
Homocysteine Metabolites, Endothelial Dysfunction, and Cardiovascular Disease



Abstract
:1. Introduction
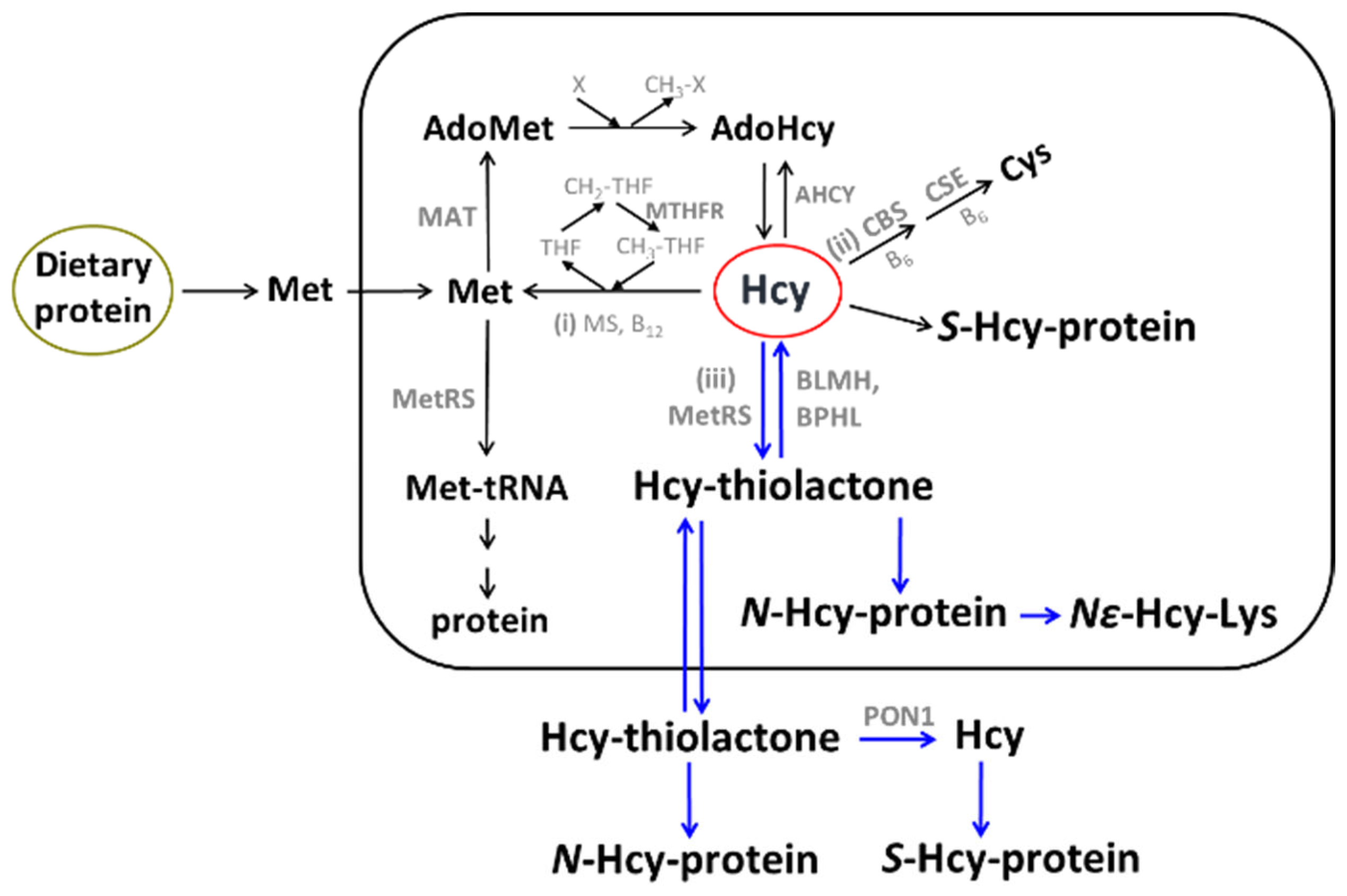
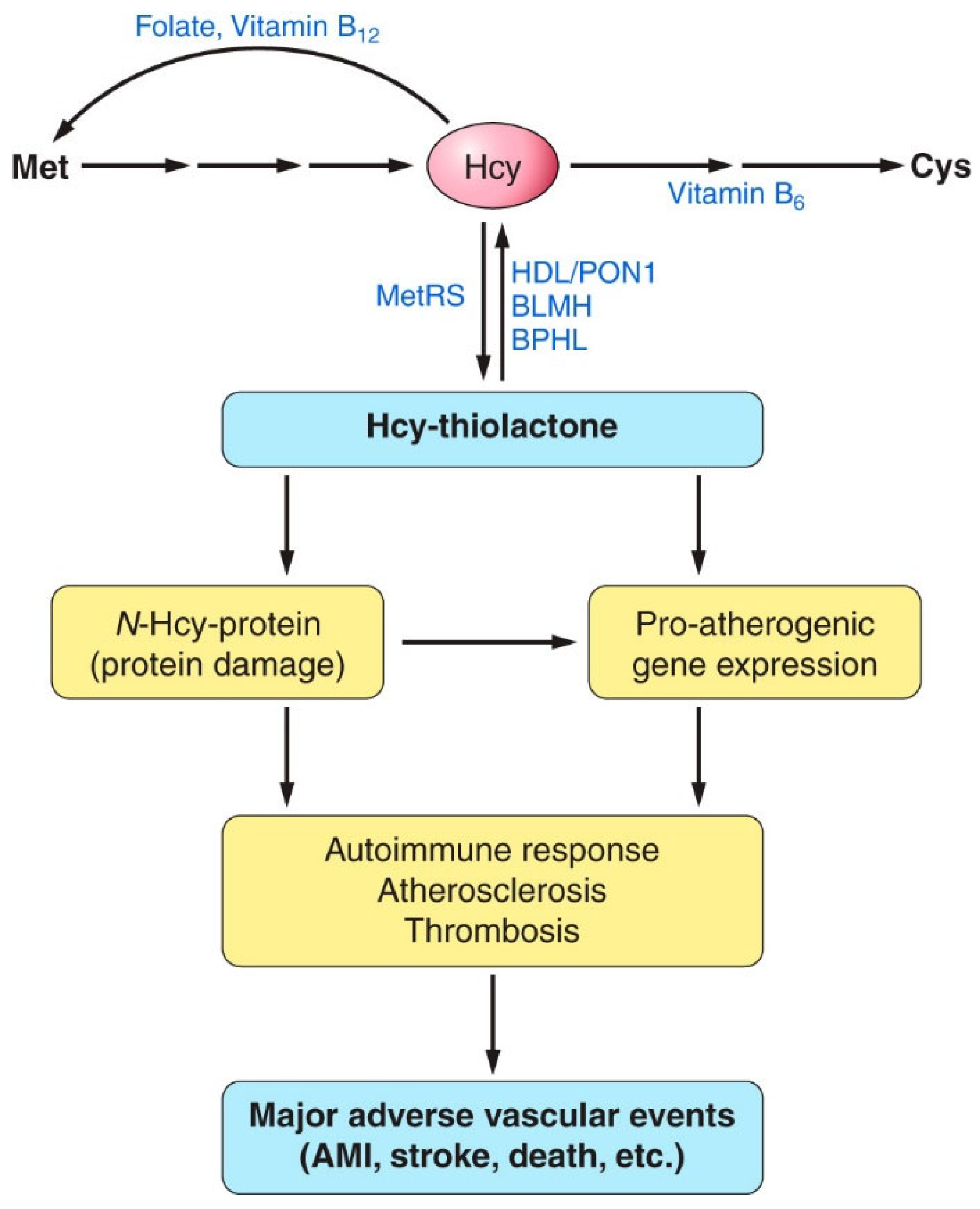
2. Hcy and Related Metabolites
3. Hcy Metabolites, CVD, and Stroke
3.1. AdoHcy
Hcy-Lowering B-Vitamins Do Not Lower Plasma AdoHcy
3.2. Free Reduced Hcy
3.2.1. Physiological Increments of Plasma tHcy Induce Vascular Endothelial Dysfunction in Healthy Humans
3.2.2. Reduced Hcy Is Associated with Vascular Endothelial Dysfunction in Healthy Humans
4. Hcy Is Metabolized to Hcy-Thiolactone and N-Hcy-Protein in Human Endothelial Cells
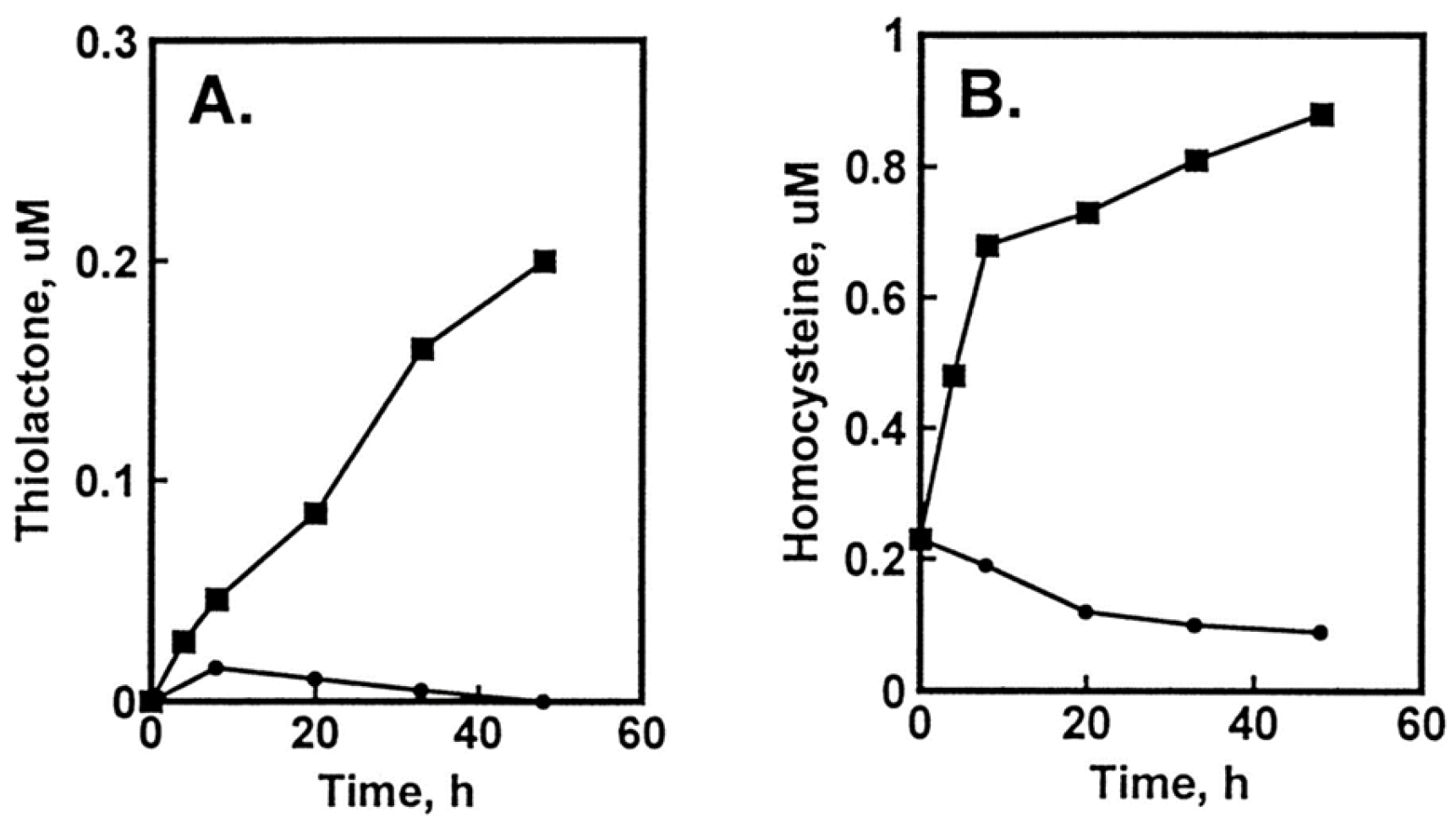
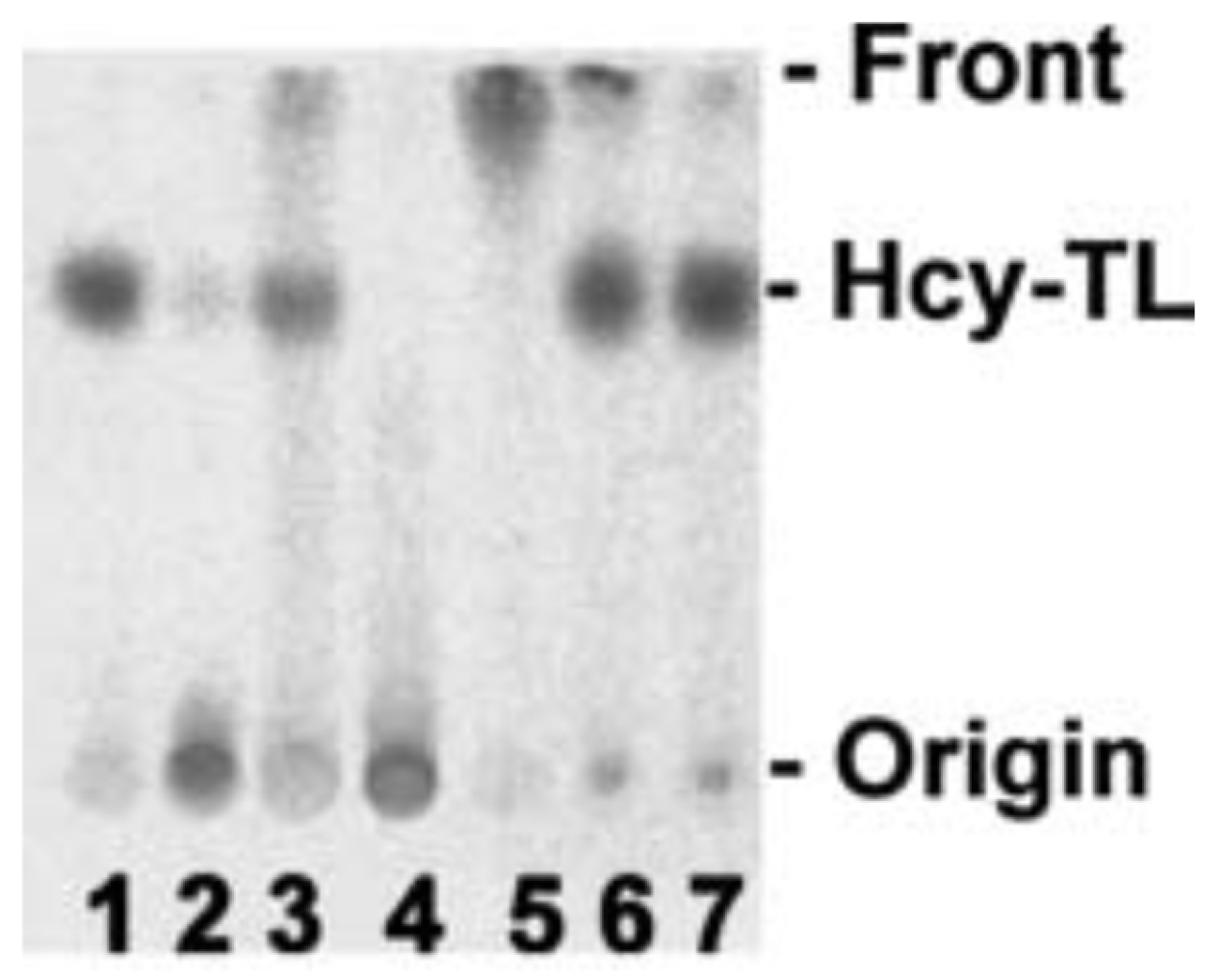
Hcy-Thiolactone Turnover in HUVEC Culture Media
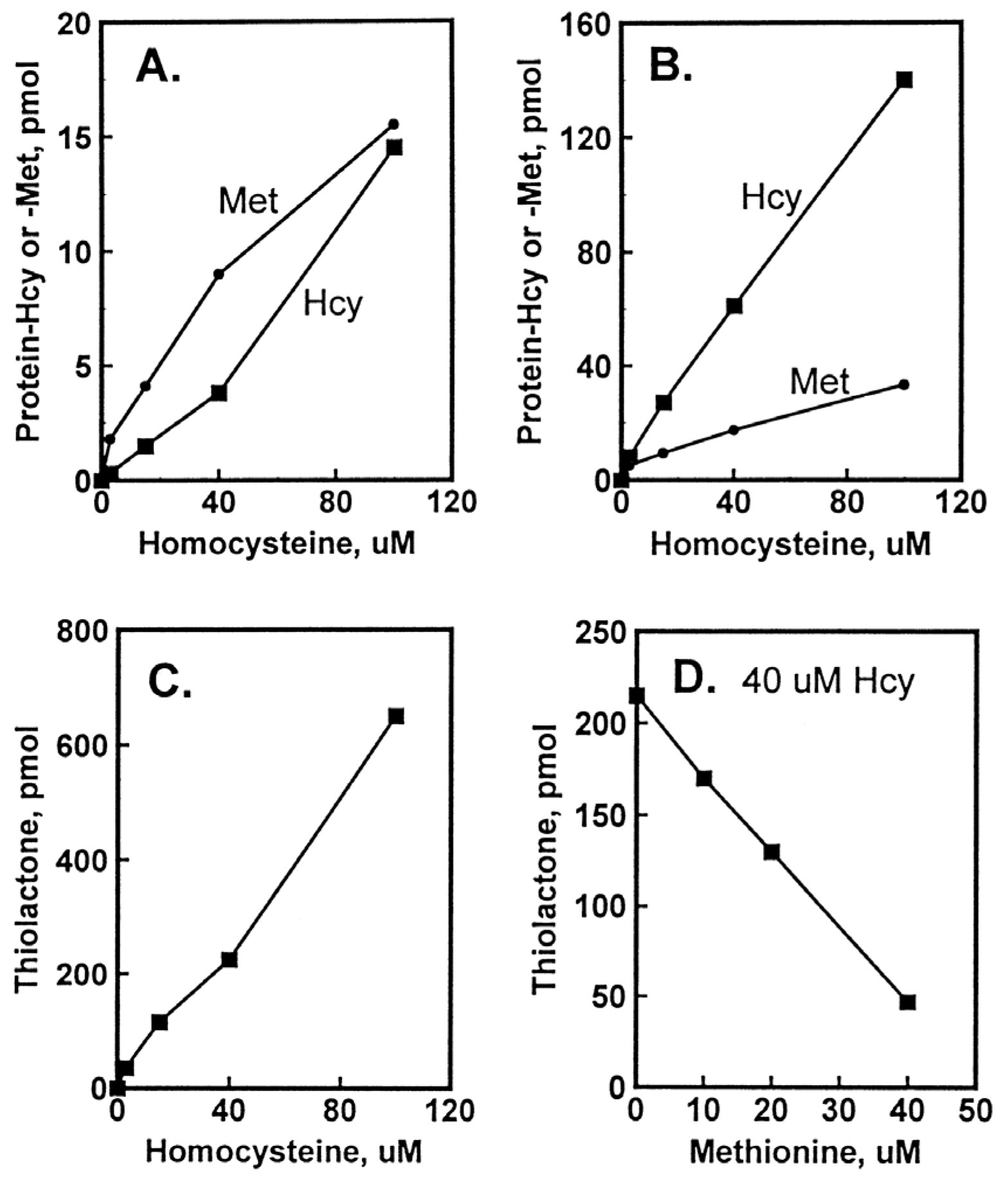
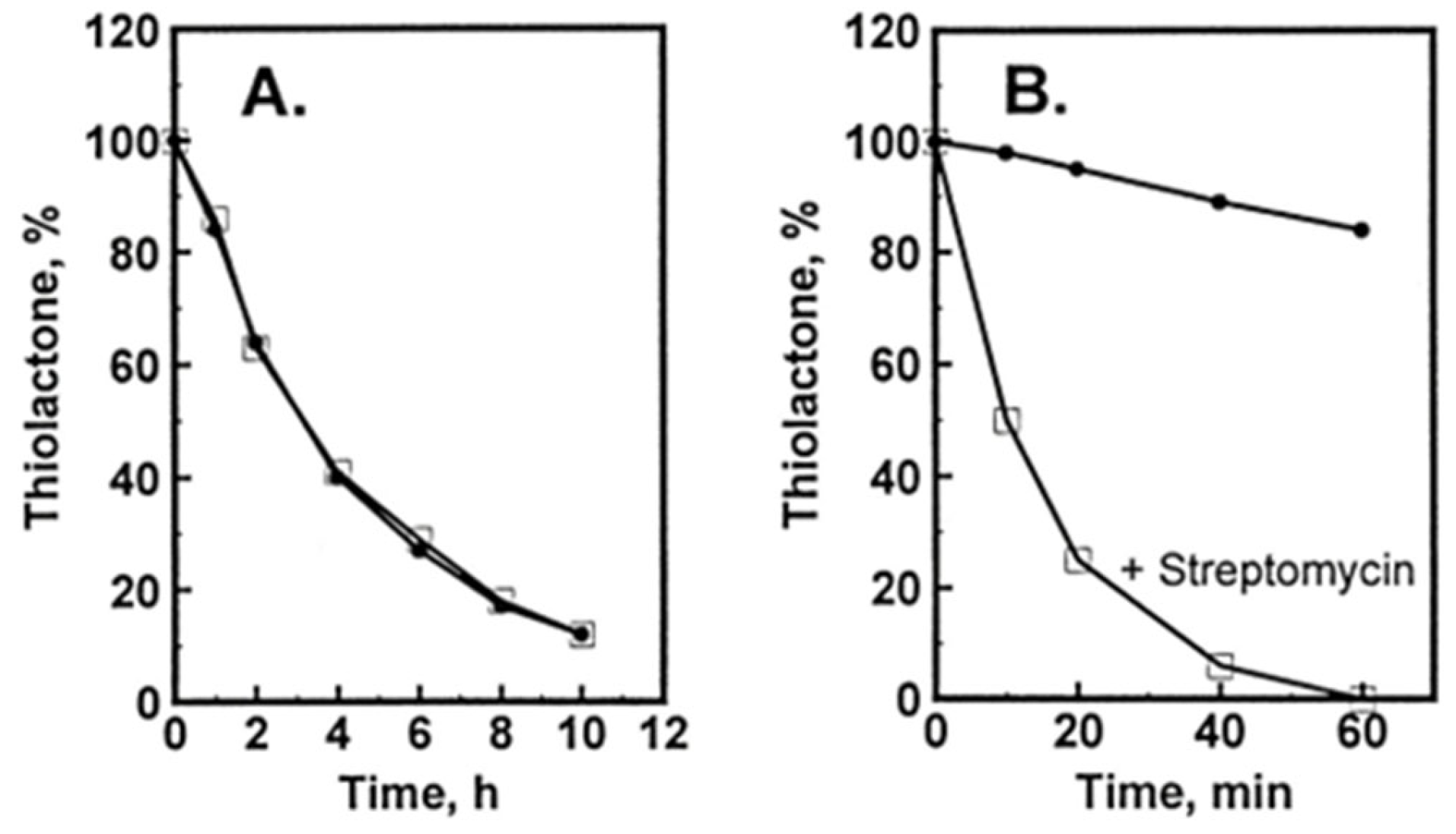
5. Factors Affecting the Accumulation of Hcy-Thiolactone and N-HCY-Protein in HUVEC Cultures
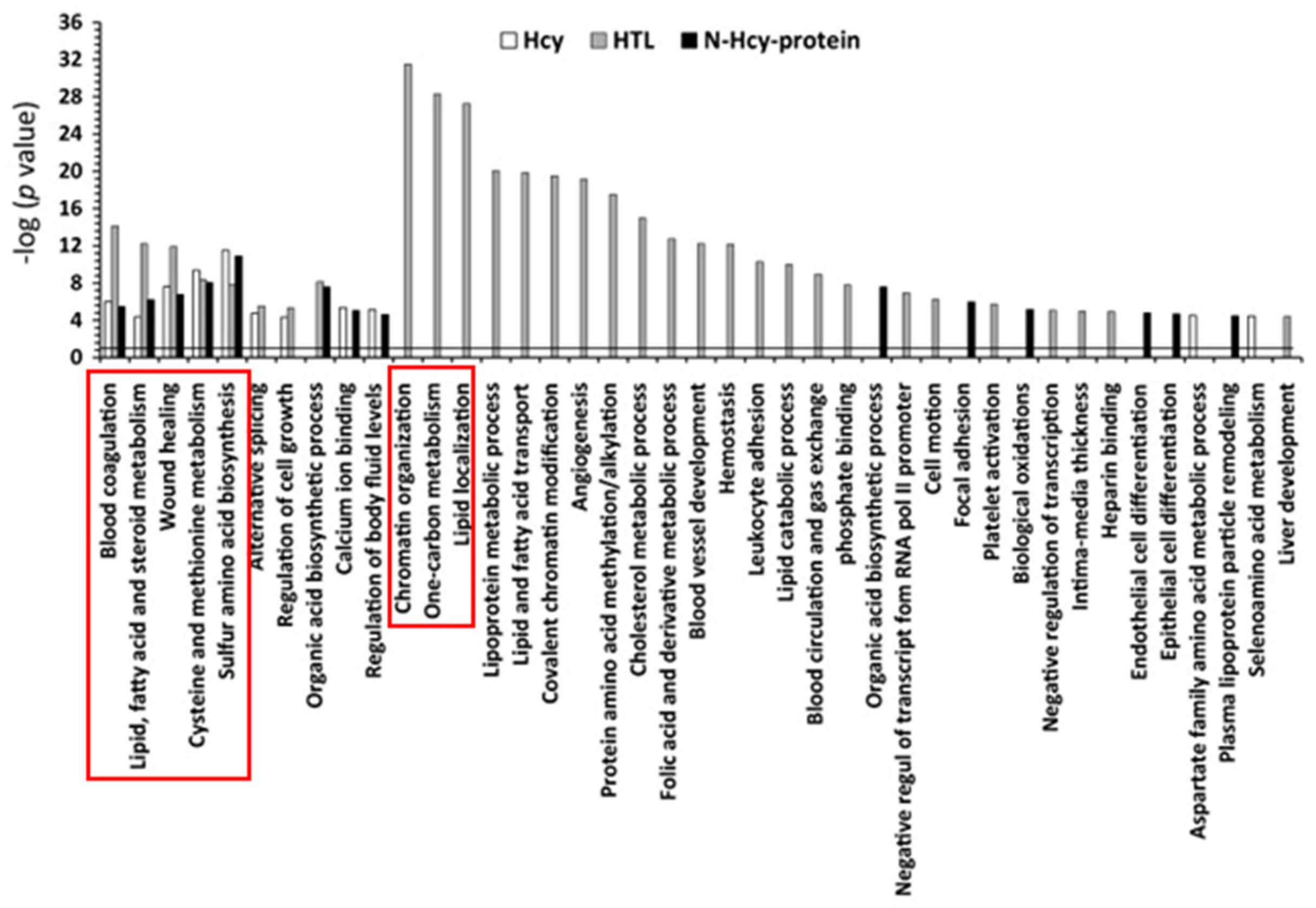
6. Hcy-Thiolactone and N-Hcy Protein Induce Proatherogenic Changes in Gene Expression in Human Vascular Endothelial Cells
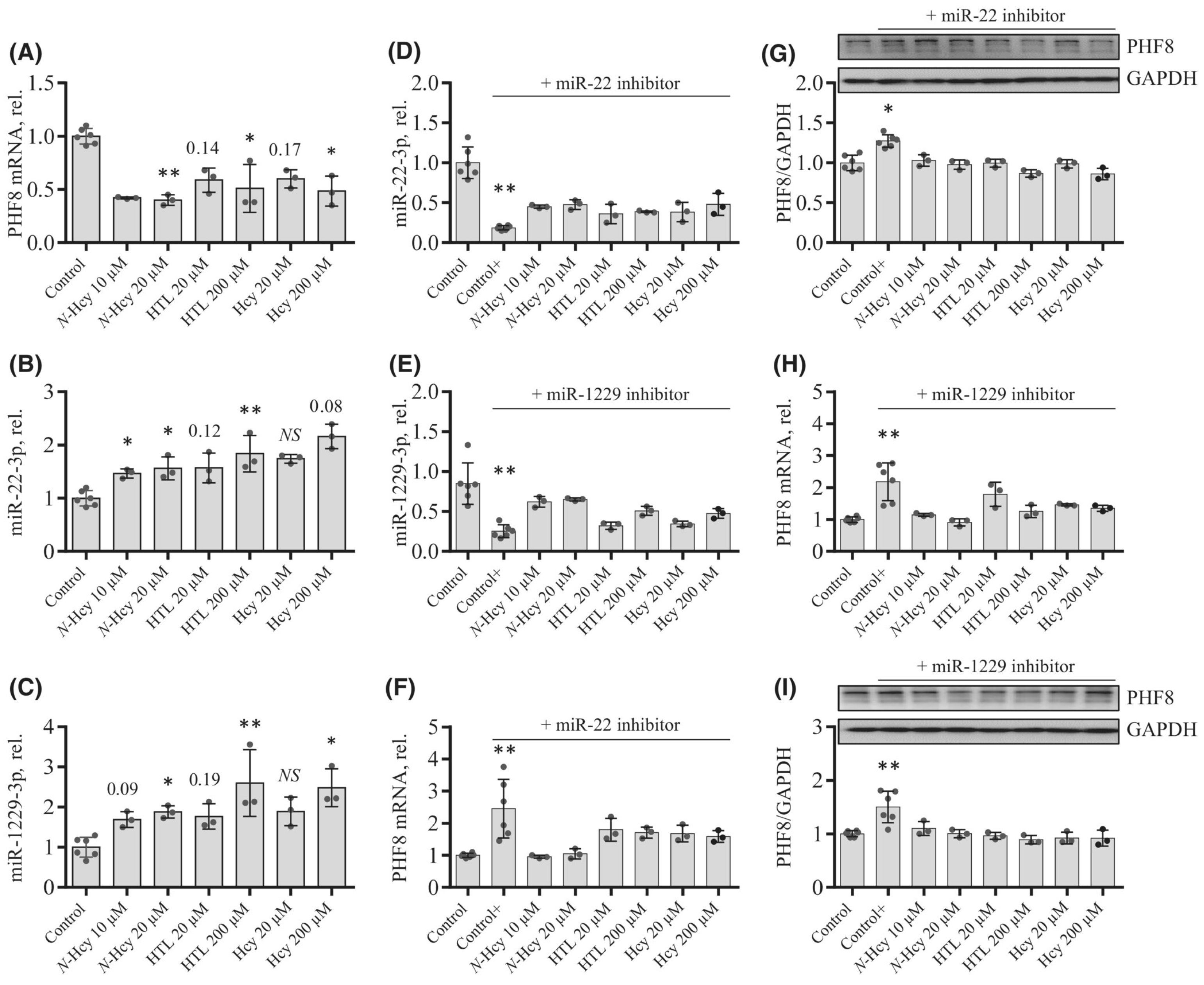
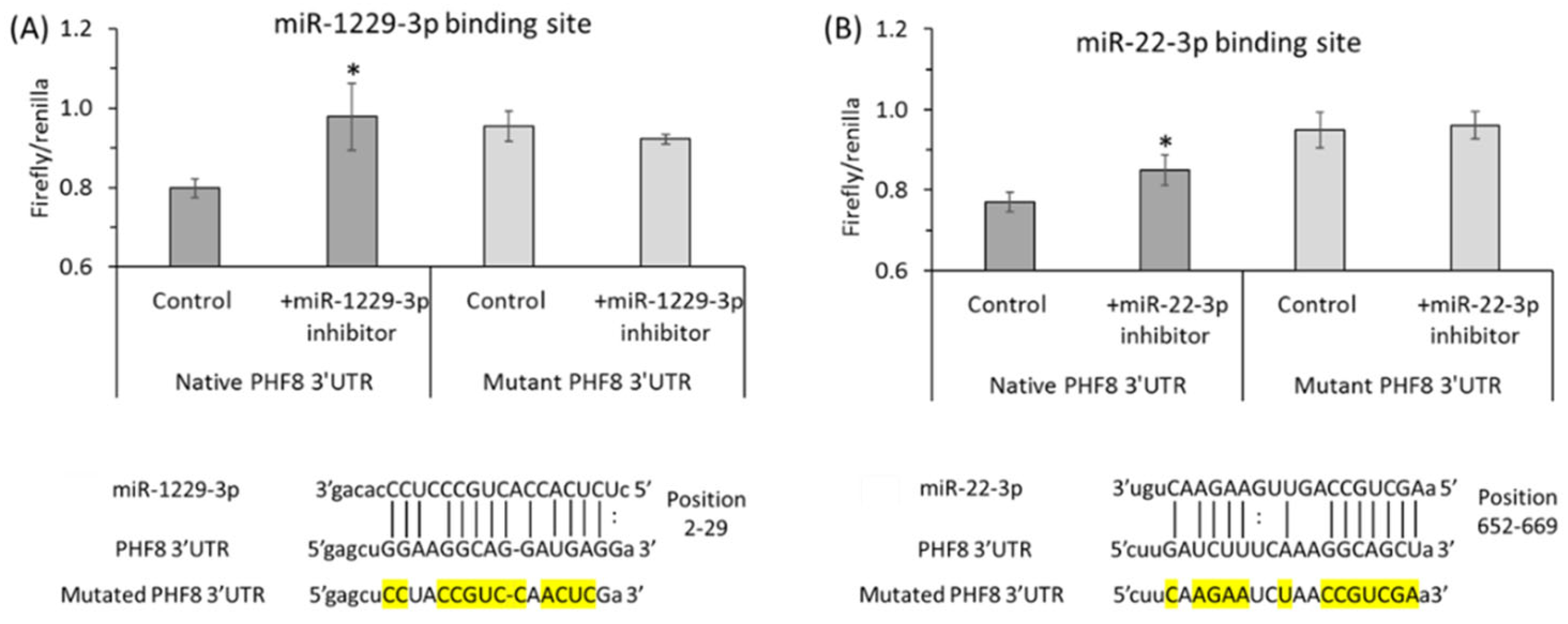
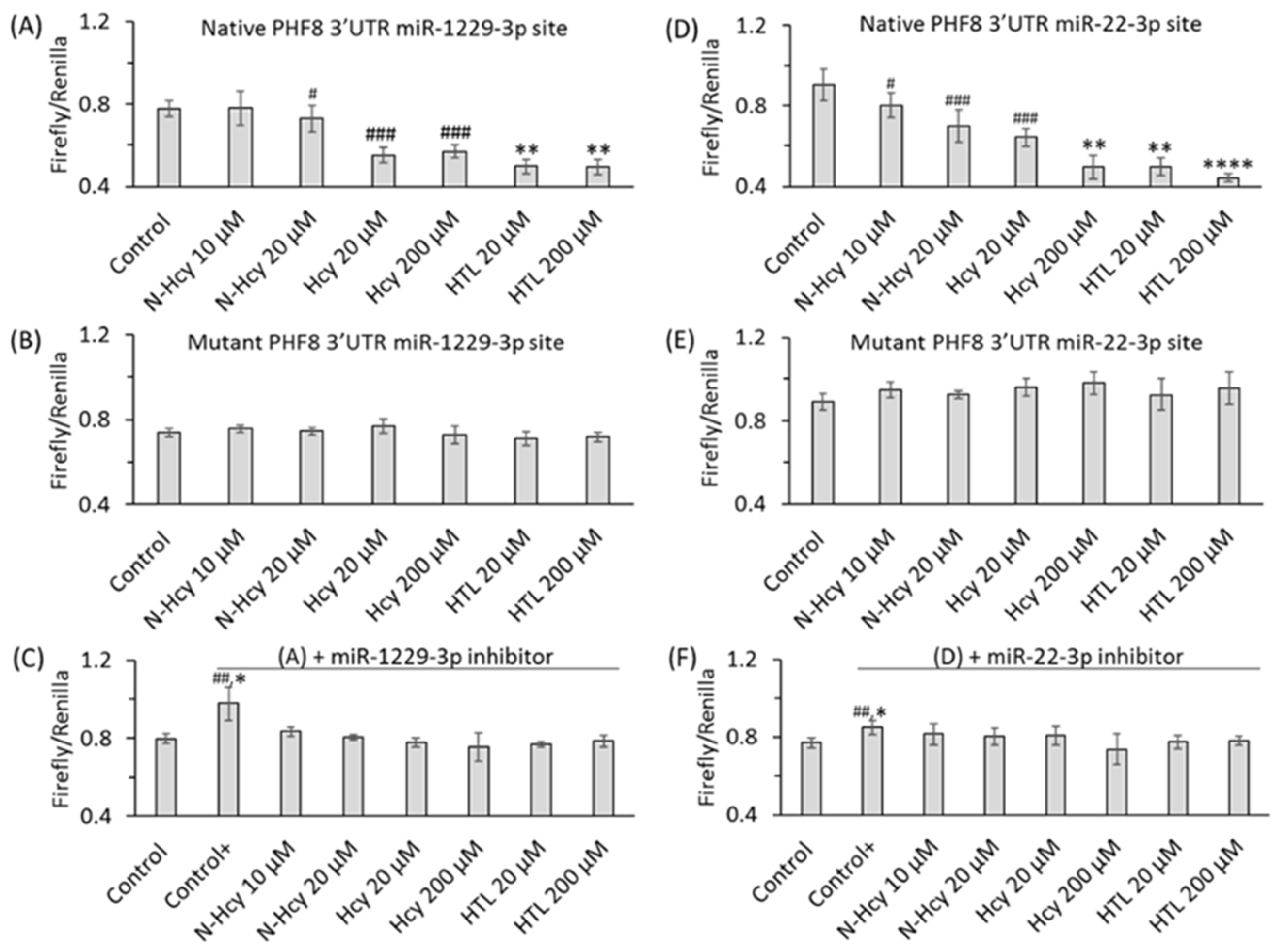
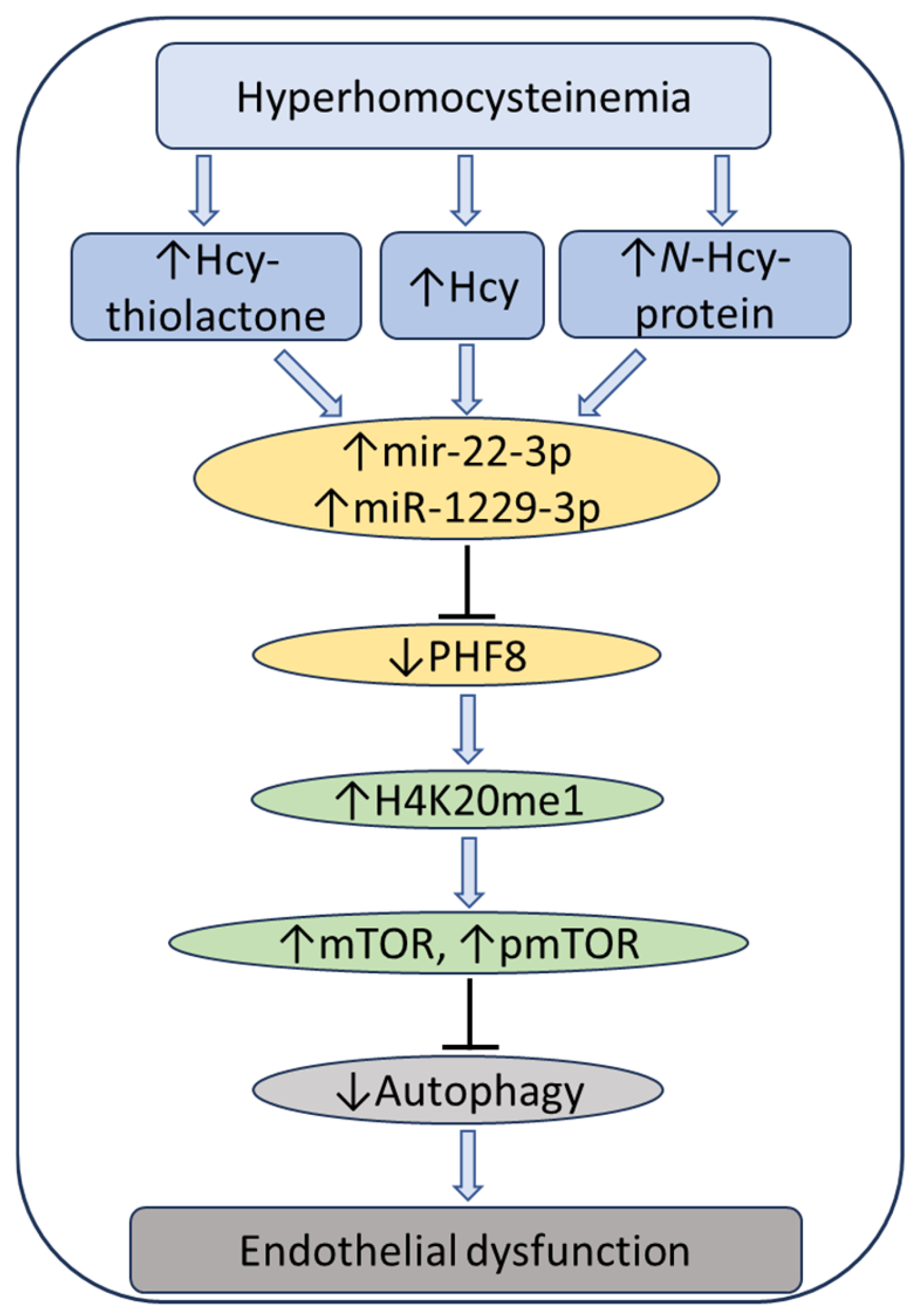
7. Hcy Metabolites Impair mTOR Signaling and Autophagy via Microrna-Mediated Mechanism in Human Endothelial Cells and Cbs−/− Mice
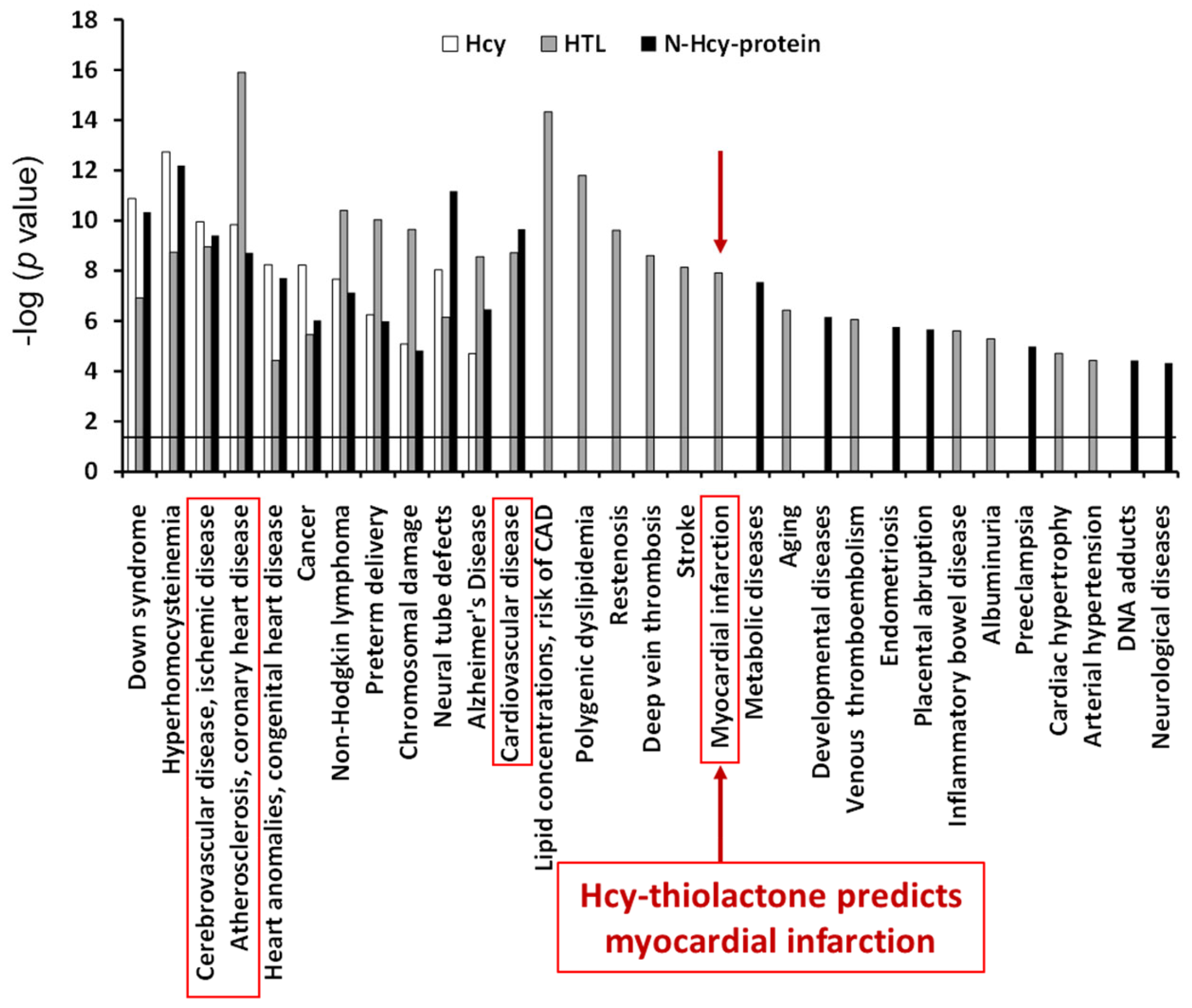
8. Hcy-Thiolactone Predicts Acute Myocardial Infarction in CAD Patients
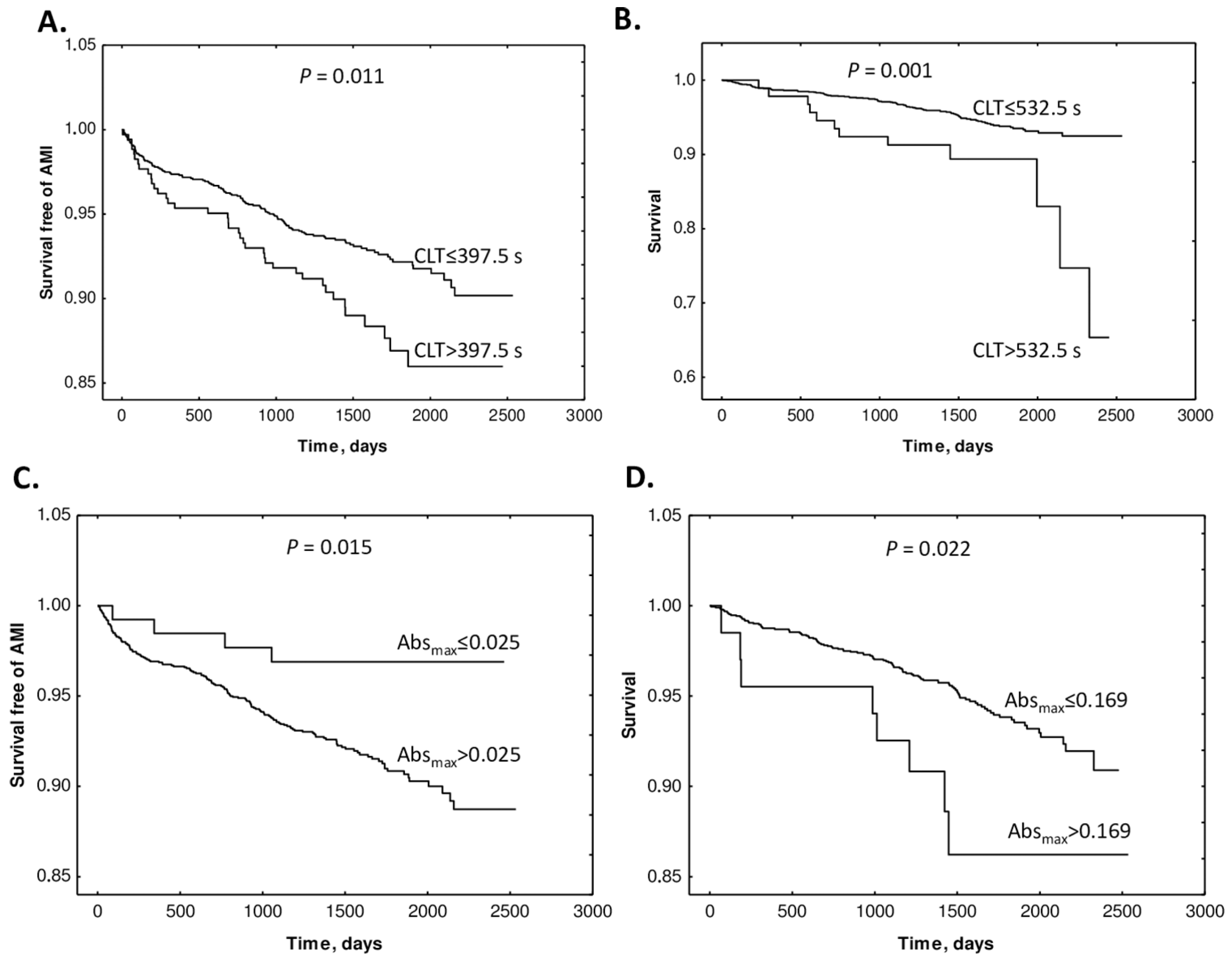
9. Hcy-Thiolactone Influences Prognostic Value of Fibrin Clot Structure/Function in CAD Patients
10. Hcy-Thiolactone Is Associated with Macro Vasculopathy
11. Hcy-Thiolactonase Activity of PON1 Is Associated with CAD Mortality
12. Hcy-Thiolactone and Other Sulfur-Containing Amino Acid Metabolites Are Associated with Fibrin Clot Properties and the Risk of Ischemic Stroke
13. Hcy, Hcy-Thiolactone, and Related Metabolites Are Severely Elevated in CBS Deficiency
14. Protein N-Homocysteinylation in CBS Deficiency
15. CBS Deficiency, Thromboembolism, N-Hcy-Fibrinogen, and Stroke
15.1. Pro-Thrombotic N-Hcy-Fibrinogen Is Elevated in CBS Deficiency
15.2. Stroke in CBS Deficiency vs. Stroke in the General Population
16. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AdoHcy | S-adenosylhomocysteine |
| AdoMet | S-adenosylmethionine |
| AHCY | Adenosylhomocysteinase |
| AMI | Acute myocardial infarction |
| ApoE | Apolipoprotein E |
| Blmh | Bleomycin hydrolase |
| Bphl | Biphenyl hydrolase-like |
| CBS | Cystathionine β-synthase |
| CLT | Clot lysis time |
| CSE | Cystathionine γ-lyase |
| CKD | Chronic kidney disease |
| CVD | Cardiovascular disease |
| Cys | Cysteine |
| Hcy | Homocysteine |
| HDL | High-density lipoprotein |
| HHcy | Hyperhomocysteinemia |
| HR | Hazard ratio |
| HUVEC | Human umbilical vein endothelial cells |
| LDL | Low-density lipoprotein |
| MAVP | Macro vasculopathy |
| Met | Mehionine |
| MetRS | Methionyl-tRNA synthetase |
| MI | Myocardial infarction |
| miR | MicroRNA |
| MTR | Methyltetrahydrofolate-homocysteine methyltransferase |
| MTRR | 5-Methyltetrahydrofolate-homocysteine methyltransferase reductase |
| N-Hcy-protein | N-homocysteinylated protein |
| pCys | Plasma cysteinę |
| pCysGly | Plasma cysteinylglycine |
| Phf8 | Plant homeodomain finger protein 8 |
| Pon1 | Paraoxonase 1 |
| SAM | S-adenosylmethionine |
| tHcy | Total homocysteine |
| uGSH | Urinary glutathione |
| uHcy | Urinary homocysteine |
| WENBIT | Western Norway B Vitamin Intervention Trial |
References
- Homem, F.; Reveles, A.; Amaral, A.; Coutinho, V.; Gonçalves, L. Improving Transitional Care after Acute Myocardial Infarction: A Scoping Review. Health Care Sci. 2024, 3, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Rallidis, S.; Jovanovic, A.; Rallidis, L. Distinctive characteristics, risk factors, and prevention of premature myocardial infarction: A narrative review. J. Family Med. Prim. Care 2024, 13, 3509–3517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, Y.; Wang, K.; Wang, N. Regulated Cell Death in Acute Myocardial Infarction: Molecular Mechanisms and Therapeutic Implications. Ageing Res. Rev. 2024, 104, 102629. [Google Scholar] [CrossRef]
- Manzo-Silberman, S.; Hawranek, M.; Banerjee, S.; Kaluzna-Oleksy, M.; Alasnag, M.; Paradies, V.; Parapid, B.; Sabouret, P.; Wolczenko, A.; Kunadian, V.; et al. Call to Action for Acute Myocardial Infarction in Women: International Multi-Disciplinary Practical Roadmap. Eur. Heart J. Open 2024, 4, oeae087. [Google Scholar] [CrossRef]
- Yadava, S.; Reddy, D.H.; Nakka, V.P.; Anusha, V.L.; Dumala, N.; Viswanadh, M.K.; Chakravarthi, G.; Nalluri, B.N.; Ramakrishna, K. Unravelling Neuroregenerative and Neuroprotective Roles of Wnt/β-Catenin Pathway in Ischemic Stroke: Insights into Molecular Mechanisms. Neuroscience 2025, 565, 527–547. [Google Scholar] [CrossRef] [PubMed]
- Mai, L.M.; Joundi, R.A.; Katsanos, A.H.; Selim, M.; Shoamanesh, A. Pathophysiology of Intracerebral Hemorrhage: Recovery Trajectories. Stroke 2024. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, L.; Peng, W.; Mao, X. Glial Polarization in Neurological Diseases: Molecular Mechanisms and Therapeutic Opportunities. Ageing Res. Rev. 2024, 104, 102638. [Google Scholar] [CrossRef]
- Huang, Z.; Xu, P.; Hess, D.C.; Zhang, Q. Cellular Senescence as a Key Contributor to Secondary Neurodegeneration in Traumatic Brain Injury and Stroke. Transl. Neurodegener. 2024, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Xiong, Y.; He, H.; Deng, Y. The Mechanism of Acetylation-Mediated Fusion of Lysosomes with Autophagosomes in Neurons after Ischemic Stroke. Life Sci. 2024, 362, 123305. [Google Scholar] [CrossRef] [PubMed]
