.; McCord, B.; Kalamara, V.; Gauthier, Q.; et al. A collaborative exercise on DNA methylation-based age prediction and body fluid typing. Forensic Sci. Int. Genet. 2022, 57, 102656. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.M.; Wagner, W. Epigenetic-aging-signature to determine age in different tissues. Aging 2011, 3, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Koch, Z.; Ideker, T. Epigenetic aging: Biological age prediction and informing a mechanistic theory of aging. J. Intern. Med. 2022, 292, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wilson, R.; Heiss, J.; Breitling, L.P.; Saum, K.U.; Schöttker, B.; Holleczek, B.; Waldenberger, M.; Peters, A.; Brenner, H. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun. 2017, 8, 14617. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Belsky, D.W.; Moffitt, T.E.; Cohen, A.A.; Corcoran, D.L.; Levine, M.E.; Prinz, J.A.; Schaefer, J.; Sugden, K.; Williams, B.; Poulton, R.; et al. Eleven Telomere, Epigenetic Clock, and Biomarker-Composite Quantifications of Biological Aging: Do They Measure the Same Thing? Am. J. Epidemiol. 2018, 187, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, W.; Wang, T.; Ran, D.; Davalos, V.; Planas-Serra, L.; Pujol, A.; Esteller, M.; Wang, X.; Yu, H. Accelerated biological aging in COVID-19 patients. Nat. Commun. 2022, 13, 2135. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Kimura, R.; Otsuka, I.; Funabiki, Y.; Murai, T.; Hishimoto, A. Epigenetic clock analysis and increased plasminogen activator inhibitor-1 in high-functioning autism spectrum disorder. PLoS ONE 2022, 17, e0263478. [Google Scholar] [CrossRef] [PubMed]
- Protsenko, E.; Yang, R.; Nier, B.; Reus, V.; Hammamieh, R.; Rampersaud, R.; Wu, G.W.Y.; Hough, C.M.; Epel, E.; Prather, A.A.; et al. “GrimAge,” an epigenetic predictor of mortality, is accelerated in major depressive disorder. Transl. Psychiatry 2021, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Katrinli, S.; Stevens, J.; Wani, A.H.; Lori, A.; Kilaru, V.; van Rooij, S.J.H.; Hinrichs, R.; Powers, A.; Gillespie, C.F.; Michopoulos, V.; et al. Evaluating the impact of trauma and PTSD on epigenetic prediction of lifespan and neural integrity. Neuropsychopharmacology 2020, 45, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Binder, A.M.; Zhang, J.; Yan, Q.; Reiner, A.P.; Cox, S.R.; Corley, J.; Harris, S.E.; Kuo, P.L.; Moore, A.Z.; et al. DNA methylation GrimAge version 2. Aging 2022, 14, 9484–9549. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, J.; Liu, B.; Shi, J. The use of DNA methylation clock in aging research. Exp. Biol. Med. 2021, 246, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Belsky, D.W.; Caspi, A.; Arseneault, L.; Baccarelli, A.; Corcoran, D.L.; Gao, X.; Hannon, E.; Harrington, H.L.; Rasmussen, L.J.H.; Houts, R.; et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. eLife 2020, 9, e54870. [Google Scholar] [CrossRef]
- Belsky, D.W.; Caspi, A.; Corcoran, D.L.; Sugden, K.; Poulton, R.; Arseneault, L.; Baccarelli, A.; Chamarti, K.; Gao, X.; Hannon, E.; et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. eLife 2022, 11, e73420. [Google Scholar] [CrossRef]
- Dobberstein, R.C.; Huppertz, J.; von Wurmb-Schwark, N.; Ritz-Timme, S. Degradation of biomolecules in artificially and naturally aged teeth: Implications for age estimation based on aspartic acid racemization and DNA analysis. Forensic Sci. Int. 2008, 179, 181–191. [Google Scholar] [CrossRef]
