
Detection of Circulating Tumor DNA in Liquid Biopsy: Current Techniques and Potential Applications in Melanoma


 , ,
, ,  , and
, and
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Stage and TNM AJCCv8 | Melanoma Specific Survival (MSS) 5 Years and 10 Years | Standard Local Treatment | (Neo)Adjuvant or Advanced Setting Treatment |
|---|---|---|---|
| I-IIA (pTbN0-pT3a) | IA 5 y 99% 10 y 98% IB 5 y 97% 10 y 94% IIA 5 y 94% 10 y 88% | WLE of primary plus SLN dissection. CLND is not recommended for patients with a positive SLN. Standard follow up | Clinical trial |
| IIB-IIC (T3b-T4bN0) | IIB 5 y 87% 10 y 82% IIC 5 y 82% 10 y 75% | WLE of primary plus SLN dissection. CLND is not recommended for patients with a positive SLN. | Adjuvant therapy with either pembrolizumab or nivolumab for 12 months should be considered. Clinical trial |
| Resectable IIIA-IIID -IV | IIIA 5 y 93% 10 y 88% IIIB 5 y 83% 10 y 77% IIIC 5 y 69% 10 y 60% IIID 5 y 32% 10 y 24% | WLE of primary CLND is not recommended for patients with a positive SLN. Patients with resectable ITMs should undergo WLE Stage III: upfront resection or after neoadjuvant treatment Resectable stage IV: Metastasectomy or local ablative | Adjuvant anti-PD-1 therapy (nivolumab for resected stage IIIB-IV or pembrolizumab for resected stage III) or dabrafenib and trametinib for patients with resected stage III BRAFV600E-mutant melanoma (not authorized in Spain). For patients with AJCC8 stage IIIA and SLN < 1 mm, adjuvant treatment is generally not recommended. Other options not EMA or FDA approved: Neoadjuvant nivolumab plus ipilimumab followed by adjuvant therapy based on pathological response and BRAF status. Neoadjuvant plus adjuvant pembrolizumab. Clinical trial |
| Non-resectable III and IV | IV OS 5 y 59–68% 10 y 43% | First-line Ipilimumab and nivolumab is a preferred option for all patients regardless of BRAF status. First-line nivolumab or pembrolizumab is also recommended. BRAF/MEKi combination therapy is also an option in the first line for patients with BRAFV600-mutant melanoma. Clinical trial |
1.1. Genomic Alterations Defining Melanoma
1.2. ctDNA, Rational for Clinical Implementation in Melanoma
1.3. cfDNA and ctDNA as Biological Material
1.4. Analytical Techniques for ctDNA
1.5. Emerging NGS Technologies
1.6. Clinical Applications of Current ctDNA Techniques
1.7. Preanalytical Factors for Current ctDNA Techniques
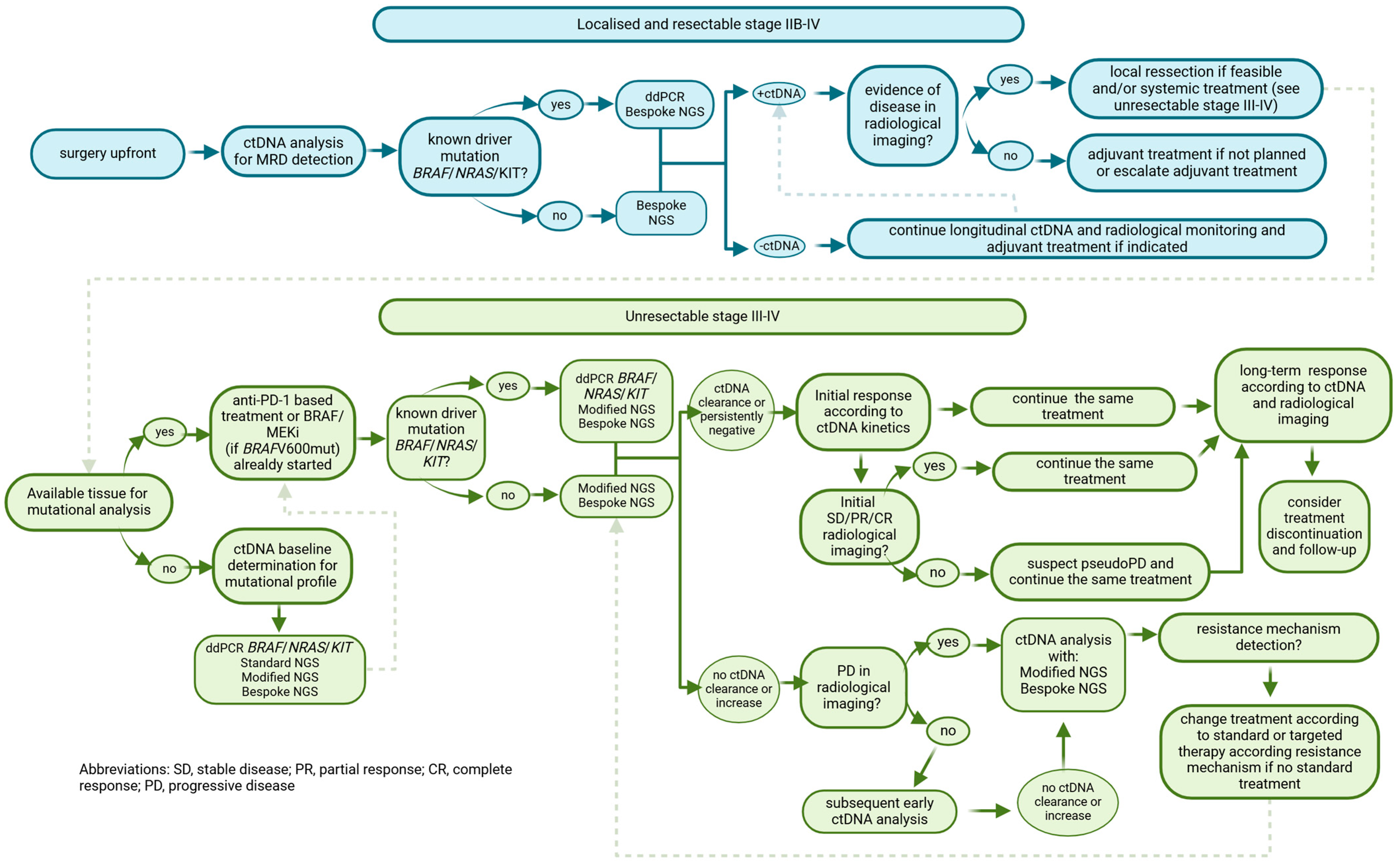
2. ctDNA Applications in Melanoma Patients
2.1. Resected High-Risk Melanoma: Stages IIB to IVD
2.1.1. ddPCR
2.1.2. NGS
2.2. Unresectable Stage III to IV Melanoma
2.2.1. Clinical Utility for Diagnostic
qPCR
ddPCR
NGS
2.2.2. Monitoring Disease Following Systemic Treatment Initiation
MM Treated with PD-1-Based Therapy
- ddPCR
- NGS
MM Treated with BRAF/MEK Inhibitors
- qPCR
- ddPCR and BEAMing
- NGS
MM Treated with ICI or BRAF/MEKi
- ddPCR
- NGS
Other Treatments
- qPCR and ddPCR
| Author Publication Date [Ref.] | N. Pts | Stage | FUP | Treatment | Age Median (Range) | Sex (M/F) (%) | Mutation | Method | Analytical Sensitivity (LoD) | Detection Rate (%) | Associated Variables | Cut Off: Positive Value or Prognostic |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lee 2018 [82] | 161 | II III | 5-year | adjuvant bevacizumab vs. placebo (AVAST-M trial) | 52 y (19–87) | 48/52 | BRAF, NRAS | ddPCR | 0.01% | 12 | OS, DFI, DMFI | Positive value: ≥1 copy/mL |
| Tan 2019 [83] | 99 | III | 20 mo | anti-PD-1 adjuvant | 57 y (22–93) | 71/29 | BRAF, NRAS, TERT | ddPCR | NR | 37 | RFS, DMFS | Positive value: ≥1 copy/mL |
| Lee 2019 [81] | 119 | III | 26 mo | NR | 64 y (20–90) | 66/34 | BRAF, NRAS | ddPCR | NR | 34 | MSS | Positive value: ≥1 positive droplets |
| Long 2022 [84] | 1127 | IIIB-D/IV | NR | adjuvant nivo + ipi vs. nivo | 56 y (45–67) | 57.5/42.5 | tumor specific alterations | WES PCR | NR | 16 | RFS, DMFS | NR |
| Genta 2024 [85] | 66 | II–IV | 39 mo | (neo) adjuvant anti-PD-1 +/− anti-CTLA-4 or BRAF/MEKi | 65 y (27–87) | 29/71 | Tumor specific alterations | WES and personalized ddPCR-NGS | NR | 29 | OS, RFS | ctDNA+: pre-set threshold defined in assays’ analytical development |
| Eroglu 2023 [108] | 30 (cohort A) | III | 19.6 mo | adjuvant nivo | 72 y (21–90) | 53/47 | tumor specific alterations | WES and personalized ddPCR-NGS | 0.004% | 17 | MRD, DMFS | Positive value: 0.07 MTM |
| Author Publication Date [Ref.] | N. Pts | Stage | FUP | Treatment | Age Median (Range) | Sex (M/F) (%) | Mutation | Method | Analytical Sensitivity (LoD) | Detection Rate (%) | Associated Variables | Cut Off: Positive Value or Prognostic |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Long-mira 2018 [86] | 19 | IV | NR | BRAF/MEKi or anti-PD-1 +/− anti-CTLA-4 or ChT | 61.63 y (43–78) | 84/16 | BRAF NRAS | allele-specific qPCR | 0.1% | 88 | ctDNA concentration and presence of BRAF/NRAS mutation | NR |
