
TGFB2 Gene Methylation in Tumors with Low CD8+ T-Cell Infiltration Drives Positive Prognostic Overall Survival Responses in Pancreatic Ductal Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Positive Prognostic Impact of High TGFB2/3 and IFI27, and Negative Prognostic Impact of TGFB1 Gene Methylation Levels on Overall Survival (OS) in Adult PDAC Patients
2.2. TGFB1/2/3, and IFI27 Gene Methylation OS Impacts on PDAC Patients, Controlling for Age and Sex
2.3. Identification of Prognostic Marker Genes from Enriched Reactome Pathways Correlating mRNA Levels with TGFB1/2/3 and IFI27 Gene Methylations
2.4. Low Levels of Marker Gene mRNA Expression Amplify the Positive Prognostic OS Impact of TGFB2 Gene Methylation
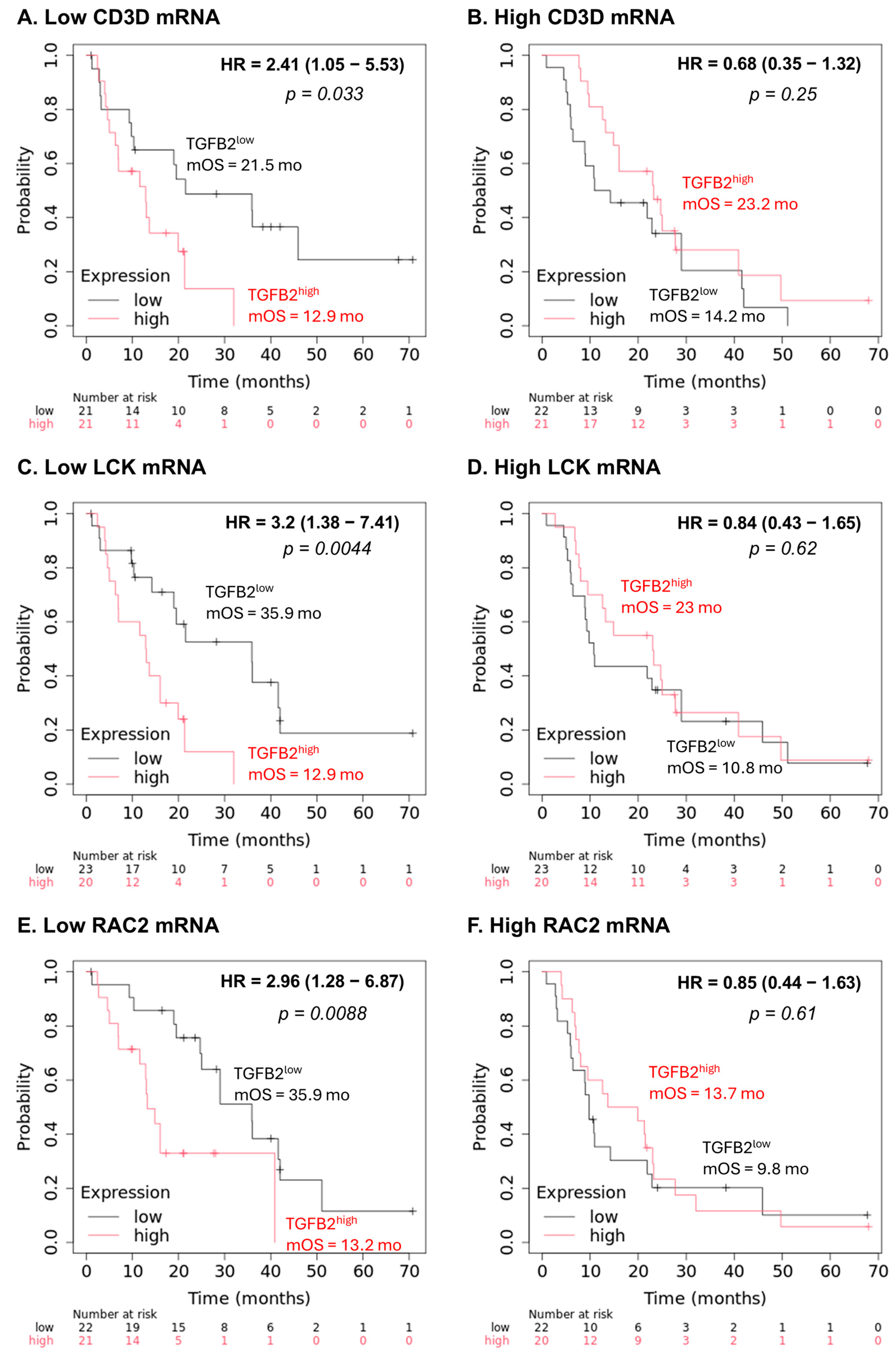
2.5. Low Levels of Marker Gene mRNA Expression Amplify the Negative Prognostic OS Impact of TGFB2 mRNA Expression
3. Discussion
3.1. Reactome Pathways Correlated to TGFB2 Gene Methylation
3.2. Reactome Pathways Correlated to IFI27 Gene Methylation
3.3. Potential T-Cell-Associated Biomarkers Amplify the Impact of TGFB2 Gene Methylation
4. Materials and Methods
4.1. Domain-Specific Identification of PubMed Articles Augmented by Artificial Intelligence
4.2. Demethylation Impact of TGFB1/2/3 and IFI27 Genes on Overall Survival (OS) for Pancreatic Adenocarcinoma Patients (PDAC)
4.3. Hazard Ratio (HR) Comparisons for PDAC Patients to Determine the Independent Effects of High Levels of TGFB1/2/3, IFI27 Gene Methylation Levels, Age, Sex
4.4. Geneset Enrichment Analysis (GSEA) in Reactome Pathways to Identify Negatively Correlated Pathways to TGB1/2/3 and IFI27 Methylation Levels
4.5. Differential Expression of mRNA Comparing PDAC Tumors Versus Normal Pancreatic Tissue Samples
4.6. Using Multivariate Cox Proportional Hazards Models, TGFB2 Gene Methylation Dependency on Marker Gene mRNA
4.7. Marker Gene mRNA Expression Correlation with T-Cell Infiltration into PDAC Tumors
4.8. Confirmation of the Prognostic Impact of Marker Genes Identified from the TCGA Dataset
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Feng, Z.; Miao, R.; Liu, X.; Liu, C.; Liu, Z. Prognosis and Survival Analysis of Patients with Pancreatic Cancer: Retrospective Experience of a Single Institution. World J. Surg. Oncol. 2022, 20, 11. [Google Scholar] [CrossRef]
- Bratlie, S.O.; Wennerblom, J.; Vilhav, C.; Persson, J.; Rangelova, E. Resectable, Borderline, and Locally Advanced Pancreatic Cancer—“the Good, the Bad, and the Ugly” Candidates for Surgery? J. Gastrointest. Oncol. 2021, 12, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Gbolahan, O.B.; Tong, Y.; Sehdev, A.; O’Neil, B.; Shahda, S. Overall Survival of Patients with Recurrent Pancreatic Cancer Treated with Systemic Therapy: A Retrospective Study. BMC Cancer 2019, 19, 468. [Google Scholar] [CrossRef]