- Liga, R.; Hostalrich, A.; Gimelli, A.; Ricco, J.B. Guidelines at a Crossroad: Comparing European and American Guidelines Regarding the Use of Imaging in Peripheral Vascular Arterial Disease and Aortic Disease. Eur. Heart J. Imaging Methods Pract. 2024, 2, qyae123. [Google Scholar] [CrossRef]
- de Klerk, P.S.; Pacor, J.M.; Soukas, P. Shockwave for Peripheral Artery Disease: An Update on Trial Data. J. Cardiovasc. Surg. 2024, 65, 420–438. [Google Scholar] [CrossRef] [PubMed]
- Poredoš, P.; Mikhailidis, D.P.; Paraskevas, K.I.; Blinc, A.; Antignani, P.L.; Stanek, A.; Mansilha, A.; Cevc, M. Management of Arterial Hypertension in Patients with Peripheral Arterial Disease. Int. Angiol. 2024, 43, 541–547. [Google Scholar] [CrossRef]
- Thomas, V.E.; Beckman, J.A. Racial and Socioeconomic Health Disparities in Peripheral Artery Disease. J. Am. Heart Assoc. 2024, 13, e031446. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Barone Gibbs, B.; Beaton, A.Z.; Boehme, A.K.; et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2024, 149, e347–e913. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in Atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M.; Nikolai, N. Anitschkow and the Lipid Hypothesis of Atherosclerosis. Cardiovasc. Pathol. 2014, 23, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F. Role of Oxidative Modifications in Atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef]
- Leopold, J.A. Antioxidants and Coronary Artery Disease: From Pathophysiology to Preventive Therapy. Coron. Artery Dis. 2015, 26, 176–183. [Google Scholar] [CrossRef]
- Maron, B.A.; Loscalzo, J. The Treatment of Hyperhomocysteinemia. Annu. Rev. Med. 2009, 60, 39–54. [Google Scholar] [CrossRef]
- Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H. Homocysteine—From Disease Biomarker to Disease Prevention. J. Intern. Med. 2021, 290, 826–854. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.; Ryalls, M.; Robinson, J.; Thomas, O.; Leonard, J.V.; Deanfield, J.E. Impaired Endothelial Function Occurs in the Systemic Arteries of Children with Homozygous Homocystinuria but Not in Their Heterozygous Parents. J. Am. Coll. Cardiol. 1993, 22, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Tawakol, A.; Omland, T.; Gerhard, M.; Wu, J.T.; Creager, M.A. Hyperhomocyst(e)Inemia Is Associated with Impaired Endothelium-Dependent Vasodilation in Humans. Circulation 1997, 95, 1119–1121. [Google Scholar] [CrossRef]
- Woo, K.S.; Chook, P.; Lolin, Y.I.; Cheung, A.S.; Chan, L.T.; Sun, Y.Y.; Sanderson, J.E.; Metreweli, C.; Celermajer, D.S. Hyperhomocyst(e)Inemia Is a Risk Factor for Arterial Endothelial Dysfunction in Humans. Circulation 1997, 96, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Salvio, G.; Ciarloni, A.; Cordoni, S.; Cutini, M.; Delli Muti, N.; Finocchi, F.; Firmani, F.; Giovannini, L.; Perrone, M.; Balercia, G. Homocysteine Levels Correlate with Velocimetric Parameters in Patients with Erectile Dysfunction Undergoing Penile Duplex Ultrasound. Andrology 2022, 10, 733–739. [Google Scholar] [CrossRef]
- Salvio, G.; Ciarloni, A.; Cutini, M.; Balercia, G. Hyperhomocysteinemia: Focus on Endothelial Damage as a Cause of Erectile Dysfunction. Int. J. Mol. Sci. 2021, 22, 418. [Google Scholar] [CrossRef] [PubMed]
- Keating, A.K.; Freehauf, C.; Jiang, H.; Brodsky, G.L.; Stabler, S.P.; Allen, R.H.; Graham, D.K.; Thomas, J.A.; Van Hove, J.L.K.; Maclean, K.N. Constitutive Induction of Pro-Inflammatory and Chemotactic Cytokines in Cystathionine Beta-Synthase Deficient Homocystinuria. Mol. Genet. Metab. 2011, 103, 330–337. [Google Scholar] [CrossRef]
- Sikora, M.; Lewandowska, I.; Marczak, Ł.; Bretes, E.; Jakubowski, H. Cystathionine β-Synthase Deficiency: Different Changes in Proteomes of Thrombosis-Resistant Cbs-/- Mice and Thrombosis-Prone CBS-/- Humans. Sci. Rep. 2020, 10, 10726. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Lewandowska, I.; Kupc, M.; Kubalska, J.; Graban, A.; Marczak, Ł.; Kaźmierski, R.; Jakubowski, H. Serum Proteome Alterations in Human Cystathionine β-Synthase Deficiency and Ischemic Stroke Subtypes. Int. J. Mol. Sci. 2019, 20, 3096. [Google Scholar] [CrossRef]
- Mudd, S.H.; Skovby, F.; Levy, H.L.; Pettigrew, K.D.; Wilcken, B.; Pyeritz, R.E.; Andria, G.; Boers, G.H.; Bromberg, I.L.; Cerone, R. The Natural History of Homocystinuria Due to Cystathionine Beta-Synthase Deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar] [PubMed]
- Majtan, T.; Park, I.; Carrillo, R.S.; Bublil, E.M.; Kraus, J.P. Engineering and Characterization of an Enzyme Replacement Therapy for Classical Homocystinuria. Biomacromolecules 2017, 18, 1747–1761. [Google Scholar] [CrossRef]
- Orendác, M.; Zeman, J.; Stabler, S.P.; Allen, R.H.; Kraus, J.P.; Bodamer, O.; Stöckler-Ipsiroglu, S.; Kvasnicka, J.; Kozich, V. Homocystinuria Due to Cystathionine Beta-Synthase Deficiency: Novel Biochemical Findings and Treatment Efficacy. J. Inherit. Metab. Dis. 2003, 26, 761–773. [Google Scholar] [CrossRef]
- Utyro, O.; Perła-Kaján, J.; Jakubowski, H. The Cbs Locus Affects the Expression of Senescence Markers and mtDNA Copy Number, but Not Telomere Dynamics in Mice. Int. J. Mol. Sci. 2020, 21, 2520. [Google Scholar] [CrossRef]
- Lentz, S.R. Mechanisms of Homocysteine-Induced Atherothrombosis. J. Thromb. Haemost. 2005, 3, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Lentz, S.R. Murine Models of Hyperhomocysteinemia and Their Vascular Phenotypes. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1596–1605. [Google Scholar] [CrossRef]