| Sobczuk 2022 [87] | 46 | III IV | ≥12 mo | BRAF/MEKi | NR | 54/46 | BRAF | qPCR | >1% | 72.4 | NR | NR |
| Giunta 2022 [88] | 56 | III IV | 18.7 mo | BRAF/MEKi or anti-PD-1 +/− anti-CTLA-4 | 62 y (34–86) | 53.3/46.7 | BRAF NRAS | qPCR | NR | 60 | tumor burden, OS | NR |
| McEvoy 2018 [89] | 32 | IV | 64.4 w | BRAF/MEKi or anti-PD-1 +/− anti-CTLA-4 | 57 y (25–83) | 62.5/37.5 | BRAF | ddPCR | NR | 71.8 | MTB, PFS | NR |
| Marcynski 2020 [91] | 19 | III IV | 130 d | NR | <61 y | 35.3/64.7 | BRAF NRAS TERT | ddPCR | 0.13–0.37% | 41.2 | PFS | NR |
| McEvoy 2017 [92] | 22 | IV | NR | treatment naïve | 51 y (24–81) | NR | TERT | ddPCR | 0.17% | 68 | PFS | NR |
| Calapre 2019 [93] | 24 | IV | NR | anti-PD-1 +/− anti-CTLA-4 | 51–70 y | 70/21 | BRAF NRAS TERT | ddPCR | NR | 70 | NR | NR |
| Lee 2017 [96] | 76 | IV | 17.5 | nivo or nivo + ipi | 65 y | 60/40 | BRAF NRAS KIT | ddPCR | NR | 53 | PFS, OS | Positive value: >2 positive droplets |
| Lee 2018 [97] | 29 | IV | 84 w | anti-PD-1 +/− anti-CTLA-4 | 65 y | 62/38 | BRAF NRAS | ddPCR | NR | 93.1 | PFS, OS | NR |
| Seremet 2019 [98] | 85 | III IV | 84 w | anti-PD-1 | 57 y (27–82) | 43.5/56.5 | BRAF NRAS | ddPCR | 0.01% | 44.4 | PFS, OS, TMTV | Positive value: >2 mutant copies per PCR Prognostic stratification: >500 copies/mL |
| Lee 2020 [99] | 72 | IVD | 35.6 mo | anti-PD-1 +/− anti-CTLA-4 | 65 y | 68/32 | BRAF NRAS KIT | ddPCR | NR | 52.7 | response PFS, OS, tumor burden | Positive value: >2.5 mutant copies/mL |
| Herbreteau 2020 [100] | 53 (exploratori cohort) 49 (validation cohort) | IIIC- IV | NR | anti-PD-1 +/− anti-CTLA-4 or BRAF/MEKi | 62 y (52.5–72.4) | 54.4/45.5 | BRAF NRAS | ddPCR | NR | 50 | PFS, OS | Positive value: >8 mutant copies/mL |
| Schreuer 2016 [110] | 36 | IV | NR | dabrafenib or dabrafenib + trametinib or vemurafenib | 52 y | 33/67 | BRAF | allele specific qPCR | NR | 75 | DCR, PFS | NR |
| Gonzalez-Cao 2015 [111] | 22 | IV | NR | BRAFi | 62 y (35–83) | 63/27 | BRAF | allele specific qPCR | 0.005% | 57.7 | PFS, OS | Positive value: BRAFV600 allele amplified in 2 of 4 quadriplicates |
| Gonzalez-Cao 2018 [112] | 66 | IV | NR | BRAF/MEKi or ipior ChT | 58 y (28–44) | 48/52 | BRAF | allele specific qPCR | 0.005% | 66.7 | tumor burden, PFS, OS | Prognostic stratification: High > 10.5 pg/µL Low–indetectable 0–10.5 pg/µL |
| Sanmamed 2015 [52] | 20 | IV | NR | BRAFi | 50 y | 65/35 | BRAF | ddPCR | 0.005% | 84.3 | tumor burden, PFS, OS | Positive value: ≥1 mutant copies/mL Prognostic stratification: >216 copies/mL |
| Forschner 2020 [113] | 19 | IV | NR | BRAF/MEKi | 51 y (32–79) | 42/58 | BRAF | ddPCR | NR | 68 | OS | NR |
| Santiago-walker 2016 [47] | 732 | IV | NR | dabrafenib (BREAK-2 trial) dabrafenib vs. DTIC (BREAK-3 trial) dabrafenib (BREAK-MB trial) trametinib vs. ChT (METRIC trial) | NR | NR | BRAF | BEAMing | 0.01% | 81 | ORR, PFS, OS | NR |
| Syeda 2021 [115] | 383 | IIIC- IV | 20 mo | Dabrafenib + trametinib (COMBI-d, COMBI-MB trials) | 56 y (45–65) | 53/47 | BRAF | ddPCR | 0.019–0.022% | 89–93 | PFS, OS, BOR | Positive value: 0.28 mutant copies/mL if BRAF V600E and 0.34 mutant copies/mL if BRAFV600K Prognostic stratification: >64 copies/mL |
| Gray 2015 [42] | 48 | IV | NR | Vemurafenib or dabrafenibdab + trametinib or pemor nivo + ipi | NR | NR | BRAF NRAS | ddPCR | 0.01% | 65 | ORR, PFS | Positive value: ≥1 mutant copies/mL Prognostic stratification: ≥10 copies/mL |
| Warburton 2020 [117] | 13 | IV | 57 mo | BRAF+/−MEKi at discontinuation | 61 y (38–71) | 54/46 | BRAF | ddPCR | NR | 15 | MRD | Positive value: ≥1 positive triplicate |
| Di guardo 2021 [118] | 24 | IV | 37.8 mo | BRAF+/−MEKi at discontinuation | 56 y (43–63) | 50/50 | BRAF | ddPCR | 0.1% | NR | PFS after treatment discontinuation | NR |
| Valpione 2018 [120] | 43 | IV | 11.9 mo | Ipi or BRAF/MEKi anti-PD-1 or DTIC | 58.1 y (18–85.1) | 58/42 | BRAF NRAS KIT | ddPCR | NR | 70 | tumor burden, OS | Prognostic stratification: ≥89 pg/μL |
| Varaljai 2019 [121] | 96 | III IV | NR | BRAF/MEKi or anti-PD-1 +/− anti-CTLA-4 | NR | NR | BRAF NRAS TERT | ddPCR | NR | NR | response, PFS, OS | NR |
| Marsavela 2020 [122] | 110 | IV | 95 w | BRAF/MEKi or anti-PD-1 +/− anti-CTLA-4 | 65 y | 65/35 | BRAF NRAS | ddPCR | NR | NR | PFS (only 1 L ICI patients) OS | Prognostic stratification: ≤20 copies/mL |
| Marsavela 2020 [123] | 142 | IV | 113 w | anti-PD-1 +/− anti-CTLA-4 or BRAF/MEKi | NR | NR | BRAF NRAS | ddPCR | NR | 65 | response, PFS, OS | NR |
| Wong 2017 [90] | 52 | IV | 391 days | BRAF/MEKi or immunotherapy | 61 y (24–83) | NR | BRAF NRAS TERT | ddPCR | 0.1% | 77 | tumor burden, PFS | NR |
| Xi 2016 [127] | 48 | IV | NR | TILs | NR | NR | BRAF | allele-specific qPCR | 0.05% | NR | CR after 1–2 years | NR |
| Forthum 2019 [128] | 26 | IV | NR | bevacizumab | 63 y (29–77) | 58/42 | BRAF NRAS | ddPCR | 0.05% | 88 | response, PFS, OS | Positive value: >1% BRAF/NRASmut-positive droplets |