- Liaki, V.; Rosas-Perez, B.; Guerra, C. Unlocking the Genetic Secrets of Pancreatic Cancer: KRAS Allelic Imbalances in Tumor Evolution. Cancers 2025, 17, 1226. [Google Scholar] [CrossRef]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From State-of-the-Art Treatments to Novel Therapies for Advanced-Stage Pancreatic Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Di Costanzo, F.; Di Costanzo, F.; Antonuzzo, L.; Mazza, E.; Giommoni, E. Optimizing First-Line Chemotherapy in Metastatic Pancreatic Cancer: Efficacy of FOLFIRINOX versus Nab-Paclitaxel Plus Gemcitabine. Cancers 2023, 15, 416. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Melisi, D.; Macarulla, T.; Pazo Cid, R.; Chandana, S.R.; De La Fouchardière, C.; Dean, A.; Kiss, I.; Lee, W.J.; Goetze, T.O.; et al. NALIRIFOX versus Nab-Paclitaxel and Gemcitabine in Treatment-Naive Patients with Metastatic Pancreatic Ductal Adenocarcinoma (NAPOLI 3): A Randomised, Open-Label, Phase 3 Trial. Lancet 2023, 402, 1272–1281. [Google Scholar] [CrossRef]
- Kang, H.; Jo, J.H.; Lee, H.S.; Chung, M.J.; Bang, S.; Park, S.W.; Song, S.Y.; Park, J.Y. Comparison of Efficacy and Safety between Standard-Dose and Modified-Dose FOLFIRINOX as a First-Line Treatment of Pancreatic Cancer. World J. Gastrointest. Oncol. 2018, 10, 421–430. [Google Scholar] [CrossRef]
- Stein, S.M.; James, E.S.; Deng, Y.; Cong, X.; Kortmansky, J.S.; Li, J.; Staugaard, C.; Indukala, D.; Boustani, A.M.; Patel, V.; et al. Final Analysis of a Phase II Study of Modified FOLFIRINOX in Locally Advanced and Metastatic Pancreatic Cancer. Br. J. Cancer 2016, 114, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; O’Reilly, E.M. Therapeutic Developments in Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 7–24. [Google Scholar] [CrossRef]
- Turpin, A.; Neuzillet, C.; Colle, E.; Dusetti, N.; Nicolle, R.; Cros, J.; de Mestier, L.; Bachet, J.-B.; Hammel, P. Therapeutic Advances in Metastatic Pancreatic Cancer: A Focus on Targeted Therapies. Ther. Adv. Med. Oncol. 2022, 14, 17588359221118019. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Q.; Castaneda, C.; Cook, S. Targeted Therapies in Pancreatic Cancer: A New Era of Precision Medicine. Biomedicines 2024, 12, 2175. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Korc, M.; Kamath, S.D.; Munshi, H.G.; Rana, A. Trials and Tribulations of Pancreatic Cancer Immunotherapy. Cancer Lett. 2021, 504, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational Advances in Pancreatic Ductal Adenocarcinoma Therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Jacob, R.; Manne, U.; Paluri, R. Advances in Pancreatic Cancer Biomarkers. Oncol. Rev. 2019, 13, 410. [Google Scholar] [CrossRef]