- Poddar, R.; Sivasubramanian, N.; DiBello, P.M.; Robinson, K.; Jacobsen, D.W. Homocysteine Induces Expression and Secretion of Monocyte Chemoattractant Protein-1 and Interleukin-8 in Human Aortic Endothelial Cells: Implications for Vascular Disease. Circulation 2001, 103, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Kerkeni, M.; Tnani, M.; Chuniaud, L.; Miled, A.; Maaroufi, K.; Trivin, F. Comparative Study on in Vitro Effects of Homocysteine Thiolactone and Homocysteine on HUVEC Cells: Evidence for a Stronger Proapoptotic and Proinflammative Homocysteine Thiolactone. Mol. Cell. Biochem. 2006, 291, 119–126. [Google Scholar] [CrossRef]
- Stamler, J.S.; Osborne, J.A.; Jaraki, O.; Rabbani, L.E.; Mullins, M.; Singel, D.; Loscalzo, J. Adverse Vascular Effects of Homocysteine Are Modulated by Endothelium-Derived Relaxing Factor and Related Oxides of Nitrogen. J. Clin. Investig. 1993, 91, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, S.; Wang, Y.; Li, Y.; Bai, L.; Liu, R.; Fan, J.; Liu, E. Homocysteine Reduces Protein S-Nitrosylation in Endothelium. Int. J. Mol. Med. 2014, 34, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Carluccio, M.A.; Ancora, M.A.; Massaro, M.; Carluccio, M.; Scoditti, E.; Distante, A.; Storelli, C.; De Caterina, R. Homocysteine Induces VCAM-1 Gene Expression through NF-kappaB and NAD(P)H Oxidase Activation: Protective Role of Mediterranean Diet Polyphenolic Antioxidants. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2344–H2354. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Yusuf, S.; Arnold, M.J.; Sheridan, P.; Pogue, J.; Micks, M.; McQueen, M.J.; Probstfield, J.; Fodor, G.; Held, C.; et al. Homocysteine Lowering with Folic Acid and B Vitamins in Vascular Disease. N. Engl. J. Med. 2006, 354, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.D.; Bang, H.; Chambless, L.E.; Stampfer, M.J. Vitamin Intervention for Stroke Prevention Trial: An Efficacy Analysis. Stroke 2005, 36, 2404–2409. [Google Scholar] [CrossRef] [PubMed]
- Saposnik, G.; Ray, J.G.; Sheridan, P.; McQueen, M.; Lonn, E.; HOPE 2 Investigators. Homocysteine-lowering therapy and stroke risk, severity, and disability: Additional findings from the HOPE 2 trial. Stroke 2009, 40, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, M.; Bleie, Ø.; Ueland, P.M.; Nordrehaug, J.E.; Nilsen, D.W.; Vollset, S.E.; Refsum, H.; Pedersen, E.K.R.; Nygård, O. Mortality and Cardiovascular Events in Patients Treated with Homocysteine-Lowering B Vitamins After Coronary Angiography: A Randomized Controlled Trial. JAMA 2008, 300, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, M.; Bønaa, K.H.; Arnesen, E.; Ueland, P.M.; Nordrehaug, J.E.; Rasmussen, K.; Njølstad, I.; Nilsen, D.W.; Refsum, H.; Tverdal, A.; et al. Combined Analyses and Extended Follow-up of Two Randomized Controlled Homocysteine-Lowering B-Vitamin Trials. J. Intern. Med. 2010, 268, 367–382. [Google Scholar] [CrossRef]
- Bønaa, K.H.; Njølstad, I.; Ueland, P.M.; Schirmer, H.; Tverdal, A.; Steigen, T.; Wang, H.; Nordrehaug, J.E.; Arnesen, E.; Rasmussen, K.; et al. Homocysteine Lowering and Cardiovascular Events after Acute Myocardial Infarction. N. Engl. J. Med. 2006, 354, 1578–1588. [Google Scholar] [CrossRef]
- Wald, D.S.; Morris, J.K.; Wald, N.J. Reconciling the Evidence on Serum Homocysteine and Ischaemic Heart Disease: A Meta-Analysis. PLoS ONE 2011, 6, e16473. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J. Anticoagulant Therapy for Patients with Ischaemic Stroke. Nat. Rev. Neurol. 2012, 8, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J.; Eikelboom, J.W.; Yi, Q.; Lees, K.R.; Chen, C.; Xavier, D.; Navarro, J.C.; Ranawaka, U.K.; Uddin, W.; Ricci, S.; et al. Antiplatelet Therapy and the Effects of B Vitamins in Patients with Previous Stroke or Transient Ischaemic Attack: A Post-Hoc Subanalysis of VITATOPS, a Randomised, Placebo-Controlled Trial. Lancet Neurol. 2012, 11, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Saposnik, G.; Ovbiagele, B.; Markovic, D.; Towfighi, A. Effect of B-Vitamins on Stroke Risk among Individuals with Vascular Disease Who Are Not on Antiplatelets: A Meta-Analysis. Int. J. Stroke 2016, 11, 206–211. [Google Scholar] [CrossRef]
- Arshi, B.; Ovbiagele, B.; Markovic, D.; Saposnik, G.; Towfighi, A. Differential Effect of B-Vitamin Therapy by Antiplatelet Use on Risk of Recurrent Vascular Events after Stroke. Stroke 2015, 46, 870–873. [Google Scholar] [CrossRef]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, Folate, and the Methionine Remethylation Cycle-Biochemistry, Pathways, and Regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Pellanda, H. Betaine Homocysteine Methyltransferase (BHMT)-Dependent Remethylation Pathway in Human Healthy and Tumoral Liver. Clin. Chem. Lab. Med. 2013, 51, 617–621. [Google Scholar] [CrossRef]
- Lan, X.; Field, M.S.; Stover, P.J. Cell Cycle Regulation of Folate-Mediated One-Carbon Metabolism. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1426. [Google Scholar] [CrossRef]
- Román, G.C.; Mancera-Páez, O.; Bernal, C. Epigenetic Factors in Late-Onset Alzheimer’s Disease: MTHFR and CTH Gene Polymorphisms, Metabolic Transsulfuration and Methylation Pathways, and B Vitamins. Int. J. Mol. Sci. 2019, 20, 319. [Google Scholar] [CrossRef] [PubMed]