| Diefenbach 2020 [95] | 74 | III IV | NR | treatment naïve | 61 y (23–88) | 74/26 | 30-genes melanoma custom panel BRAF, NRAS,-KIT | NGS ddPCR | 0.2% (NGS) | 84 | stage with cfDNA input | Positive value: >1% BRAF/NRAS/KIT mutation-positive droplets |
| Khagi 2017 [103] | 69 (10 melanoma) | IV | NR | anti-PD-1 | 56 y (21–85) | 62.3/37.7 | 73-genes panel | NGS | 0.1% | 91 | PFS, OS, response (SD ≥ 6, PR, CR) | Prognostic stratification: VUS > 3 alterations better outcome |
| Forschner 2019 [105] | 35 | IV | 213 d | anti-PD-1 +/− anti-CTLA-4 | 55 y (17–79) | 54/46 | BRAF 710-tumor associated genes | NGS ddPCR | NR | NR | response, OS | Prognostic stratification: TMB high > 23.1 better outcome |
| Bratman 2020 [62] | 94 (10 melanoma) | IV | 13.8 mo | anti-PD-1 | 59 y | 48/62 | tumor specific alterations | WES and personalized ddPCR-NGS | 0.004% | 98 | PFS, OS, CBR | Positive value: 0.07 MTM |
| Eroglu 2023 [108] | 29 (cohort B) 10 (cohort C) | III IV | 14.2 mo (cohort B) 14.67 mo (cohort C) | cohort B: nivo +/− ipi or ICI + agent cohort C: after planned completion of ICI for MM disease | 64 y (39–89) (cohort B) 66 y (51–85) (cohort C) | 69/31 (cohort B) 70/30 (cohort C) | tumor specific alterations | WES and personalized ddPCR-NGS | 0.004% | 90 (cohort B) 10 (cohort C) | PFS | Positive value: 0.07 MTM |
| Schroeder 2024 [109] | 87 | III-IV resected n = 22 III-IV unresectable n = 65 | NR | adjuvant nivo/pem n = 22 Systemic treatment nivo + ipi n = 65 | 64 y (56–76) | 44/56 | 700-gene panel | targeted NGS | NR | 87 | LDH, S100, PET/CT MTV, PFS, OS | Positive: ≥3 tumor variants |
| Gangadhar 2018 [125] | 25 | III IV | NR | BRAF/MEKi or anti-PD-1 +/− anti-CTLA-4 | 57.6 y | 72/28 | 61-gene panel | NGS | 1% | 48 | tumor burden | NR |
| Olbryt 2021 [126] | 22 | IV | NR | BRAF/MEKi or anti-PD-1 +/− anti-CTLA-4 | 52 y | 40/60 | 52-gene panel | NGS | 0.1% | NR | LDH | NR |
2.3. ctDNA in the Cerebrospinal Fluid for Monitoring Intracranial Response
3. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients with Previously Untreated BRAF Wild-Type Advanced Melanoma Treated with Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Carlino, M.S.; McNeil, C.; Ribas, A.; Grob, J.J.; Schachter, J.; Nyakas, M.; Kee, D.; Petrella, T.M.; Blaustein, A.; et al. Seven-Year Follow-Up of the Phase III KEYNOTE-006 Study: Pembrolizumab Versus Ipilimumab in Advanced Melanoma. J. Clin. Oncol. 2023, 41, 3998–4003. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Rutkowski, P.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Queirolo, P.; Dummer, R.; Butler, M.O.; Hill, A.G.; et al. CheckMate 067 Investigators. Final, 10-Year Outcomes with Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2024, 392, 11–22. [Google Scholar] [CrossRef]
- Long, G.V.; Stephen Hodi, F.; Lipson, E.J.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Salman, P.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; et al. Overall Survival and Response with Nivolumab and Relatlimab in Advanced Melanoma. NEJM Evid. 2023, 2, EVIDoa2200239, Erratum in NEJM Evid. 2023, 2, EVIDx2300104. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised con-trolled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Long, G.; Flaherty, K.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2019, 30, 1848, Erratum in Ann. Oncol. 2017, 28, 1631–1639. https://doi.org/10.1093/annonc/mdx176. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsová, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients with BRAF V600-Mutant Melanoma. Clin. Oncol. 2023, 41, 2301. [Google Scholar] [CrossRef]
- Luke, J.J.; Ascierto, P.A.; Khattak, M.A.; Merino, L.d.l.C.; Del Vecchio, M.; Rutkowski, P.; Spagnolo, F.; Mackiewicz, J.; Chiarion-Sileni, V.; Kirkwood, J.M.; et al. Pembrolizumab versus placebo as adjuvant therapy in resected stage IIB or IIC melanoma: Final analysis of distant metastasis-free survival in the phase III KEYNOTE-716 study. J. Clin. Oncol. 2024, 42, 1619–1624. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Del Vecchio, M.; Weber, J.; Hoeller, C.; Grob, J.-J.; Mohr, P.; Loquai, C.; Dutriaux, C.; Chiarion-Sileni, V.; Mackiewicz, J.; et al. Adjuvant nivolumab in resected stage IIB/C melanoma: Primary results from the randomized, phase 3 CheckMate 76K trial. Nat. Med. 2023, 29, 2835–2843, Erratum in Nat. Med. 2023, 30, 607; Erratum in Nat. Med. 2024, 30, 906. https://doi.org/10.1038/s41591-023-02775-w. [Google Scholar] [CrossRef]