- Ubelaker, D.H.; Khosrowshahi, H. Estimation of age in forensic anthropology: Historical perspective and recent methodological advances. Forensic Sci. Res. 2019, 4, 1–9. [Google Scholar] [CrossRef]
- Zapico, S.C.; Ubelaker, D.H. Applications of physiological bases of ageing to forensic sciences. Estimation of age-at-death. Ageing Res. Rev. 2013, 12, 605–617. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ohtani, S. Estimation of chronological age from the racemization rate of L- and D-aspartic acid: How to completely separate enantiomers from dentin. Methods Mol. Biol. 2012, 794, 265–272. [Google Scholar] [CrossRef]
- Paton, B.; Suarez, M.; Herrero, P.; Canela, N. Glycosylation Biomarkers Associated with Age-Related Diseases and Current Methods for Glycan Analysis. Int. J. Mol. Sci. 2021, 22, 5788. [Google Scholar] [CrossRef]
- Zubakov, D.; Liu, F.; Kokmeijer, I.; Choi, Y.; van Meurs, J.B.J.; van IJcken, W.F.; Uitterlinden, A.G.; Hofman, A.; Broer, L.; van Duijn, C.M.; et al. Human age estimation from blood using mRNA, DNA methylation, DNA rearrangement, and telomere length. Forensic Sci. Int. Genet. 2016, 24, 33–43. [Google Scholar] [CrossRef]
- Amorim, A.; Fernandes, T.; Taveira, N. Mitochondrial DNA in human identification: A review. PeerJ 2019, 7, e7314. [Google Scholar] [CrossRef]
- Syndercombe Court, D. Mitochondrial DNA in forensic use. Emerg. Top. Life Sci. 2021, 5, 415–426. [Google Scholar] [CrossRef]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage-how and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef]
- Cunha, E.; Baccino, E.; Martrille, L.; Ramsthaler, F.; Prieto, J.; Schuliar, Y.; Lynnerup, N.; Cattaneo, C. The problem of aging human remains and living individuals: A review. Forensic Sci. Int. 2009, 193, 1–13. [Google Scholar] [CrossRef]
- Gopalan, S.; Gaige, J.; Henn, B.M. DNA methylation-based forensic age estimation in human bone. bioRxiv 2019, 801647. [Google Scholar] [CrossRef]
- Franceschetti, L.; Merelli, V.G.; Corona, S.; Magli, F.; Maggioni, L.; Cummaudo, M.; Tritella, S.; De Angelis, D.; Cattaneo, C. Analysis of interrater reliability in age assessment of minors: How does expertise influence the evaluation? Int. J. Leg. Med. 2022, 136, 279–285. [Google Scholar] [CrossRef]
- Cummaudo, M.; Obertova, Z.; Lynnerup, N.; Petaros, A.; de Boer, H.; Baccino, E.; Steyn, M.; Cunha, E.; Ross, A.; Adalian, P.; et al. Age assessment in unaccompanied minors: Assessing uniformity of protocols across Europe. Int. J. Leg. Med. 2024, 138, 983–995. [Google Scholar] [CrossRef]
- McEwen, L.M.; O’Donnell, K.J.; McGill, M.G.; Edgar, R.D.; Jones, M.J.; MacIsaac, J.L.; Lin, D.T.S.; Ramadori, K.; Morin, A.; Gladish, N.; et al. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc. Natl. Acad. Sci. USA 2020, 117, 23329–23335. [Google Scholar] [CrossRef]
- Dammering, F.; Martins, J.; Dittrich, K.; Czamara, D.; Rex-Haffner, M.; Overfeld, J.; de Punder, K.; Buss, C.; Entringer, S.; Winter, S.M.; et al. The pediatric buccal epigenetic clock identifies significant ageing acceleration in children with internalizing disorder and maltreatment exposure. Neurobiol. Stress 2021, 15, 100394. [Google Scholar] [CrossRef]