- Corona-Trejo, A.; Gonsebatt, M.E.; Trejo-Solis, C.; Campos-Peña, V.; Quintas-Granados, L.I.; Villegas-Vázquez, E.Y.; Daniel Reyes-Hernández, O.; Hernández-Abad, V.J.; Figueroa-González, G.; Silva-Adaya, D. Transsulfuration Pathway: A Targeting Neuromodulator in Parkinson’s Disease. Rev. Neurosci. 2023, 34, 915–932. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Zinellu, A. Transsulfuration and Folate Pathways in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Eur. J. Clin. Investig. 2024, 54, e14158. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Thiolactone Detoxifying Enzymes and Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 8095. [Google Scholar] [CrossRef]
- Mudd, S.H.; Finkelstein, J.D.; Refsum, H.; Ueland, P.M.; Malinow, M.R.; Lentz, S.R.; Jacobsen, D.W.; Brattström, L.; Wilcken, B.; Wilcken, D.E.; et al. Homocysteine and Its Disulfide Derivatives: A Suggested Consensus Terminology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1704–1706. [Google Scholar] [CrossRef]
- Chwatko, G.; Jakubowski, H. Urinary Excretion of Homocysteine-Thiolactone in Humans. Clin. Chem. 2005, 51, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Chwatko, G.; Boers, G.H.J.; Strauss, K.A.; Shih, D.M.; Jakubowski, H. Mutations in Methylenetetrahydrofolate Reductase or Cystathionine Beta-Synthase Gene, or a High-Methionine Diet, Increase Homocysteine Thiolactone Levels in Humans and Mice. FASEB J. 2007, 21, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Perla-Kaján, J.; Finnell, R.H.; Cabrera, R.M.; Wang, H.; Gupta, S.; Kruger, W.D.; Kraus, J.P.; Shih, D.M. Genetic or Nutritional Disorders in Homocysteine or Folate Metabolism Increase Protein N-Homocysteinylation in Mice. FASEB J. 2009, 23, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Boers, G.H.J.; Strauss, K.A. Mutations in Cystathionine β-Synthase or Methylenetetrahydrofolate Reductase Gene Increase N-Homocysteinylated Protein Levels in Humans. FASEB J. 2008, 22, 4071–4076. [Google Scholar] [CrossRef]
- Dhar, I.; Svingen, G.F.T.; Ueland, P.M.; Lysne, V.; Svenningsson, M.M.; Tell, G.S.; Nygård, O.K. Plasma Cystathionine and Risk of Incident Stroke in Patients with Suspected Stable Angina Pectoris. J. Am. Heart Assoc. 2018, 7, e008824. [Google Scholar] [CrossRef] [PubMed]
- Dhar, I.; Svingen, G.F.T.; Pedersen, E.R.; DeRatt, B.; Ulvik, A.; Strand, E.; Ueland, P.M.; Bønaa, K.H.; Gregory, J.F.; Nygård, O.K. Plasma Cystathionine and Risk of Acute Myocardial Infarction among Patients with Coronary Heart Disease: Results from Two Independent Cohorts. Int. J. Cardiol. 2018, 266, 24–30. [Google Scholar] [CrossRef]
- Stabler, S.P.; Lindenbaum, J.; Savage, D.G.; Allen, R.H. Elevation of Serum Cystathionine Levels in Patients with Cobalamin and Folate Deficiency. Blood 1993, 81, 3404–3413. [Google Scholar] [CrossRef]
- Stabler, S.P.; Korson, M.; Jethva, R.; Allen, R.H.; Kraus, J.P.; Spector, E.B.; Wagner, C.; Mudd, S.H. Metabolic Profiling of Total Homocysteine and Related Compounds in Hyperhomocysteinemia: Utility and Limitations in Diagnosing the Cause of Puzzling Thrombophilia in a Family. JIMD Rep. 2013, 11, 149–163. [Google Scholar] [CrossRef]
- Gupta, S.; Kühnisch, J.; Mustafa, A.; Lhotak, S.; Schlachterman, A.; Slifker, M.J.; Klein-Szanto, A.; High, K.A.; Austin, R.C.; Kruger, W.D. Mouse Models of Cystathionine Beta-Synthase Deficiency Reveal Significant Threshold Effects of Hyperhomocysteinemia. FASEB J. 2009, 23, 883–893. [Google Scholar] [CrossRef]
- Wagner, C.; Koury, M.J. S-Adenosylhomocysteine: A Better Indicator of Vascular Disease than Homocysteine? Am. J. Clin. Nutr. 2007, 86, 1581–1585. [Google Scholar] [CrossRef]
- Dhar, I.; Lysne, V.; Svingen, G.F.T.; Ueland, P.M.; Gregory, J.F.; Bønaa, K.H.; Nygård, O.K. Elevated Plasma Cystathionine Is Associated with Increased Risk of Mortality among Patients with Suspected or Established Coronary Heart Disease. Am. J. Clin. Nutr. 2019, 109, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
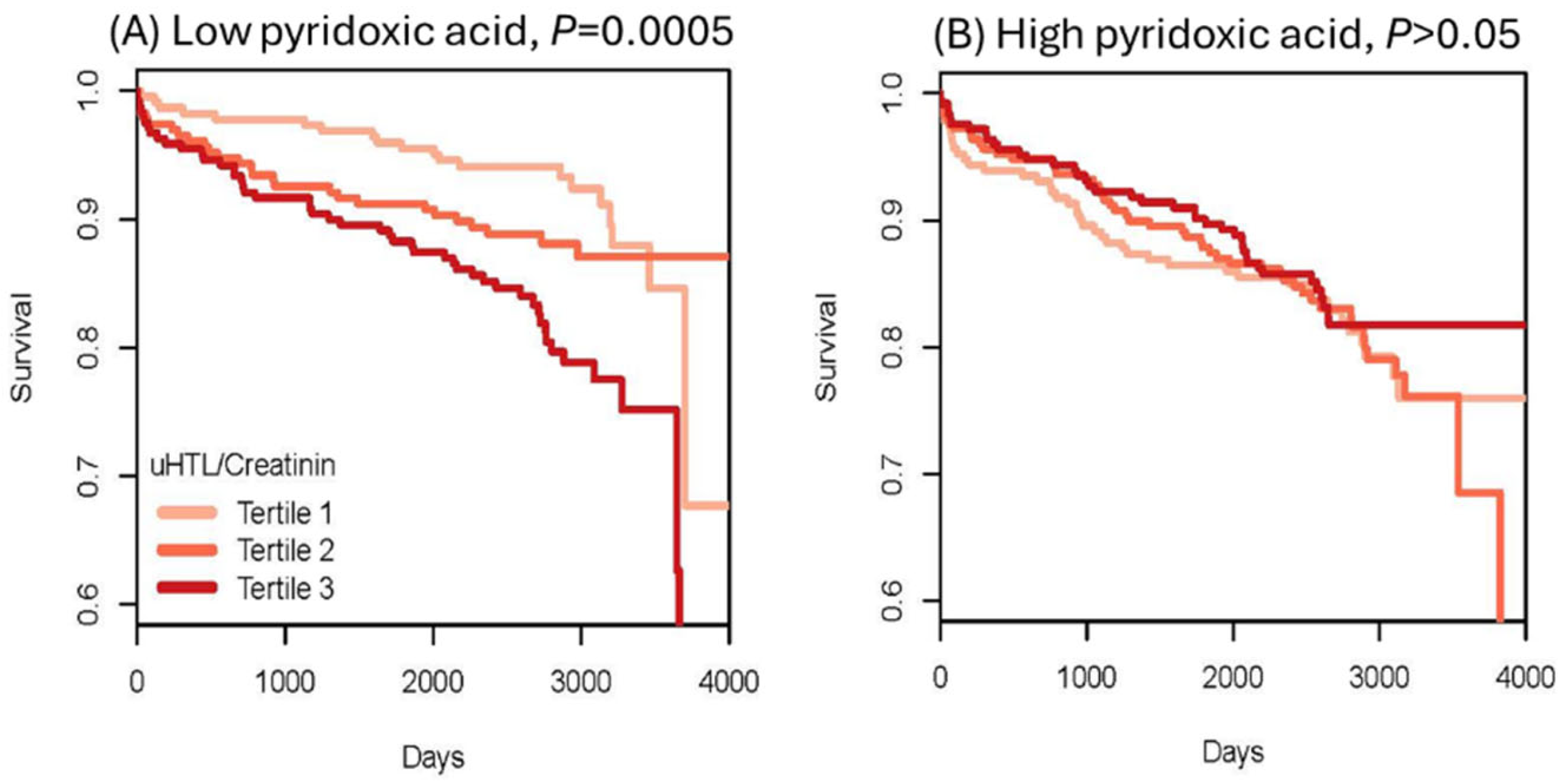
- Borowczyk, K.; Piechocka, J.; Głowacki, R.; Dhar, I.; Midtun, Ø.; Tell, G.S.; Ueland, P.M.; Nygård, O.; Jakubowski, H. Urinary Excretion of Homocysteine Thiolactone and the Risk of Acute Myocardial Infarction in Coronary Artery Disease Patients: The WENBIT Trial. J. Intern. Med. 2019, 285, 232–244. [Google Scholar] [CrossRef]
- Undas, A.; Perła, J.; Lacinski, M.; Trzeciak, W.; Kaźmierski, R.; Jakubowski, H. Autoantibodies against N-Homocysteinylated Proteins in Humans: Implications for Atherosclerosis. Stroke 2004, 35, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Jankowski, M.; Twardowska, M.; Padjas, A.; Jakubowski, H.; Szczeklik, A. Antibodies to N-Homocysteinylated Albumin as a Marker for Early-Onset Coronary Artery Disease in Men. Thromb. Haemost. 2005, 93, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Stepień, E.; Glowacki, R.; Tisończyk, J.; Tracz, W.; Jakubowski, H. Folic Acid Administration and Antibodies against Homocysteinylated Proteins in Subjects with Hyperhomocysteinemia. Thromb. Haemost. 2006, 96, 342–347. [Google Scholar] [CrossRef] [PubMed]
- De La Haba, G.; Cantoni, G.L. The Enzymatic Synthesis of S-Adenosyl-L-Homocysteine from Adenosine and Homocysteine. J. Biol. Chem. 1959, 234, 603–608. [Google Scholar] [CrossRef]
- Kusakabe, Y.; Ishihara, M.; Umeda, T.; Kuroda, D.; Nakanishi, M.; Kitade, Y.; Gouda, H.; Nakamura, K.T.; Tanaka, N. Structural Insights into the Reaction Mechanism of S-Adenosyl-L-Homocysteine Hydrolase. Sci. Rep. 2015, 5, 16641. [Google Scholar] [CrossRef] [PubMed]