- Larkin, J.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Fernandez, A.M.A.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.-J.; Chiarion-Sileni, V.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III/IV Melanoma: 5-Year Efficacy and Biomarker Results from CheckMate 238. Clin. Cancer Res. 2023, 29, 3352–3361. [Google Scholar] [CrossRef]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Kicinski, M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Meshcheryakov, A.; Khattak, A.; et al. Five-Year Analysis of Adjuvant Pembrolizumab or Placebo in Stage III Melanoma. NEJM Evid. 2022, 1, EVIDoa2200214. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Kirkwood, J.M.; Chiarion Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; et al. Five-Year Analysis of Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N. Engl. J. Med. 2020, 383, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Othus, M.; Prieto, V.; Lowe, M.; Buchbinder, E.; Chen, Y.; Hyngstrom, J.; Lao, C.D.; Truong, T.G.; Chandra, S.; et al. LBA6 Neoadjuvant versus adjuvant pembrolizumab for resected stage III-IV melanoma (SWOG S1801). Ann. Oncol. 2022, 33 (Suppl. S7), S1408. [Google Scholar] [CrossRef]
- Blank, C.U.; Lucas, M.W.; Scolyer, R.A.; van de Wiel, B.A.; Menzies, A.M.; Lopez-Yurda, M.; Hoeijmakers, L.L.; Saw, R.P.; Lijnsvelt, J.M.; Maher, N.G.; et al. Neoadjuvant Nivolumab and Ipilimumab in Resectable Stage III Melanoma. N. Engl. J. Med. 2024, 391, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Amaral, T.; Ottaviano, M.; Arance, A.; Blank, C.; Chiarion-Sileni, V.; Donia, M.; Dummer, R.; Garbe, C.; Gershenwald, J.; Gogas, H.; et al. ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2024, 36, 10–30. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Borczuk, A.C.; Allen, T.C. PD-L1 and Lung Cancer: The Era of Precision-ish Medicine? Arch. Pathol. Lab. Med. 2016, 140, 351–354. [Google Scholar] [CrossRef]
- Schmidt, L.H.; Kümmel, A.; Görlich, D.; Mohr, M.; Bröckling, S.; Mikesch, J.H.; Hartmann, W. PD-1 and PD-L1 Expression in NSCLC Indicate a Favorable Prognosis in Defined Subgroups. PLoS ONE 2015, 10, e0136023. [Google Scholar] [CrossRef]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Claps, G.; Faouzi, S.; Quidville, V.; Chehade, F.; Shen, S.; Vagner, S.; Robert, C. The multiple roles of LDH in cancer. Nat. Rev. Clin. Oncol. 2022, 19, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Vanni, I.; Tanda, E.T.; Spagnolo, F.; Andreotti, V.; Bruno, W.; Ghiorzo, P. The current state of molecular testing in the BRAF-mutated melanoma landscape. Front. Mol. Biosci. 2020, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A. Melanoma Staging: American Joint Committee on Cancer (AJCC) 8th Edition and Beyond. Ann. Surg. Oncol. 2018, 25, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Weigelt, B.; Reis-Filho, J.S. Going with the flow: From circulating tumor cells to DNA. Sci. Transl. Med. 2013, 5, 207ps14. [Google Scholar] [CrossRef]
- Molina-Vila, M.A.; de-Las-Casas, C.M.; Bertran-Alamillo, J.; Jordana-Ariza, N.; González-Cao, M.; Rosell, R. cfDNA analysis from blood in melanoma. Ann. Transl. Med. 2015, 3, 309. [Google Scholar] [CrossRef] [PubMed]
- Castrejon, N.; Martin, R.; Carrasco, A.; Castillo, P.; Garcia, A.; Albero-González, R.; García, M.; Marginet, M.; Palau, N.; Hernández, M.; et al. Feasibility and Impact of Embedding an Extended DNA and RNA Tissue-Based Sequencing Panel for the Routine Care of Patients with Advanced Melanoma in Spain. Int. J. Mol. Sci. 2024, 25, 6942. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; Fujimoto, A.; Morton, D.L.; Hoon, D.S.B. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin. Cancer Res. 2004, 10, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.L.; Aplin, A.E. Targeting mutant NRAS signaling pathways in melanoma. Pharmacol. Res. 2016, 107, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Jakob, J.A.; Bassett, R.L.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2023, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Ekedahl, H.; Lauss, M.; Olsson, H.; Griewank, K.G.; Schadendorf, D.; Ingvar, C.; Jönsson, G. High TERT promoter mutation frequency in non-acral cutaneous metastatic melanoma. Pigment. Cell Melanoma Res. 2016, 29, 598–600. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2021, 61, 1659–1665. [Google Scholar] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef]
- Jenkins, S.; Yang, J.C.-H.; Ramalingam, S.S.; Yu, K.; Patel, S.; Weston, S.; Hodge, R.; Cantarini, M.; Jänne, P.A.; Mitsudomi, T.; et al. Plasma ctDNA analysis for detection of the EGFR T790M mutation in patients with advanced non–small cell lung cancer. J. Thorac. Oncol. 2017, 12, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Digital PCR hits its stride. Nat. Methods 2012, 9, 541–544. [Google Scholar] [CrossRef]