- Yuan, J.; Xu, B.; Su, Y.; Zhang, P.; Zhang, X.; Gong, P. Identification of USP39 as a Prognostic and Predictive Biomarker for Determining the Response to Immunotherapy in Pancreatic Cancer. BMC Cancer 2025, 25, 758. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhou, Q.; Sun, Y.; Lai, F.; Wang, Z.; Hao, Z.; Li, G. MethMarkerDB: A Comprehensive Cancer DNA Methylation Biomarker Database. Nucleic Acids Res. 2024, 52, D1380–D1392. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, Y. Comparison of the Classifiers Based on MRNA, MicroRNA and LncRNA Expression and DNA Methylation Profiles for the Tumor Origin Detection. Front. Genet. 2024, 15, 1383852. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Lin, X.; Cai, L.; Wang, Y.; Chang, Z. Classification and Prognosis Analysis of Pancreatic Cancer Based on DNA Methylation Profile and Clinical Information. Genes 2022, 13, 1913. [Google Scholar] [CrossRef]
- Yang, J.; Tang, Y.; Wang, J.; Yu, C.; Li, H.; Yi, B.; Li, Y.; Zhou, J.; Zhang, Z. Development and Validation of a Tissue-Based DNA Methylation Risk-Score Model to Predict the Prognosis of Surgically Resected Pancreatic Cancer Patients. Gland. Surg. 2022, 11, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Rashid, S.; Rashid, S.; Dash, N.R.; Gupta, S.; Saraya, A. Clinical Significance of Promoter Methylation Status of Tumor Suppressor Genes in Circulating DNA of Pancreatic Cancer Patients. J. Cancer Res. Clin. Oncol. 2020, 146, 897–907. [Google Scholar] [CrossRef]
- Gu, M.; Sun, J.; Zhang, S.; Chen, J.; Wang, G.; Ju, S.; Wang, X. A Novel Methylation Signature Predicts Inferior Outcome of Patients with PDAC. Aging 2021, 13, 2851–2863. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, R.; Sun, W.; Wang, J.; Liu, J. Analysis of Methylation-driven Genes in Pancreatic Ductal Adenocarcinoma for Predicting Prognosis. J. Cancer 2021, 12, 6507–6518. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Fujimoto, M.; Ito, N.; Takahashi, Y.; Kitago, M.; Gotoh, M.; Hiraoka, N.; Yoshida, T.; Kitagawa, Y.; Kanai, Y.; et al. Clinicopathological Impacts of DNA Methylation Alterations on Pancreatic Ductal Adenocarcinoma: Prediction of Early Recurrence Based on Genome-Wide DNA Methylation Profiling. J. Cancer Res. Clin. Oncol. 2021, 147, 1341–1354. [Google Scholar] [CrossRef]
- Qazi, S.; Potts, M.; Myers, S.; Richardson, S.; Trieu, V. Positive Prognostic Overall Survival Impacts of Methylated TGFB2 and MGMT in Adult Glioblastoma Patients. Cancers 2025, 17, 1122. [Google Scholar] [CrossRef]