- Brzezinski, K.; Dauter, Z.; Jaskolski, M. High-Resolution Structures of Complexes of Plant S-Adenosyl-L-Homocysteine Hydrolase (Lupinus Luteus). Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 218–231. [Google Scholar] [CrossRef]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine Thiolactone and N-Homocysteinylated Protein Induce pro-Atherogenic Changes in Gene Expression in Human Vascular Endothelial Cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D.; Kyle, W.E.; Harris, B.J. Methionine Metabolism in Mammals: Regulatory Effects of S-Adenosylhomocysteine. Arch. Biochem. Biophys. 1974, 165, 774–779. [Google Scholar] [CrossRef]
- Loehrer, F.M.; Angst, C.P.; Brunner, F.P.; Haefeli, W.E.; Fowler, B. Evidence for Disturbed S-Adenosylmethionine: S-Adenosylhomocysteine Ratio in Patients with End-Stage Renal Failure: A Cause for Disturbed Methylation Reactions? Nephrol. Dial. Transplant. 1998, 13, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Kerins, D.M.; Koury, M.J.; Capdevila, A.; Rana, S.; Wagner, C. Plasma S-Adenosylhomocysteine Is a More Sensitive Indicator of Cardiovascular Disease than Plasma Homocysteine. Am. J. Clin. Nutr. 2001, 74, 723–739. [Google Scholar] [CrossRef]
- Jabs, K.; Koury, M.J.; Dupont, W.D.; Wagner, C. Relationship between Plasma S-Adenosylhomocysteine Concentration and Glomerular Filtration Rate in Children. Metabolism 2006, 55, 252–257. [Google Scholar] [CrossRef]
- van Guldener, C.; Donker, A.J.; Jakobs, C.; Teerlink, T.; de Meer, K.; Stehouwer, C.D. No Net Renal Extraction of Homocysteine in Fasting Humans. Kidney Int. 1998, 54, 166–169. [Google Scholar] [CrossRef]
- Becker, A.; Smulders, Y.M.; Teerlink, T.; Struys, E.A.; de Meer, K.; Kostense, P.J.; Jakobs, C.; Dekker, J.M.; Nijpels, G.; Heine, R.J.; et al. S-Adenosylhomocysteine and the Ratio of S-Adenosylmethionine to S-Adenosylhomocysteine Are Not Related to Folate, Cobalamin and Vitamin B6 Concentrations. Eur. J. Clin. Investig. 2003, 33, 17–25. [Google Scholar] [CrossRef]
- Valli, A.; Carrero, J.J.; Qureshi, A.R.; Garibotto, G.; Bárány, P.; Axelsson, J.; Lindholm, B.; Stenvinkel, P.; Anderstam, B.; Suliman, M.E. Elevated Serum Levels of S-Adenosylhomocysteine, but Not Homocysteine, Are Associated with Cardiovascular Disease in Stage 5 Chronic Kidney Disease Patients. Clin. Chim. Acta 2008, 395, 106–110. [Google Scholar] [CrossRef]
- Huang, X.; Lv, X.; Song, H.; Yang, Q.; Sun, Y.; Zhang, W.; Yu, X.; Dong, S.; Yao, W.; Li, Y.; et al. The Relationship between S-Adenosylhomocysteine and Coronary Artery Lesions: A Case Control Study. Clin. Chim. Acta 2017, 471, 314–320. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, Y.; Wang, M.; Li, X.; Su, D.; Qiu, J.; Li, D.; Yang, Y.; Xia, M.; Ling, W. Plasma S-Adenosylhomocysteine Is Associated with the Risk of Cardiovascular Events in Patients Undergoing Coronary Angiography: A Cohort Study. Am. J. Clin. Nutr. 2013, 98, 1162–1169. [Google Scholar] [CrossRef]
- Xiao, Y.; Xia, J.; Cheng, J.; Huang, H.; Zhou, Y.; Yang, X.; Su, X.; Ke, Y.; Ling, W. Inhibition of S-Adenosylhomocysteine Hydrolase Induces Endothelial Dysfunction via Epigenetic Regulation of P66shc-Mediated Oxidative Stress Pathway. Circulation 2019, 139, 2260–2277. [Google Scholar] [CrossRef]
- Green, T.J.; Skeaff, C.M.; McMahon, J.A.; Venn, B.J.; Williams, S.M.; Devlin, A.M.; Innis, S.M. Homocysteine-Lowering Vitamins Do Not Lower Plasma S-Adenosylhomocysteine in Older People with Elevated Homocysteine Concentrations. Br. J. Nutr. 2010, 103, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Krupková-Meixnerová, L.; Veselá, K.; Vitová, A.; Janosíková, B.; Andel, M.; Kozich, V. Methionine-Loading Test: Evaluation of Adverse Effects and Safety in an Epidemiological Study. Clin. Nutr. 2002, 21, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Daly, L.; Robinson, K.; Naughten, E.; Cahalane, S.; Fowler, B.; Graham, I. Hyperhomocysteinemia: An Independent Risk Factor for Vascular Disease. N. Engl. J. Med. 1991, 324, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Obeid, O.A.; Kooner, J.S. Physiological Increments in Plasma Homocysteine Induce Vascular Endothelial Dysfunction in Normal Human Subjects. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2922–2927. [Google Scholar] [CrossRef]
- Chambers, J.C.; Ueland, P.M.; Wright, M.; Doré, C.J.; Refsum, H.; Kooner, J.S. Investigation of Relationship between Reduced, Oxidized, and Protein-Bound Homocysteine and Vascular Endothelial Function in Healthy Human Subjects. Circ. Res. 2001, 89, 187–192. [Google Scholar] [CrossRef]
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine Thiolactone and Protein Homocysteinylation in Human Endothelial Cells: Implications for Atherosclerosis. Circ. Res. 2000, 87, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Quantification of Urinary S- and N-Homocysteinylated Protein and Homocysteine-Thiolactone in Mice. Anal. Biochem. 2016, 508, 118–123. [Google Scholar] [CrossRef]
- Borowczyk, K.; Shih, D.M.; Jakubowski, H. Metabolism and Neurotoxicity of Homocysteine Thiolactone in Mice: Evidence for a Protective Role of Paraoxonase 1. J. Alzheimer’s Dis. 2012, 30, 225–231. [Google Scholar] [CrossRef]
- Borowczyk, K.; Tisończyk, J.; Jakubowski, H. Metabolism and Neurotoxicity of Homocysteine Thiolactone in Mice: Protective Role of Bleomycin Hydrolase. Amino. Acids 2012, 43, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- van Aerts, L.A.; Klaasboer, H.H.; Postma, N.S.; Pertijs, J.C.; Copius Peereboom, J.H.; Eskes, T.K.; Noordhoek, J. Stereospecific in Vitro Embryotoxicity of L-Homocysteine in Pre- and Post-Implantation Rodent Embryos. Toxicol. In Vitro 1993, 7, 743–749. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Is a Protein Amino Acid in Humans: IMPLICATIONS FOR HOMOCYSTEINE-LINKED DISEASE. J. Biol. Chem. 2002, 277, 30425–30428. [Google Scholar] [CrossRef]