- Richardson, A.L.; Iglehart, J.D. BEAMing up personalized medicine: Mutation detection in blood. Clin. Cancer Res. 2012, 18, 3209–3211. [Google Scholar] [CrossRef]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.S. Digital assays part I: Partition ing statistics and digital PCR. SLAS Technol. 2017, 22, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.; Emslie, K.R. Basic concepts and validation of digital PCR measurements. In Digital PCR; Springer: New York, NY, USA, 2018; pp. 11–24. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Milbury, C.A.; Zhong, Q.; Lin, J.; Williams, M.; Olson, J.; Link, D.R.; Hutchison, B. Determining lower limits of detection of digital PCR assays for cancer-related gene mutations. Biomol. Detect. Quantif. 2014, 1, 8–22. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative cell- free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Shu, Y.; Wu, X.; Tong, X.; Wang, X.; Chang, Z.; Mao, Y.; Chen, X.; Sun, J.; Wang, Z.; Hong, Z.; et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci. Rep. 2017, 7, 583. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Monitoring Melanoma Using Circulating Free DNA. Am. J. Clin. Dermatol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khier, S.; Lohan, L. Kinetics of circulating cell-free DNA for biomedical applications: Critical appraisal of the literature. Future Sci. OA 2018, 4, FSO295. [Google Scholar] [CrossRef] [PubMed]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.B.; Kopetz, E.S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451, Erratum in Nature 2018, 554, 264. https://doi.org/10.1038/nature25161. [Google Scholar] [CrossRef] [PubMed]
- Coombes, R.C.; Page, K.; Salari, R.; Hastings, R.K.; Armstrong, A.C.; Ahmed, S.; Ali, S.; Cleator, S.J.; Kenny, L.M.; Stebbing, J.; et al. Personalized Detection of Circulating Tumor DNA Antedates Breast Cancer Metastatic Recurrence. Clin. Cancer Res. 2019, 25, 4255–4263. [Google Scholar] [CrossRef]
- Bratman, S.V.; Yang, S.Y.C.; Iafolla, M.A.J.; Liu, Z.; Hansen, A.R.; Bedard, P.L.; Lheureux, S.; Spreafico, A.; Razak, A.A.; Shchegrova, S.; et al. Personalised circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat. Cancer 2020, 38, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Flach, S.; Howarth, K.; Hackinger, S.; Pipinikas, C.; Ellis, P.; McLay, K.; Marsico, G.; Forshew, T.; Walz, C.; Reichel, C.A.; et al. Liquid BIOpsy for MiNimal RESidual DiSease Detection in Head and Neck Squamous Cell Carcinoma (LIONESS)-a personalised circulating tumour DNA analysis in head and neck squamous cell carcinoma. Br. J. Cancer 2022, 126, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Trigg, R.M.; Martinson, L.J.; Parpart-Li, S.; Shaw, J.A. Factors that influence quality and yield of circulating-free DNA: A systematic review of the methodology literature. Heliyon 2018, 4, e00699. [Google Scholar] [CrossRef]
- Thierry, A.R.; Mouliere, F.; Gongora, C.; Ollier, J.; Robert, B.; Ychou, M.; Del Rio, M.; Molina, F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010, 38, 6159–6175. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Devonshire, A.S.; Whale, A.S.; Gutteridge, A.; Jones, G.; Cowen, S.; Foy, C.A.; Huggett, J.F. Towards standardisation of cell-free DNA measurement in plasma: Controls for extraction efficiency, fragment size bias and quantification. Anal. Bioanal. Chem. 2014, 406, 6499–6512. [Google Scholar] [CrossRef] [PubMed]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.; Spittle, C.; Karlin-Neumann, G.; Polsky, D. Validation of Circulating Tumor DNA Assays for Detection of Metastatic Melanoma. Methods Mol. Biol. 2020, 2055, 155–180. [Google Scholar] [CrossRef]
- Barra, G.B.; Santa Rita, T.H.; de Almeida Vasques, J.; Chianca, C.F.; Nery, L.F.; Santana Soares Costa, S. EDTA-mediated inhibition of DNases protects circulating cell-free DNA from ex vivo degradation in blood samples. Clin. Biochem. 2015, 48, 976–981. [Google Scholar] [CrossRef] [PubMed]
- van Ginkel, J.H.; van den Broek, D.A.; van Kuik, J.; Linders, D.; de Weger, R.; Willems, S.M.; Huibers, M.M.H. Preanalytical blood sample workup for cell-free DNA analysis using Droplet Digital PCR for future molecular cancer diagnostics. Cancer Med. 2017, 6, 2297–2307. [Google Scholar] [CrossRef]
- Lam, N.Y.; Rainer, T.H.; Chiu, R.W.; Lo, Y.M. EDTA is a better anticoagulant than heparin or citrate for delayed blood processing for plasma DNA analysis. Clin. Chem. 2004, 50, 256–257. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S.; Blenkiron, C.; Stephens, R.; Mathy, J.A.; Somers-Edgar, T.; Rolfe, G.; Martin, R.; Jackson, C.; Eccles, M.; Robb, T.; et al. Dynamic ctDNA Mutational Complexity in Patients with Melanoma Receiving Immunotherapy. Mol. Diagn. Ther. 2023, 27, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.W.; Poon, L.L.; Lau, T.K.; Leung, T.N.; Wong, E.M.; Lo, Y.D. Effects of blood-processing protocols on fetal and total DNA quantification in maternal plasma. Clin. Chem. 2001, 47, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. NCI Biospecimen Evidence-Based Practices (BEBP)—Cell-Free DNA: Biospecimen Collection and Processing V1; National Cancer Institute: Singapore, 2024. [Google Scholar] [CrossRef]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.-K.; Jänne, P.A.; et al. False positive plasma genotyping due to clonal hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed]