- Moo-Young, T.A.; Larson, J.W.; Belt, B.A.; Tan, M.C.; Hawkins, W.G.; Eberlein, T.J.; Goedegebuure, P.S.; Linehan, D.C. Tumor-Derived TGF-β Mediates Conversion of CD4+Foxp3+ Regulatory T Cells in a Murine Model of Pancreas Cancer. J. Immunother. 2009, 32, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Timbers, K.E.; Atia, L.G.; Koch, R.M.; Rana, A. TGFβ Signaling in the Pancreatic Tumor Microenvironment. Cancers 2021, 13, 5086. [Google Scholar] [CrossRef]
- Birnbaum, D.J.; Mamessier, E.; Birnbaum, D. The Emerging Role of the TGFβ Tumor Suppressor Pathway in Pancreatic Cancer. Cell Cycle 2012, 11, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.D.; Pillarisetty, V.G. T-Cell Programming in Pancreatic Adenocarcinoma: A Review. Cancer Gene Ther. 2017, 24, 106–113. [Google Scholar] [CrossRef]
- von Bernstorff, W.; Voss, M.; Freichel, S.; Schmid, A.; Vogel, I.; Jöhnk, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Systemic and Local Immunosuppression in Pancreatic Cancer Patients. Clin. Cancer Res. 2001, 7, 925s–932s. [Google Scholar] [PubMed]
- Mukherjee, P.; Ginardi, A.R.; Madsen, C.S.; Tinder, T.L.; Jacobs, F.; Parker, J.; Agrawal, B.; Longenecker, B.M.; Gendler, S.J. MUC1-Specific CTLs Are Non-Functional within a Pancreatic Tumor Microenvironment. Glycoconj. J. 2001, 18, 931–942. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-Cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune Therapies in Pancreatic Ductal Adenocarcinoma: Where Are We Now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef]
- Qazi, S.; Trieu, V. TGFB2 MRNA Levels Prognostically Interact with Interferon-Alpha Receptor Activation of IRF9 and IFI27, and an Immune Checkpoint LGALS9 to Impact Overall Survival in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2024, 25, 11221. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The Role of TGF-β/SMAD4 Signaling in Cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Ma, M.-J.; Shi, Y.-H.; Liu, Z.-D.; Zhu, Y.-Q.; Zhao, G.-Y.; Ye, J.-Y.; Li, F.-X.; Huang, X.-T.; Wang, X.-Y.; Wang, J.-Q.; et al. N6-Methyladenosine Modified TGFB2 Triggers Lipid Metabolism Reprogramming to Confer Pancreatic Ductal Adenocarcinoma Gemcitabine Resistance. Oncogene 2024, 43, 2405–2420. [Google Scholar] [CrossRef] [PubMed]
- Porciello, N.; Franzese, O.; D’Ambrosio, L.; Palermo, B.; Nisticò, P. T-Cell Repertoire Diversity: Friend or Foe for Protective Antitumor Response? J. Exp. Clin. Cancer Res. 2022, 41, 356. [Google Scholar] [CrossRef] [PubMed]