- Hortin, G.L.; Seam, N.; Hoehn, G.T. Bound Homocysteine, Cysteine, and Cysteinylglycine Distribution between Albumin and Globulins. Clin. Chem. 2006, 52, 2258–2264. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of Fibrinogen by Homocysteine Thiolactone Increases Resistance to Fibrinolysis: A Potential Mechanism of the Thrombotic Tendency in Hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef]
- Paoli, P.; Sbrana, F.; Tiribilli, B.; Caselli, A.; Pantera, B.; Cirri, P.; De Donatis, A.; Formigli, L.; Nosi, D.; Manao, G.; et al. Protein N-Homocysteinylation Induces the Formation of Toxic Amyloid-like Protofibrils. J. Mol. Biol. 2010, 400, 889–907. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, Ł.; Kubalska, J.; Graban, A.; Jakubowski, H. Identification of N-Homocysteinylation Sites in Plasma Proteins. Amino Acids 2014, 46, 235–244. [Google Scholar] [CrossRef]
- Witucki, Ł.; Jakubowski, H. Homocysteine Metabolites Inhibit Autophagy, Elevate Amyloid Beta, and Induce Neuropathy by Impairing Phf8/H4K20me1-Dependent Epigenetic Regulation of mTOR in Cystathionine β-Synthase-Deficient Mice. J. Inherit. Metab. Dis. 2023, 46, 1114–1130. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, R.; Qu, Y.Y.; Mei, X.Y.; Zhang, X.; Zhou, Q.; Li, Y.; Yang, S.B.; Zuo, Z.G.; Chen, Y.M.; et al. Colonic Lysine Homocysteinylation Induced by High-Fat Diet Suppresses DNA Damage Repair: Cell Reports. Cell Rep. 2018, 25, 398–412. [Google Scholar] [CrossRef]
- Jakubowski, H. Protein N-Homocysteinylation and Colorectal Cancer. Trends Cancer 2019, 5, 7–10. [Google Scholar] [CrossRef]
- Dudman, N.P.; Hicks, C.; Lynch, J.F.; Wilcken, D.E.; Wang, J. Homocysteine Thiolactone Disposal by Human Arterial Endothelial Cells and Serum in Vitro. Arterioscler. Thromb. 1991, 11, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y. Manual Micro-Sequence Analysis of Polypeptides Using Dimethylaminoazobenzene Isothiocyanate. Methods Enzymol. 1983, 91, 455–466. [Google Scholar] [CrossRef]
- Jakubowski, H. Mechanism of the Condensation of Homocysteine Thiolactone with Aldehydes. Chem. Eur. J. 2006, 12, 8039–8043. [Google Scholar] [CrossRef]
- Jakubowski, H. Calcium-Dependent Human Serum Homocysteine Thiolactone Hydrolase. A Protective Mechanism against Protein N-Homocysteinylation. J. Biol. Chem. 2000, 275, 3957–3962. [Google Scholar] [CrossRef]
- Jakubowski, H. Molecular Basis of Homocysteine Toxicity in Humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef]
- Jakubowski, H. Metabolism of Homocysteine Thiolactone in Human Cell Cultures. Possible Mechanism for Pathological Consequences of Elevated Homocysteine Levels. J. Biol. Chem. 1997, 272, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, M.; Urbanek, M.O.; Jaworska, E.; Witucki, L.; Szczesniak, M.W.; Makalowska, I.; Krzyzosiak, W.J. Sequence-Non-Specific Effects Generated by Various Types of RNA Interference Triggers. Biochim. Biophys. Acta 2016, 1859, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Minjares, M.; Wu, W.; Wang, J.-M. Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders. Cells 2023, 12, 1341. [Google Scholar] [CrossRef]
- Zhou, S.S.; Jin, J.P.; Wang, J.Q.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in Cardiovascular Diseases: Potential Biomarkers, Therapeutic Targets and Challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Mens, M.M.J.; Heshmatollah, A.; Fani, L.; Ikram, M.A.; Ikram, M.K.; Ghanbari, M. Circulatory MicroRNAs as Potential Biomarkers for Stroke Risk: The Rotterdam Study. Stroke 2021, 52, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, S.; Zhou, Y.; Han, Y.; Li, S.; Xu, Q.; Xu, L.; Zhu, Z.; Deng, Y.; Yu, L.; et al. Phf8 Histone Demethylase Deficiency Causes Cognitive Impairments through the mTOR Pathway. Nat. Commun. 2018, 9, 114. [Google Scholar] [CrossRef]
- Laumonnier, F.; Holbert, S.; Ronce, N.; Faravelli, F.; Lenzner, S.; Schwartz, C.E.; Lespinasse, J.; Esch, H.V.; Lacombe, D.; Goizet, C.; et al. Mutations in PHF8 Are Associated with X Linked Mental Retardation and Cleft Lip/Cleft Palate. J. Med. Genet. 2005, 42, 780–786. [Google Scholar] [CrossRef]
- Sobering, A.K.; Bryant, L.M.; Li, D.; McGaughran, J.; Maystadt, I.; Moortgat, S.; Graham, J.M.; van Haeringen, A.; Ruivenkamp, C.; Cuperus, R.; et al. Variants in PHF8 Cause a Spectrum of X-Linked Neurodevelopmental Disorders and Facial Dysmorphology. HGG Adv. 2022, 3, 100102. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.-Z.; Wen, S.-Y.; Wang, X.-J.; Liu, Y.; Liang, H. MYC Regulates PHF8, Which Promotes the Progression of Gastric Cancer by Suppressing miR-22-3p. Technol. Cancer Res. Treat. 2020, 19, 1533033820967472. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human miRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Witucki, Ł.; Jakubowski, H. Depletion of Paraoxonase 1 (Pon1) Dysregulates mTOR, Autophagy, and Accelerates Amyloid Beta Accumulation in Mice. Cells 2023, 12, 746. [Google Scholar] [CrossRef]
- Kaldirim, M.; Lang, A.; Pfeiler, S.; Fiegenbaum, P.; Kelm, M.; Bönner, F.; Gerdes, N. Modulation of mTOR Signaling in Cardiovascular Disease to Target Acute and Chronic Inflammation. Front. Cardiovasc. Med. 2022, 9, 907348. [Google Scholar] [CrossRef] [PubMed]
- Samidurai, A.; Kukreja, R.C.; Das, A. Emerging Role of mTOR Signaling-Related miRNAs in Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 41902. [Google Scholar] [CrossRef]
- Mei, X.; Qi, D.; Zhang, T.; Zhao, Y.; Jin, L.; Hou, J.; Wang, J.; Lin, Y.; Xue, Y.; Zhu, P.; et al. Inhibiting MARSs Reduces Hyperhomocysteinemia-Associated Neural Tube and Congenital Heart Defects. EMBO Mol. Med. 2020, 12, e9469. [Google Scholar] [CrossRef]