- Garlan, F.; Blanchet, B.; Kramkimel, N.; Puszkiel, A.; Golmard, J.-L.; Noe, G.; Dupin, N.; Laurent-Puig, P.; Vidal, M.; Taly, V.; et al. Circulating Tumor DNA Measurement by Picoliter Droplet-Based Digital PCR and Vemurafenib Plasma Concentrations in Patients with Advanced BRAF-Mutated Melanoma. Target. Oncol. 2017, 12, 365–371. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response and re-sistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cellfree plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Kalashnikova, E.; Aushev, V.N.; Malashevich, A.K.; Tin, A.; Krinshpun, S.; Salari, R.; Scalise, C.B.; Ram, R.; Malhotra, M.; Ravi, H.; et al. Correlation between variant allele frequency and mean tumor molecules with tumor burden in patients with solid tumors. Mol. Oncol. 2023, 18, 2649–2657. [Google Scholar] [CrossRef]
- Lee, J.; Saw, R.; Thompson, J.; Lo, S.; Spillane, A.; Shannon, K.; Stretch, J.; Howle, J.; Menzies, A.; Carlino, M.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef]
- Lee, R.; Gremel, G.; Marshall, A.; Myers, K.; Fisher, N.; Dunn, J.; Dhomen, N.; Corrie, P.; Middleton, M.; Lorigan, P.; et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Sandhu, S.; Lee, R.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef]
- Long, G.; Desai, K.; Tang, T.; Weber, J.; Dolfi, S.; Ritchings, C.; Huang, S.-P.; Bolisetty, M.; Sausen, M.; Del Vecchio, M.; et al. 788O—Association of pre-treatment ctDNA with disease recurrence and clinical and translational factors in patients with stage IIIB-D/IV melanoma treated with adjuvant immunotherapy(CheckMate 915). Ann. Oncol. 2022, 33 (Suppl. S7), S356–S409. [Google Scholar] [CrossRef]
- Genta, S.; Araujo, D.; Hueniken, K.; Pipinikas, C.; Ventura, R.; Rojas, P.; Jones, G.; Butler, M.; Saibil, S.; Yu, C.; et al. Bespoke ctDNA for longitudinal detection of molecular residual disease in high-risk melanoma patients. ESMO Open 2024, 9, 103978. [Google Scholar] [CrossRef] [PubMed]
- Long-Mira, E.; Ilie, M.; Chamorey, E.; Leduff-Blanc, F.; Montaudié, H.; Tanga, V.; Allégra, M.; Lespinet-Fabre, V.; Bordone, O.; Bonnetaud, C.; et al. Monitoring BRAF and NRAS mutations with cell-free cir-culating tumor DNA from metastatic melanoma patients. Oncotarget 2018, 9, 36238–36249. [Google Scholar] [CrossRef] [PubMed]
- Sobczuk, P.; Kozak, K.; Kopeć, S.; Rogala, P.; Świtaj, T.; Koseła-Paterczyk, H.; Gos, A.; Tysarowski, A.; Rutkowski, P. The Use of ctDNA for BRAF Mutation Testing in Routine Clinical Practice in Patients with Advanced Melanoma. Cancers 2022, 14, 777. [Google Scholar] [CrossRef]
- Giunta, E.F.; De Falco, V.; Vitiello, P.P.; Guerrera, L.P.; Suarato, G.; Napolitano, R.; Perrone, A.; Argenziano, G.; Franco, R.; Caraglia, M.; et al. Clinical Utility of Liquid Biopsy to Detect BRAF and NRAS Mutations in Stage III/IV Melanoma Patients by Using Real-Time PCR. Cancers 2022, 14, 3053. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, A.C.; Warburton, L.; Al-Ogaili, Z.; Celliers, L.; Calapre, L.; Pereira, M.R.; Khattak, M.A.; Meniawy, T.M.; Millward, M.; Ziman, M.; et al. Correlation between circulating tumor DNA and metabolic tumor burden in metastatic melanoma patients. BMC Cancer 2018, 18, 726. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marczynski, G.T.; Laus, A.C.; Dos Reis, M.B.; Reis, R.M.; Vazquez, V.L. Circulating tumor DNA (ctDNA) detection is associated with shorter progression-free survival in advanced melanoma patients. Sci. Rep. 2020, 10, 18682. [Google Scholar] [CrossRef]
- McEvoy, A.C.; Calapre, L.; Pereira, M.R.; Giardina, T.; Robinson, C.; Khattak, M.A.; Meniawy, T.M.; Pritchard, A.L.; Hayward, N.K.; Amanuel, B.; et al. Sensitive droplet digital PCR method for detection of TERT promoter mutations in cell free DNA from patients with metastatic melanoma. Oncotarget 2017, 8, 78890–78900. [Google Scholar] [CrossRef] [PubMed]
- Calapre, L.; Giardina, T.; Robinson, C.; Reid, A.L.; Al-Ogaili, Z.; Pereira, M.R.; McEvoy, A.C.; Warburton, L.; Hayward, N.K.; Khattak, M.A.; et al. Locus-specific concordance of genomic alterations between tissue and plasma circulating tumor DNA in metastatic melanoma. Mol. Oncol. 2019, 13, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Huang, S.K.; Huynh, K.T.; Salomon, M.P.; Chang, S.-C.; Marzese, D.M.; Lanman, R.B.; Talasaz, A.; Hoon, D.S. Multiplex Gene Profiling of Cell-Free DNA in Patients with Metastatic Melanoma for Monitoring Disease. JCO Precis. Oncol. 2018, 2, PO.17.00225. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Saw, R.P.M.; Howle, J.R.; Spillane, A.J.; Scolyer, R.A.; Kefford, R.F.; et al. Design and Testing of a Custom Melanoma Next Generation Sequencing Panel for Analysis of Circulating Tumor DNA. Cancers 2020, 12, 2228. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumor DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients with Metastatic Melanoma Treated with Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef]