- Javadrashid, D.; Baghbanzadeh, A.; Derakhshani, A.; Leone, P.; Silvestris, N.; Racanelli, V.; Solimando, A.G.; Baradaran, B. Pancreatic Cancer Signaling Pathways, Genetic Alterations, and Tumor Microenvironment: The Barriers Affecting the Method of Treatment. Biomedicines 2021, 9, 373. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, X.; Huang, N.; Yuan, S.; Li, J.; Liu, X.; Jiang, Q.; Wu, S.; Ju, Y.; Kleeff, J.; et al. Chimeric Antigen Receptor Macrophages Targeting C-MET(CAR-M-c-MET) Inhibit Pancreatic Cancer Progression and Improve Cytotoxic Chemotherapeutic Efficacy. Mol. Cancer 2024, 23, 270. [Google Scholar] [CrossRef]
- Kim, J.; Lee, T.S.; Lee, M.H.; Cho, I.R.; Ryu, J.K.; Kim, Y.-T.; Lee, S.H.; Paik, W.H. Pancreatic Cancer Treatment Targeting the HGF/c-MET Pathway: The MEK Inhibitor Trametinib. Cancers 2024, 16, 1056. [Google Scholar] [CrossRef]
- Greil, C.; Engelhardt, M.; Wäsch, R. The Role of the APC/C and Its Coactivators Cdh1 and Cdc20 in Cancer Development and Therapy. Front. Genet. 2022, 13, 941565. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhu, Q.; Wu, Q.; Zhang, Z.; Guo, J.; Wu, G.; Zheng, C.; Xu, Q.; Huang, D.; Cui, D. Prognostic and Immune Landscape Analysis of Ubiquitination-Related Genes in Hepatocellular Carcinoma: Based on Bulk and Single-Cell RNA Sequencing Data. J. Cancer 2024, 15, 2580–2600. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8+ T Cell Differentiation and Dysfunction in Cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T Cells in Cancer and Cancer Immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Koh, C.-H.; Lee, S.; Kwak, M.; Kim, B.-S.; Chung, Y. CD8 T-Cell Subsets: Heterogeneity, Functions, and Therapeutic Potential. Exp. Mol. Med. 2023, 55, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- Klintman, M.; Buus, R.; Cheang, M.C.U.; Sheri, A.; Smith, I.E.; Dowsett, M. Changes in Expression of Genes Representing Key Biologic Processes after Neoadjuvant Chemotherapy in Breast Cancer, and Prognostic Implications in Residual Disease. Clin. Cancer Res. 2016, 22, 2405–2416. [Google Scholar] [CrossRef]
- Shi, M.-J.; Meng, X.-Y.; Wu, Q.-J.; Zhou, X.-H. High CD3D/CD4 Ratio Predicts Better Survival in Muscle-Invasive Bladder Cancer. Cancer Manag. Res. 2019, 11, 2987–2995. [Google Scholar] [CrossRef]
- Wei, Z.; Shen, Y.; Zhou, C.; Cao, Y.; Deng, H.; Shen, Z. CD3D: A Prognostic Biomarker Associated with Immune Infiltration and Immunotherapeutic Response in Head and Neck Squamous Cell Carcinoma. Bioengineered 2022, 13, 13784–13800. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, C.K.; Tye, G.J.; Balasubramaniam, S.D.; Kaur, G. CD74 and HLA-DRA in Cervical Carcinogenesis: Potential Targets for Antitumour Therapy. Medicina (B Aires) 2022, 58, 190. [Google Scholar] [CrossRef]
- Samuels, S.; Spaans, V.M.; Osse, M.; Peters, L.A.W.; Kenter, G.G.; Fleuren, G.J.; Jordanova, E.S. Human Leukocyte Antigen-DR Expression Is Significantly Related to an Increased Disease-Free and Disease-Specific Survival in Patients With Cervical Adenocarcinoma. Int. J. Gynecol. Cancer 2016, 26, 1503–1509. [Google Scholar] [CrossRef]