- Witucki, Ł.; Jakubowski, H. Homocysteine Metabolites Impair the PHF8/H4K20me1/mTOR/Autophagy Pathway by Upregulating the Expression of Histone Demethylase PHF8-Targeting microRNAs in Human Vascular Endothelial Cells and Mice. FASEB J. 2024, 38, e70072. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Borowczyk, K.; Głowacki, R.; Nygård, O.; Jakubowski, H. Paraoxonase 1 Q192R Genotype and Activity Affect Homocysteine Thiolactone Levels in Humans. FASEB J. 2018, 32, 6019–6024. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Bakker, S.J.L.; James, R.W.; Dullaart, R.P.F. Serum Paraoxonase-1 Activity and Risk of Incident Cardiovascular Disease: The PREVEND Study and Meta-Analysis of Prospective Population Studies. Atherosclerosis 2016, 245, 143–154. [Google Scholar] [CrossRef]
- Hassan, A.; Dohi, T.; Miyauchi, K.; Ogita, M.; Kurano, M.; Ohkawa, R.; Nakamura, K.; Tamura, H.; Isoda, K.; Okazaki, S.; et al. Prognostic Impact of Homocysteine Levels and Homocysteine Thiolactonase Activity on Long-Term Clinical Outcomes in Patients Undergoing Percutaneous Coronary Intervention. J. Cardiol. 2017, 69, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Ariëns, R.A. Fibrin Clot Structure and Function: A Role in the Pathophysiology of Arterial and Venous Thromboembolic Diseases. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e88–e99. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M.; Cymbalista, C.M.; Spector, T.D.; Grant, P.J. EuroCLOT Investigators Heritability of Clot Formation, Morphology, and Lysis: The EuroCLOT Study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2783–2789. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Skrzydlewski, P.; Perła-Kaján, J.; Jakubowski, H. Homocysteine Thiolactone Contributes to the Prognostic Value of Fibrin Clot Structure/Function in Coronary Artery Disease. PLoS ONE 2022, 17, e0275956. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Lu, J.; Yang, G.; Dou, J.; Mu, Y.; Meng, J.; Pan, C. Plasma Homocysteine Thiolactone Associated with Risk of Macrovasculopathy in Chinese Patients with Type 2 Diabetes Mellitus. Adv. Ther. 2008, 25, 914–924. [Google Scholar] [CrossRef]
- Barathi, S.; Angayarkanni, N.; Pasupathi, A.; Natarajan, S.K.; Pukraj, R.; Dhupper, M.; Velpandian, T.; Muralidharan, C.; Sivashanmugham, M. Homocysteinethiolactone and Paraoxonase. Diabetes Care 2010, 33, 2031–2037. [Google Scholar] [CrossRef]
- Sikora, M.; Bretes, E.; Perła-Kaján, J.; Utyro, O.; Borowczyk, K.; Piechocka, J.; Głowacki, R.; Wojtasz, I.; Kaźmierski, R.; Jakubowski, H. Homocysteine Thiolactone and Other Sulfur-Containing Amino Acid Metabolites Are Associated with Fibrin Clot Properties and the Risk of Ischemic Stroke. Sci. Rep. 2024, 14, 11222. [Google Scholar] [CrossRef]
- Stabler, S.P.; Allen, R.H. Quantification of Serum and Urinary S-Adenosylmethionine and S-Adenosylhomocysteine by Stable-Isotope-Dilution Liquid Chromatography-Mass Spectrometry. Clin. Chem. 2004, 50, 365–372. [Google Scholar] [CrossRef]
- Carson, N.A.; Dent, C.E.; Field, C.M.; Gaull, G.E. Homocystinuria: Clinical and Pathological Review of Ten Cases. J. Pediatr. 1965, 66, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Protein Homocysteinylation: Possible Mechanism Underlying Pathological Consequences of Elevated Homocysteine Levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Homocysteine in Protein Structure/Function and Human Disease: Chemical Biology of Homocysteine-Containing Proteins; Springer: Vienna, Austria, 2013; ISBN 978-3-7091-1409-4. [Google Scholar]
- Glowacki, R.; Jakubowski, H. Cross-Talk between Cys34 and Lysine Residues in Human Serum Albumin Revealed by N-Homocysteinylation. J. Biol. Chem. 2004, 279, 10864–10871. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, L.; Twardowski, T.; Stobiecki, M.; Jakubowski, H. Direct Monitoring of Albumin Lysine-525 N-Homocysteinylation in Human Serum by Liquid Chromatography/Mass Spectrometry. Anal. Biochem. 2010, 405, 132–134. [Google Scholar] [CrossRef]
- Marczak, L.; Sikora, M.; Stobiecki, M.; Jakubowski, H. Analysis of Site-Specific N-Homocysteinylation of Human Serum Albumin in Vitro and in Vivo Using MALDI-ToF and LC-MS/MS Mass Spectrometry. J. Proteomics 2011, 74, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Marczak, Ł.; Perła-Kajan, J.; Jakubowski, H. Sex Affects N-Homocysteinylation at Lysine Residue 212 of Albumin in Mice. Scientific Reports. Sci. Rep. 2019, 9, 2669. [Google Scholar] [CrossRef] [PubMed]
- Maclean, K.N.; Sikora, J.; Kožich, V.; Jiang, H.; Greiner, L.S.; Kraus, E.; Krijt, J.; Crnic, L.S.; Allen, R.H.; Stabler, S.P.; et al. Cystathionine Beta-Synthase Null Homocystinuric Mice Fail to Exhibit Altered Hemostasis or Lowering of Plasma Homocysteine in Response to Betaine Treatment. Mol. Genet. Metab. 2010, 101, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Homocysteine Thiolactone: Metabolic Origin and Protein Homocysteinylation in Humans. J. Nutr. 2000, 130, 377S–381S. [Google Scholar] [CrossRef]
- Genoud, V.; Lauricella, A.M.; Kordich, L.C.; Quintana, I. Impact of Homocysteine-Thiolactone on Plasma Fibrin Networks. J. Thromb. Thrombolysis. 2014, 38, 540–545. [Google Scholar] [CrossRef]
- Ząbczyk, M.; Ariëns, R.A.S.; Undas, A. Fibrin Clot Properties in Cardiovascular Disease: From Basic Mechanisms to Clinical Practice. Cardiovasc. Res. 2023, 119, 94–111. [Google Scholar] [CrossRef]
- Blombäck, B. Fibrinogen and Fibrin--Proteins with Complex Roles in Hemostasis and Thrombosis. Thromb. Res. 1996, 83, 1–75. [Google Scholar] [CrossRef]