- Lee, J.H.; Menzies, A.M.; Carlino, M.S.; McEvoy, A.C.; Sandhu, S.; Weppler, A.M.; Diefenbach, R.J.; Dawson, S.-J.; Kefford, R.F.; Millward, M.J.; et al. Longitudinal Monitoring of ctDNA in Patients with Melanoma and Brain Metastases Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2020, 26, 4064–4071. [Google Scholar] [CrossRef] [PubMed]
- Herbreteau, G.; Vallée, A.; Knol, A.-C.; Théoleyre, S.; Quéreux, G.; Frénard, C.; Varey, E.; Hofman, P.; Khammari, A.; Dréno, B.; et al. Circulating Tumour DNA Is an Independent Prognostic Biomarker for Survival in Metastatic BRAF or NRAS-Mutated Mela-noma Patients. Cancers 2020, 12, 1871. [Google Scholar] [CrossRef]
- Martínez-Vila, C.; González-Navarro, E.A.; Teixido, C.; Martin, R.; Aya, F.; Juan, M.; Arance, A. Lymphocyte T Subsets and Outcome of Immune Check-point Inhibitors in Melanoma Patients: An Oncologist’s Perspective on Current Knowledge. Int. J. Mol. Sci. 2024, 25, 9506. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e13. [Google Scholar] [CrossRef] [PubMed]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef]
- Niknafs, N.; Balan, A.; Cherry, C.; Hummelink, K.; Monkhorst, K.; Shao, X.M.; Belcaid, Z.; Marrone, K.A.; Murray, J.; Smith, K.N.; et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat. Med. 2023, 29, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.-T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Landon, B.V.; Zaidi, A.H.; Singh, D.; Canzoniero, J.V.; Balan, A.; Hales, R.K.; Voong, K.R.; Battafarano, R.J.; Jobe, B.A.; et al. Neoadjuvant nivolumab or nivolumab plus LAG-3 inhibitor relatlimab in resectable esophageal/gastroesophageal junction cancer: A phase Ib trial and ctDNA analyses. Nat. Med. 2024, 30, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Krinshpun, S.; Kalashnikova, E.; Sudhaman, S.; Topcu, T.O.; Nichols, M.; Martin, J.; Bui, K.M.; Palsuledesai, C.C.; Malhotra, M.; et al. Circulating tumor DNA-based molecular residual disease detection for treatment monitoring in advanced melanoma patients. Cancer 2023, 129, 1723–1734. [Google Scholar] [CrossRef]
- Schroeder, C.; Gatidis, S.; Kelemen, O.; Schütz, L.; Bonzheim, I.; Muyas, F.; Martus, P.; Admard, J.; Armeanu-Ebinger, S.; Gückel, B.; et al. Tumour-informed liquid biopsies to monitor advanced melanoma patients under immune checkpoint inhibition. Nat. Commun. 2024, 15, 8750. [Google Scholar] [CrossRef] [PubMed]
- Schreuer, M.; Meersseman, G.; Van Den Herrewegen, S.; Jansen, Y.; Chevolet, I.; Bott, A.; Wilgenhof, S.; Seremet, T.; Jacobs, B.; Buyl, R.; et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J. Transl. Med. 2016, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cao, M.; Mayo-De-Las-Casas, C.; Molina-Vila, M.A.; De Mattos-Arruda, L.; Muñoz-Couselo, E.; Manzano, J.L.; Cortes, J.; Berros, J.P.; Drozdowskyj, A.; Sanmamed, M.; et al. BRAF mutation analysis in circulating free tumor DNA of melanoma patients treated with BRAF inhibitors. Melanoma Res. 2015, 25, 486–495. [Google Scholar] [CrossRef]
- Gonzalez-Cao, M.; Casas, C.M.d.L.; Ariza, N.J.; Manzano, J.L.; Molina-Vila, M.; Soriano, V.; Puertolas, T.; Balada, A.; Soria, A.; Majem, M.; et al. Early evolution of BRAFV600 status in the blood of melanoma patients correlates with clinical outcome and identifies patients refractory to therapy. Melanoma Res. 2018, 28, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Weißgraeber, S.; Hadaschik, D.; Schulze, M.; Kopp, M.; Kelkenberg, S.; Sinnberg, T.; Garbe, C.; Biskup, S.; Battke, F. Circulating Tumor DNA Correlates with Outcome in Metastatic Melanoma Treated by BRAF and MEK Inhibitors—Results of a Prospective Biomarker Study. Onco Targets Ther. 2020, 13, 5017–5032. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.C.; Belloum, Y.; Deitert, B.; Sementsov, M.; Heidrich, I.; Gebhardt, C.; Keller, L.; Pantel, K. Current and Future Clinical Applications of ctDNA in Immuno-Oncology. Cancer Res. 2022, 82, 349–358. [Google Scholar] [CrossRef]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumor DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.J.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalised Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Warburton, L.; Meniawy, T.M.; Calapre, L.; Pereira, M.; McEvoy, A.; Ziman, M.; Gray, E.S.; Millward, M. Stopping targeted therapy for complete responders in advanced BRAF mutant melanoma. Sci. Rep. 2020, 10, 18878. [Google Scholar] [CrossRef] [PubMed]
- Di Guardo, L.; Randon, G.; Corti, F.; Vallacchi, V.; Raimondi, A.; Fucà, G.; Bini, M.; Maurichi, A.; Patuzzo, R.; Gallino, G.; et al. Liquid Biopsy and Radiological Response Predict Outcomes Following Discontinuation of Targeted Therapy in Patients with BRAF Mutated Melanoma. Oncologist 2021, 26, 1079–1084. [Google Scholar] [CrossRef]