- Kling, T.; Wenger, A.; Carén, H. DNA methylation-based age estimation in pediatric healthy tissues and brain tumors. Aging 2020, 12, 21037–21056. [Google Scholar] [CrossRef]
- Heidegger, A.; Xavier, C.; Niederstätter, H.; de la Puente, M.; Pośpiech, E.; Pisarek, A.; Kayser, M.; Branicki, W.; Parson, W. Development and optimization of the VISAGE basic prototype tool for forensic age estimation. Forensic Sci. Int. Genet. 2020, 48, 102322. [Google Scholar] [CrossRef]
- Peng, F.; Feng, L.; Chen, J.; Wang, L.; Li, P.; Ji, A.; Zeng, C.; Liu, F.; Li, C. Validation of methylation-based forensic age estimation in time-series bloodstains on FTA cards and gauze at room temperature conditions. Forensic Sci. Int. Genet. 2019, 40, 168–174. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, J.; Hou, J.; Fu, X.; Li, L.; Hou, Y. Developing a DNA methylation assay for human age prediction in blood and bloodstain. Forensic Sci. Int. Genet. 2015, 17, 129–136. [Google Scholar] [CrossRef]
- Correia Dias, H.; Cordeiro, C.; Corte Real, F.; Cunha, E.; Manco, L. Age Estimation Based on DNA Methylation Using Blood Samples From Deceased Individuals. J. Forensic Sci. 2020, 65, 465–470. [Google Scholar] [CrossRef]
- Kayser, M. Forensic use of Y-chromosome DNA: A general overview. Human. Genet. 2017, 136, 621–635. [Google Scholar] [CrossRef]
- Vidaki, A.; Montiel González, D.; Planterose Jiménez, B.; Kayser, M. Male-specific age estimation based on Y-chromosomal DNA methylation. Aging 2021, 13, 6442–6458. [Google Scholar] [CrossRef]
- Lund, J.B.; Li, S.; Baumbach, J.; Svane, A.M.; Hjelmborg, J.; Christiansen, L.; Christensen, K.; Redmond, P.; Marioni, R.E.; Deary, I.J.; et al. DNA methylome profiling of all-cause mortality in comparison with age-associated methylation patterns. Clin. Epigenet. 2019, 11, 23. [Google Scholar] [CrossRef]
- Lund, J.B.; Li, S.; Christensen, K.; Mengel-From, J.; Soerensen, M.; Marioni, R.E.; Starr, J.; Pattie, A.; Deary, I.J.; Baumbach, J.; et al. Age-dependent DNA methylation patterns on the Y chromosome in elderly males. Aging Cell 2020, 19, e12907. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, K.; Wei, X.; Li, J.; Wang, S.; Wang, Z.; Zhou, Y.; Zha, L.; Luo, H.; Song, F. Developing a male-specific age predictive model based on Y-CpGs for forensic analysis. Forensic Sci. Int. 2023, 343, 111566. [Google Scholar] [CrossRef]
- Kotková, L.; Drábek, J. Age-related changes in sperm DNA methylation and their forensic and clinical implications. Epigenomics 2023, 15, 1157–1173. [Google Scholar] [CrossRef]
- Jenkins, T.G.; Aston, K.I.; Cairns, B.R.; Carrell, D.T. Paternal aging and associated intraindividual alterations of global sperm 5-methylcytosine and 5-hydroxymethylcytosine levels. Fertil. Steril. 2013, 100, 945–951. [Google Scholar] [CrossRef]
- Jenkins, T.G.; Aston, K.I.; Pflueger, C.; Cairns, B.R.; Carrell, D.T. Age-associated sperm DNA methylation alterations: Possible implications in offspring disease susceptibility. PLoS Genet. 2014, 10, e1004458. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, H.; Zhou, H.; Gu, Y.; Bai, Y.; Yu, S.; An, R.; Qi, J. Identification of potential key genes and high-frequency mutant genes in prostate cancer by using RNA-Seq data. Oncol. Lett. 2018, 15, 4550–4556. [Google Scholar] [CrossRef]