- Chen, D.; Yao, J.; Hu, B.; Kuang, L.; Xu, B.; Liu, H.; Dou, C.; Wang, G.; Guo, M. New Biomarker: The Gene HLA-DRA Associated with Low-Grade Glioma Prognosis. Chin. Neurosurg. J. 2022, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.; Jiao, W.; Yang, X.; Liang, Y.; Li, Z.; Niu, H. C3, C3AR1, HLA-DRA, and HLA-E as Potential Prognostic Biomarkers for Renal Clear Cell Carcinoma. Transl. Androl. Urol. 2020, 9, 2640–2656. [Google Scholar] [CrossRef]
- Leite, F.A.; Lira, R.C.P.; Fedatto, P.F.; Antonini, S.R.R.; Martinelli, C.E.; de Castro, M.; Neder, L.; Ramalho, L.N.Z.; Tucci, S.; Mastelaro, M.J.; et al. Low Expression of HLA-DRA, HLA-DPA1, and HLA-DPB1 Is Associated with Poor Prognosis in Pediatric Adrenocortical Tumors (ACT). Pediatr. Blood Cancer 2014, 61, 1940–1948. [Google Scholar] [CrossRef]
- Mei, J.; Jiang, G.; Chen, Y.; Xu, Y.; Wan, Y.; Chen, R.; Liu, F.; Mao, W.; Zheng, M.; Xu, J. HLA Class II Molecule HLA-DRA Identifies Immuno-Hot Tumors and Predicts the Therapeutic Response to Anti-PD-1 Immunotherapy in NSCLC. BMC Cancer 2022, 22, 738. [Google Scholar] [CrossRef]
- Pérez-Pena, J.; Tibor Fekete, J.; Páez, R.; Baliu-Piqué, M.; García-Saenz, J.Á.; García-Barberán, V.; Manzano, A.; Pérez-Segura, P.; Esparis-Ogando, A.; Pandiella, A.; et al. A Transcriptomic Immunologic Signature Predicts Favorable Outcome in Neoadjuvant Chemotherapy Treated Triple Negative Breast Tumors. Front. Immunol. 2019, 10, 2802. [Google Scholar] [CrossRef] [PubMed]
- Creeden, J.F.; Alganem, K.; Imami, A.S.; Henkel, N.D.; Brunicardi, F.C.; Liu, S.-H.; Shukla, R.; Tomar, T.; Naji, F.; McCullumsmith, R.E. Emerging Kinase Therapeutic Targets in Pancreatic Ductal Adenocarcinoma and Pancreatic Cancer Desmoplasia. Int. J. Mol. Sci. 2020, 21, 8823. [Google Scholar] [CrossRef]
- Philipsen, L.; Reddycherla, A.V.; Hartig, R.; Gumz, J.; Kästle, M.; Kritikos, A.; Poltorak, M.P.; Prokazov, Y.; Turbin, E.; Weber, A.; et al. De Novo Phosphorylation and Conformational Opening of the Tyrosine Kinase Lck Act in Concert to Initiate T Cell Receptor Signaling. Sci. Signal. 2017, 10, eaaf4736. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-S.; Han, M.-K.; Choi, Y.B.; Yun, Y.; Shin, J.; Kim, U.-H. Direct Interaction of the CD38 Cytoplasmic Tail and the Lck SH2 Domain. J. Biol. Chem. 2000, 275, 1685–1690. [Google Scholar] [CrossRef]
- Bommhardt, U.; Schraven, B.; Simeoni, L. Beyond TCR Signaling: Emerging Functions of Lck in Cancer and Immunotherapy. Int. J. Mol. Sci. 2019, 20, 3500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Di, X.; Chen, G.; Liu, J.; Zhang, B.; Feng, L.; Cheng, S.; Wang, Y. An Immune-Related Signature That to Improve Prognosis Prediction of Breast Cancer. Am. J. Cancer Res. 2021, 11, 1267–1285. [Google Scholar]