- Nakashima, F.; Shibata, T.; Kamiya, K.; Yoshitake, J.; Kikuchi, R.; Matsushita, T.; Ishii, I.; Giménez-Bastida, J.A.; Schneider, C.; Uchida, K. Structural and Functional Insights into S-Thiolation of Human Serum Albumins. Sci. Rep. 2018, 8, 932. [Google Scholar] [CrossRef] [PubMed]
- Schienle, H.W.; Seitz, R.; Rohner, I.; Lerch, L.; Krumpholz, B.; Krauss, G.; Fowler, B.; Baumgartner, R.; Willenbockel, U.; Egbring, R. Coagulation Factors and Markers of Activation of Coagulation in Homocystinuria (HOCY): A Study in Two Siblings. Blood Coagul. Fibrinolysis 1994, 5, 873–878. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable (n = 491; Stroke n = 200, Controls n = 291) | Bivariate Correlations | Logistic Regression | ||||||
|---|---|---|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 3 | ||||||
| β | p | B | p | B | p | B | p | |
| uHTL #42 | −0.16 | 0.000 | −0.01 | 0.010 | −0.01 | 0.008 | −0.01 | 0.007 |
| uHcy #39 | 0.19 | 0.000 | 0.10 | 0.035 | 0.12 | 0.031 | 0.10 | 0.078 |
| uCys #40 | 0.19 | 0.000 | 0.01 | 0.027 | 0.01 | 0.045 | 0.01 | 0.039 |
| uCysGly #41 | 0.06 | 0.189 | ns | ns | ns | |||
| uGSH #63 | −0.23 | 0.000 | −0.11 | 0.003 | −0.31 | 0.025 | −0.13 | 0.005 |
| uCreatinine #43 | −0.07 | 0.127 | ns | ns | ns | |||
| pHcy #31 | 0.24 | 0.000 | ns | ns | ns | |||
| pCys #32 | 0.26 | 0.000 | 0.01 | 0.000 | 0.01 | 0.001 | 0.01 | 0.003 |
| pCysGly #33 | −0.11 | 0.016 | −0.08 | 0.005 | −0.08 | 0.011 | −0.07 | 0.035 |
| pGSH #62 | −0.16 | 0.000 | ns | ns | ns | |||
| pCreatinine #68 | 0.32 | 0.000 | 0.04 | 0.000 | 0.04 | 0.001 | ns | |
| Age #2 | 0.53 | 0.000 | 0.06 | 0.000 | 0.05 | 0.001 | 0.05 | 0.003 |
| Sex #3 | 0.17 | 0.000 | ns | ns | ns | |||
| Anti-N-Hcy #64 | 0.14 | 0.002 | ns | ns | ns | |||
| GFR #30 | −0.45 | 0.000 | ns | |||||
| Glucose #61 | 0.24 | 0.000 | ns | |||||
| LDL cholesterol #27 | −0.18 | 0.000 | ns | |||||
| HDL cholesterol #28 | −0.28 | 0.000 | ns | |||||
| Triglycerides #29 | 0.11 | 0.008 | ns | |||||
| Hypertension #8 | 0.52 | 0.000 | 1.28 | 0.000 | 1.20 | 0.001 | ||
| Other heart disease #10 | 0.28 | 0.000 | 1.23 | 0.024 | 1.13 | 0.046 | ||
| Early CAD #5 | 0.48 | 0.000 | ns | ns | ||||
| Early MI #6 | 0.20 | 0.000 | ns | ns | ||||
| Diabetes #9 | 0.31 | 0.000 | ns | ns | ||||
| MTHFR C677T #13 | 0.07 | 0.088 | 0.54 | 0.037 | −0.67 | 0.023 | −0.69 | 0.029 |
| MTHFR A1298C #14 | 0.05 | 0.282 | ns | ns | ns | |||
| CBS T833C 844ins68 #17 | −0.06 | 0.135 | ns | ns | ns | |||
| Fibrin CLT #36 | 0.16 | 0.001 | ns | ns | ns | |||
| Fibrin Absmax #37 | 0.21 | 0.000 | 7.1 | 0.049 | 10.6 | 0.007 | 10.9 | 0.010 |
| Variables included in each model are shown by numerical or textual entries. Ischemic stroke was coded as 1, no stroke as 0. | −2 log likelihood = 311.8, Cox & Snell R2 = 0.48, Nagelkerke R2 = 0.64; % Correct 84.5 | −2 log likelihood = 266.9, Cox & Snell R2 = 0.52, Nagelkerke R2 = 0.71; % Correct 87.6 | −2 log likelihood = 250.8, Cox & Snell R2 = 0.53, Nagelkerke R2 = 0.71; % Correct 87.6 | |||||
| Metabolite | Humans | Mice | ||
|---|---|---|---|---|
| CBS−/− | CBS+/+ | Cbs−/− | Cbs+/+ | |
| μM | μM | μM | μM | |
| Hcy-thiolactone | 0.0144 | 0.0004 (0.168) † | (10.8) † | 0.0037 (0.136) † |
| Nε-Hcy-Lys | 0.56 | <0.1 | 0.40 | |
| N-Hcy-protein | 3.0 a; 12.1 a,b | 0.49 c | 16.6 (11.4 d) † | 1.89 (0.34 d) † |
| S-Hcy-protein | 9.80 c | 90 (142.0 e) † | (5.8 e) † | |
| Hcy | 0.25 | 20 | <0.2 WK2009 | |
| Hcy-S-S-Hcy + Hcy-S-S-Cys | 1.90 | 130 | 0.4 WK2009 | |
| tHcy f | 124.8; 48.5; 294.0 b (1108.7) † | 7.4; 12.0 (2.5) † | 296, 272 (4104.0) † | 5.5, 3.0 (45.0) † |
| Cystathionine, µM | 0.040 | 0.157 | 0.43 | 1 |
| Cys, µM | 136 | 289 | 80 | 100 |
| Met, µM | 160; 819.9 (1586.1) † | 22.4; 10.9 (9.7) † | 528.6 (1542.4) † | 35.4 (240.4) † |
| AdoMet, µM | 0.488 | 0.107; 0.109 (10.2) † | ||
| Cardioembolic vs. Large-Vessel Stroke (n = 10) | Cardioembolic vs. Lacunar Stroke (n = 6) | Large-Vessel vs. Lacunar Stroke (n = 6) | CBS−/− vs. Control | |
|---|---|---|---|---|
| Hcy-Dependent (n = 18) | Hcy-Independent † (n = 22) | |||
| APCS | AMBP | APOL1 | APOA1 | AFM |
| APOM | APOA4 | C5 | APOC3 | AHSG |
| C1QA | FCN3 ‡ | GSN ‡ | APOH | APOC1 |
| C4BPA | ITIH4 | GPX3 ‡ | C1R | APOM |
| CPB2 ‡ | LBP | H2AFJ | C1S | C9 |
| FBLN1 ‡ | PF4 | IGK@ ‡ | CFI | CBP2 |
| IGKV1D-12 | HEL0213 | CLU | ||
| KLKB1 ‡ | HPX | F2 | ||
| SERPINF2 ‡ | IGHV3-7 | HEL-S-51, GC | ||
| F2 ‡ | IGHD | F13B | ||
| IGHV3-7 | FBLN1 | |||
| IGH@ | FCN3 | |||
| IGJ; JCHAIN | GPX3 | |||
| IGKV2D-24 | GSN | |||
| ITIH2 | IGK@ | |||
| ORM2 | KNG | |||
| SERPINC1 | KLKB1 | |||
| cDNA FLJ53075, like KNG1 | SAA1 | |||
| HEL111, TTR | ||||
| SERPINA1 | ||||
| SERPIND1 | ||||
| SERPINF2 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakubowski, H.; Witucki, Ł. Homocysteine Metabolites, Endothelial Dysfunction, and Cardiovascular Disease. Int. J. Mol. Sci. 2025, 26, 746. https://doi.org/10.3390/ijms26020746
Jakubowski H, Witucki Ł. Homocysteine Metabolites, Endothelial Dysfunction, and Cardiovascular Disease. International Journal of Molecular Sciences. 2025; 26(2):746. https://doi.org/10.3390/ijms26020746
Chicago/Turabian StyleJakubowski, Hieronim, and Łukasz Witucki. 2025. "Homocysteine Metabolites, Endothelial Dysfunction, and Cardiovascular Disease" International Journal of Molecular Sciences 26, no. 2: 746. https://doi.org/10.3390/ijms26020746
APA StyleJakubowski, H., & Witucki, Ł. (2025). Homocysteine Metabolites, Endothelial Dysfunction, and Cardiovascular Disease. International Journal of Molecular Sciences, 26(2), 746. https://doi.org/10.3390/ijms26020746