- Lee, J.W.; Choung, C.M.; Jung, J.Y.; Lee, H.Y.; Lim, S.K. A validation study of DNA methylation-based age prediction using semen in forensic casework samples. Leg. Med. 2018, 31, 74–77. [Google Scholar] [CrossRef]
- Jenkins, T.G.; Aston, K.I.; Cairns, B.; Smith, A.; Carrell, D.T. Paternal germ line aging: DNA methylation age prediction from human sperm. BMC Genom. 2018, 19, 763. [Google Scholar] [CrossRef]
- Jenkins, T.G.; James, E.R.; Alonso, D.F.; Hoidal, J.R.; Murphy, P.J.; Hotaling, J.M.; Cairns, B.R.; Carrell, D.T.; Aston, K.I. Cigarette smoking significantly alters sperm DNA methylation patterns. Andrology 2017, 5, 1089–1099. [Google Scholar] [CrossRef]
- Aston, K.I.; Uren, P.J.; Jenkins, T.G.; Horsager, A.; Cairns, B.R.; Smith, A.D.; Carrell, D.T. Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil. Steril. 2015, 104, 1388–1397.e5. [Google Scholar] [CrossRef]
- Pilsner, J.R.; Saddiki, H.; Whitcomb, B.W.; Suvorov, A.; Buck Louis, G.M.; Mumford, S.L.; Schisterman, E.F.; Oluwayiose, O.A.; Balzer, L.B. Sperm epigenetic clock associates with pregnancy outcomes in the general population. Hum. Reprod. 2022, 37, 1581–1593. [Google Scholar] [CrossRef]
- Laurentino, S.; Cremers, J.F.; Horsthemke, B.; Tüttelmann, F.; Czeloth, K.; Zitzmann, M.; Pohl, E.; Rahmann, S.; Schröder, C.; Berres, S.; et al. A germ cell-specific ageing pattern in otherwise healthy men. Aging Cell 2020, 19, e13242. [Google Scholar] [CrossRef]
- Theda, C.; Hwang, S.H.; Czajko, A.; Loke, Y.J.; Leong, P.; Craig, J.M. Quantitation of the cellular content of saliva and buccal swab samples. Sci. Rep. 2018, 8, 6944. [Google Scholar] [CrossRef]
- Eipel, M.; Mayer, F.; Arent, T.; Ferreira, M.R.; Birkhofer, C.; Gerstenmaier, U.; Costa, I.G.; Ritz-Timme, S.; Wagner, W. Epigenetic age predictions based on buccal swabs are more precise in combination with cell type-specific DNA methylation signatures. Aging 2016, 8, 1034–1048. [Google Scholar] [CrossRef]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef]
- Alsaleh, H.; McCallum, N.A.; Halligan, D.L.; Haddrill, P.R. A multi-tissue age prediction model based on DNA methylation analysis. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e62–e64. [Google Scholar] [CrossRef]
- Naue, J.; Hoefsloot, H.C.J.; Mook, O.R.F.; Rijlaarsdam-Hoekstra, L.; van der Zwalm, M.C.H.; Henneman, P.; Kloosterman, A.D.; Verschure, P.J. Chronological age prediction based on DNA methylation: Massive parallel sequencing and random forest regression. Forensic Sci. Int. Genet. 2017, 31, 19–28. [Google Scholar] [CrossRef]
- Alghanim, H.; Wu, W.; McCord, B. DNA methylation assay based on pyrosequencing for determination of smoking status. Electrophoresis 2018, 39, 2806–2814. [Google Scholar] [CrossRef]
- Koop, B.E.; Reckert, A.; Becker, J.; Han, Y.; Wagner, W.; Ritz-Timme, S. Epigenetic clocks may come out of rhythm—Implications for the estimation of chronological age in forensic casework. Int. J. Leg. Med. 2020, 134, 2215–2228. [Google Scholar] [CrossRef]
- Horvath, S.; Levine, A.J. HIV-1 Infection Accelerates Age According to the Epigenetic Clock. J. Infect. Dis. 2015, 212, 1563–1573. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y.; Brenner, H. Associations of Helicobacter pylori infection and chronic atrophic gastritis with accelerated epigenetic ageing in older adults. Br. J. Cancer 2017, 117, 1211–1214. [Google Scholar] [CrossRef]