- Chen, Q.; Jun, H.; Yang, C.; Yang, F.; Xu, Y. The Pyroptosis-Related Risk Genes APOBEC3D, TNFRSF14, and RAC2 Were Used to Evaluate Prognosis and as Tumor Suppressor Genes in Breast Cancer. J. Oncol. 2022, 2022, 3625790. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cai, Y.; Deng, Y.; He, Y.; Wu, J.; Chang, S.; Yan, X.; Wang, J. RAC2 as a Tumor-Suppressive Biomarker Associated with T Cell Infiltration in Breast Cancer. Cancer Biother. Radiopharm. 2025, 40, 62–77. [Google Scholar] [CrossRef]
- D’Cruz, O.J.; Qazi, S.; Hwang, L.; Ng, K.; Trieu, V. Impact of Targeting Transforming Growth Factor β-2 with Antisense OT-101 on the Cytokine and Chemokine Profile in Patients with Advanced Pancreatic Cancer. OncoTargets Ther. 2018, 11, 2779–2796. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust Enumeration of Cell Subsets from Tissue Expression Profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Posta, M.; Győrffy, B. Analysis of a Large Cohort of Pancreatic Cancer Transcriptomic Profiles to Reveal the Strongest Prognostic Factors. Clin. Transl. Sci. 2023, 16, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B. Integrated Analysis of Public Datasets for the Discovery and Validation of Survival-Associated Genes in Solid Tumors. Innovation 2024, 5, 100625. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trieu, V.; Potts, M.; Myers, S.; Richardson, S.; Qazi, S. TGFB2 Gene Methylation in Tumors with Low CD8+ T-Cell Infiltration Drives Positive Prognostic Overall Survival Responses in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2025, 26, 5567. https://doi.org/10.3390/ijms26125567
Trieu V, Potts M, Myers S, Richardson S, Qazi S. TGFB2 Gene Methylation in Tumors with Low CD8+ T-Cell Infiltration Drives Positive Prognostic Overall Survival Responses in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2025; 26(12):5567. https://doi.org/10.3390/ijms26125567
Chicago/Turabian StyleTrieu, Vuong, Michael Potts, Scott Myers, Stephen Richardson, and Sanjive Qazi. 2025. "TGFB2 Gene Methylation in Tumors with Low CD8+ T-Cell Infiltration Drives Positive Prognostic Overall Survival Responses in Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 26, no. 12: 5567. https://doi.org/10.3390/ijms26125567
APA StyleTrieu, V., Potts, M., Myers, S., Richardson, S., & Qazi, S. (2025). TGFB2 Gene Methylation in Tumors with Low CD8+ T-Cell Infiltration Drives Positive Prognostic Overall Survival Responses in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 26(12), 5567. https://doi.org/10.3390/ijms26125567