- Kananen, L.; Nevalainen, T.; Jylhävä, J.; Marttila, S.; Hervonen, A.; Jylhä, M.; Hurme, M. Cytomegalovirus infection accelerates epigenetic aging. Exp. Gerontol. 2015, 72, 227–229. [Google Scholar] [CrossRef]
- Attia, M.H. A cautionary note on altered pace of aging in the COVID-19 era. Forensic Sci. Int. Genet. 2022, 59, 102724. [Google Scholar] [CrossRef]
- Vidaki, A.; Planterose Jimenez, B.; Poggiali, B.; Kalamara, V.; van der Gaag, K.J.; Maas, S.C.E.; Consortium, B.I.O.S.; Ghanbari, M.; Sijen, T.; Kayser, M. Targeted DNA methylation analysis and prediction of smoking habits in blood based on massively parallel sequencing. Forensic Sci. Int. Genet. 2023, 65, 102878. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, J.; Mao, Y.; Li, M.D. Investigation of the genetic effect of 56 tobacco-smoking susceptibility genes on DNA methylation and RNA expression in human brain. Front. Psychiatry 2022, 13, 924062. [Google Scholar] [CrossRef]
- Maas, S.C.E.; Vidaki, A.; Wilson, R.; Teumer, A.; Liu, F.; van Meurs, J.B.J.; Uitterlinden, A.G.; Boomsma, D.I.; de Geus, E.J.C.; Willemsen, G.; et al. Validated inference of smoking habits from blood with a finite DNA methylation marker set. Eur. J. Epidemiol. 2019, 34, 1055–1074. [Google Scholar] [CrossRef]
- Spólnicka, M.; Pośpiech, E.; Adamczyk, J.G.; Freire-Aradas, A.; Pepłońska, B.; Zbieć-Piekarska, R.; Makowska, Ż.; Pięta, A.; Lareu, M.V.; Phillips, C.; et al. Modified aging of elite athletes revealed by analysis of epigenetic age markers. Aging 2018, 10, 241–252. [Google Scholar] [CrossRef]
- Carroll, J.E.; Irwin, M.R.; Levine, M.; Seeman, T.E.; Absher, D.; Assimes, T.; Horvath, S. Epigenetic Aging and Immune Senescence in Women With Insomnia Symptoms: Findings From the Women’s Health Initiative Study. Biol. Psychiatry 2017, 81, 136–144. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; Kresovich, J.K.; Xu, Z.; Sandler, D.P.; Taylor, J.A. Shift work, DNA methylation and epigenetic age. Int. J. Epidemiol. 2019, 48, 1536–1544. [Google Scholar] [CrossRef]
- Fokias, K.; Dierckx, L.; Van de Voorde, W.; Bekaert, B. Age determination through DNA methylation patterns in fingernails and toenails. Forensic Sci. Int. Genet. 2023, 64, 102846. [Google Scholar] [CrossRef]
- Hao, T.; Guo, J.; Liu, J.; Wang, J.; Liu, Z.; Cheng, X.; Li, J.; Ren, J.; Li, Z.; Yan, J.; et al. Predicting human age by detecting DNA methylation status in hair. Electrophoresis 2021, 42, 1255–1261. [Google Scholar] [CrossRef]
- Gomaa, R.; Nader, L.; Jamal, J. Application of DNA methylation-based markers in identification of mixed body fluid evidences simulating crime scene scenarios. Egypt. J. Forensic Sci. 2021, 11, 12. [Google Scholar] [CrossRef]
- Jung, S.-E.; Cho, S.; Antunes, J.; Gomes, I.; Uchimoto, M.L.; Oh, Y.N.; Di Giacomo, L.; Schneider, P.M.; Park, M.S.; van der Meer, D.; et al. A collaborative exercise on DNA methylation based body fluid typing. Electrophoresis 2016, 37, 2759–2766. [Google Scholar] [CrossRef]
- Fleischer, J.G.; Schulte, R.; Tsai, H.H.; Tyagi, S.; Ibarra, A.; Shokhirev, M.N.; Huang, L.; Hetzer, M.W.; Navlakha, S. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 2018, 19, 221. [Google Scholar] [CrossRef]
- Lehallier, B.; Gate, D.; Schaum, N.; Nanasi, T.; Lee, S.E.; Yousef, H.; Moran Losada, P.; Berdnik, D.; Keller, A.; Verghese, J.; et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 2019, 25, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Greto, V.L.; Cvetko, A.; Štambuk, T.; Dempster, N.J.; Kifer, D.; Deriš, H.; Cindrić, A.; Vučković, F.; Falchi, M.; Gillies, R.S.; et al. Extensive weight loss reduces glycan age by altering IgG N-glycosylation. Int. J. Obes. 2021, 45, 1521–1531. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sample Type | Method | Accuracy in Years (Validation/Testing Set) | Authors |
|---|---|---|---|
| (1) Saliva | MS-HRM | MAD 6.25 | [84] |
| SNaPshot | MAE 3.13 | [37] | |
| MAE 3.66 | [63] | ||
| MAD 4.29 | [64] | ||
| SNaPshot/NGS | MAD 3.19 (platform independent) | [96] | |
| ddPCR | MAD 3.3 | [118] | |
| (1) Buccal Swabs | SNaPshot | MAD 3.55 | [64] |
| MAD 6.44 | [65] | ||
| Pyrosequencing | MAD 5.33 | [65] | |
| MAD 5.09–7.03 | [178] | ||
| NGS | MAE 3.7 | [62] | |
| (1) Cigarette Butts | MS-HRM | MAD 7.65 | [84] |
| (2) Semen | SNaPshot | MAD 5.4 | [97] |
| MAD 4.8–5.2 | [171] | ||
| Pyrosequencing | MAD 3.8–4.3 | [106] | |
| NGS | MAE 5.1 | [114] | |
| MAE 2.4 | [172] | ||
| (3) Teeth | Pyrosequencing | MAD 4.86 (dentin) | [57] |
| MAE 1.5–2.13 (pulp) | [59] | ||
| EpiTYPER | MAD 1.2–7.1 (dentin, pulp, cementum) | [58] | |
| Sanger sequencing | MAD 11.35 | [75] | |
| RT-MSP | MAD 8.94 | [77] | |
| MAE 6.69–8.28 | [78] | ||
| SNaPshot | MAD 7.1 | [75] | |
| Pyrosequencing | MAE 4.8–6.9 | [108] | |
| (3) Bones | Sanger sequencing | MAD 2.56 | [75] |
| SNaPshot | MAD 7.2 | [75] | |
| NGS | MAE 3.4 | [62] | |
| (4) Blood | EpiTYPER | MAD 2.49 | [88] |
| SNaPshot | MAD 3.48 | [64] | |
| MAD 5.56 | [93] | ||
| MAD 3.01 | [94] | ||
| Pyrosequencing | MAD 4.5 | [15] | |
| MAD 5.75 | [67] | ||
| SE 3.9 | [18] | ||
| MAD 3.29 | [54] | ||
| NGS | MAD 3.76 (statistical analysis from array data) | [110] | |
| MAE 3.2 | [158] | ||
| MAE 3.2 | [62] | ||
| MAD 2.8–2.93 (Bloodstains) | [56] | ||
| (4) Postmortem Blood | Sanger sequencing | MAD 8.84 (Deceased individuals) | [161] |
| MS-HRM | MAD 7.71 (Living and deceased individuals) | [83] | |
| SNaPshot | MAD 4.25 (Living individuals) | [95] | |
| MAD 5.36 (Deceased individuals) | |||
| MAD 4.97 (Living and deceased individuals) | |||
| Pyrosequencing | MAD 3.75 (Living and deceased individuals) | [57] | |
| MAD 7.42 | [104] | ||
| NGS | MAE 3.1 (Deceased individuals) | [62] | |
| (4) Blood (ChrY) | SNaPshot | MAD 5.73 | [166] |
| NGS | MAE 7.54–8.46 | [163] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castagnola, M.J.; Medina-Paz, F.; Zapico, S.C. Uncovering Forensic Evidence: A Path to Age Estimation through DNA Methylation. Int. J. Mol. Sci. 2024, 25, 4917. https://doi.org/10.3390/ijms25094917
Castagnola MJ, Medina-Paz F, Zapico SC. Uncovering Forensic Evidence: A Path to Age Estimation through DNA Methylation. International Journal of Molecular Sciences. 2024; 25(9):4917. https://doi.org/10.3390/ijms25094917
Chicago/Turabian StyleCastagnola, María Josefina, Francisco Medina-Paz, and Sara C. Zapico. 2024. "Uncovering Forensic Evidence: A Path to Age Estimation through DNA Methylation" International Journal of Molecular Sciences 25, no. 9: 4917. https://doi.org/10.3390/ijms25094917
APA StyleCastagnola, M. J., Medina-Paz, F., & Zapico, S. C. (2024). Uncovering Forensic Evidence: A Path to Age Estimation through DNA Methylation. International Journal of Molecular Sciences, 25(9), 4917. https://doi.org/10.3390/ijms25094917