- Kaisaki, P.J.; Cutts, A.; Popitsch, N.; Camps, C.; Pentony, M.M.; Wilson, G.; Page, S.; Kaur, K.; Vavoulis, D.; Henderson, S.; et al. Targeted Next-Generation Sequencing of Plasma DNA from Cancer Pa-tients: Factors Influencing Consistency with Tumor DNA and Prospective Investigation of Its Utility for Diagnosis. PLoS ONE 2016, 11, e0162809. [Google Scholar] [CrossRef]
- Valpione, S.; Gremel, G.; Mundra, P.; Middlehurst, P.; Galvani, E.; Girotti, M.R.; Lee, R.J.; Garner, G.; Dhomen, N.; Lorigan, P.C.; et al. Plasma total cell-free DNA (cfDNA) is a surrogate biomarker for tumor burden and a prognostic biomarker for survival in metastatic melanoma patients. Eur. J. Cancer 2018, 88, 1–9. [Google Scholar] [CrossRef]
- Váraljai, R.; Wistuba-Hamprecht, K.; Seremet, T.; Diaz, J.M.S.; Nsengimana, J.; Sucker, A.; Griewank, K.; Placke, J.-M.; Horn, P.A.; von Neuhoff, N.; et al. Application of Circulating Cell-Free Tumor DNA Profiles for Therapeutic Monitoring and Outcome Prediction in Genetically Heterogeneous Metastatic Melanoma. JCO Precis. Oncol. 2019, 3, PO.18.00229. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; Lee, J.; Calapre, L.; Wong, S.Q.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Abed, A.; et al. Circulating Tumor DNA Predicts Outcome from First-, but not Second-line Treatment and Identifies Melanoma Patients Who May Benefit from Combination Immunotherapy. Clin. Cancer Res. 2020, 26, 5926–5933. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; Johansson, P.A.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Khattak, M.A.; Meniawy, T.M.; Amanuel, B.; et al. The Prognostic Impact of Circulating Tumor DNA in Melanoma Patients Treated with Systemic Therapies-Beyond BRAF Mutant Detection. Cancers 2020, 12, 3793. [Google Scholar] [CrossRef]
- Feng, S.N.; Cen, X.T.; Tan, R.; Wei, S.S.; Sun, L.D. The prognostic value of circulating tumor DNA in patients with melanoma: A systematic review and meta-analysis. Transl. Oncol. 2021, 14, 101072. [Google Scholar] [CrossRef] [PubMed]
- Gangadhar, T.C.; Savitch, S.L.; Yee, S.S.; Xu, W.; Huang, A.C.; Harmon, S.; Lieberman, D.B.; Soucier, D.; Fan, R.; Black, T.A.; et al. Feasibility of monitoring advanced melanoma patients using cell-free DNA from plasma. Pigment. Cell Melanoma Res. 2018, 31, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Olbryt, M.; Rajczykowski, M.; Bal, W.; Fiszer-Kierzkowska, A.; Cortez, A.J.; Mazur, M.; Suwiński, R.; Widłak, W. NGS Analysis of Liquid Biopsy (LB) and Formalin-Fixed Paraffin-Embedded (FFPE) Melanoma Samples Using Oncomine™ Pan-Cancer Cell-Free As-say. Genes 2021, 12, 1080. [Google Scholar] [CrossRef]
- Xi, L.; Pham, T.H.; Payabyab, E.C.; Sherry, R.M.; Rosenberg, S.A.; Raffeld, M. Circulating Tumor DNA as an Early Indicator of Response to T-cell Transfer Immunotherapy in Metastatic Melanoma. Clin. Cancer Res. 2016, 22, 5480–5486. [Google Scholar] [CrossRef] [PubMed]
- Forthun, R.B.; Hovland, R.; Schuster, C.; Puntervoll, H.; Brodal, H.P.; Namløs, H.M.; Aasheim, L.B.; Meza-Zepeda, L.A.; Gjertsen, B.T.; Knappskog, S.; et al. ctDNA detected by ddPCR reveals changes in tumor load in metastatic malignant melanoma treated with bevacizumab. Sci. Rep. 2019, 9, 17471. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Butler, M.O.; Shoushtari, A.N.; Hassel, J.C.; Ikeguchi, A.; Hernandez-Aya, L.; Nathan, P.; Hamid, O.; Piulats, J.M.; Rioth, M.; et al. Clinical and molecular response to tebentafusp in previously treated patients with metastatic uveal melanoma: A phase 2 trial. Nat. Med. 2022, 28, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumor DNA better represents the genomic alterations of brain tumors than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, P.; Pentsova, E.; Abdel-Wahab, O.; Diamond, E.; Hyman, D.; Merghoub, T.; You, D.; Gasmi, B.; Viale, A.; Chapman, P.B. Quanti-fication of tumor-derived cell free DNA(cfDNA) by digital PCR (DigPCR) in cerebrospinal fluid of patients with BRAFV600 mutated malignancies. Oncotarget 2016, 7, 85430–85436. [Google Scholar] [CrossRef]
- Li, Y.; Pan, W.; Connolly, I.D.; Reddy, S.; Nagpal, S.; Quake, S.; Gephart, M.H. Tumor DNA in cerebral spinal fluid reflects clinical course in a patient with melanoma leptomeningeal brain metastases. J. Neurooncol 2016, 128, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Glitza, I.C.; Smalley, K.S.M.; Brastianos, P.K.; Davies, M.A.; McCutcheon, I.; Liu, J.K.C.; Ahmed, K.A.; Arrington, J.A.; Evernden, B.R.; Smalley, I.; et al. Leptomeningeal disease in melanoma patients: An update to treatment, challenges, and future directions. Pigment. Cell Melanoma Res. 2020, 33, 527–541. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Glitza Oliva, I.C.; Douse, D.Y.; Chen, M.M.; Lan, C.; Haydu, L.E.; Huse, J.T.; Roy-Chowdhuri, S.; Luthra, R.; Wistuba, I.I.; et al. Evaluating Circulating Tumor DNA From the Cerebrospinal Fluid of Patients with Melanoma and Leptomeningeal Disease. J. Neuropathol. Exp. Neurol. 2018, 77, 628–635. [Google Scholar] [CrossRef] [PubMed]
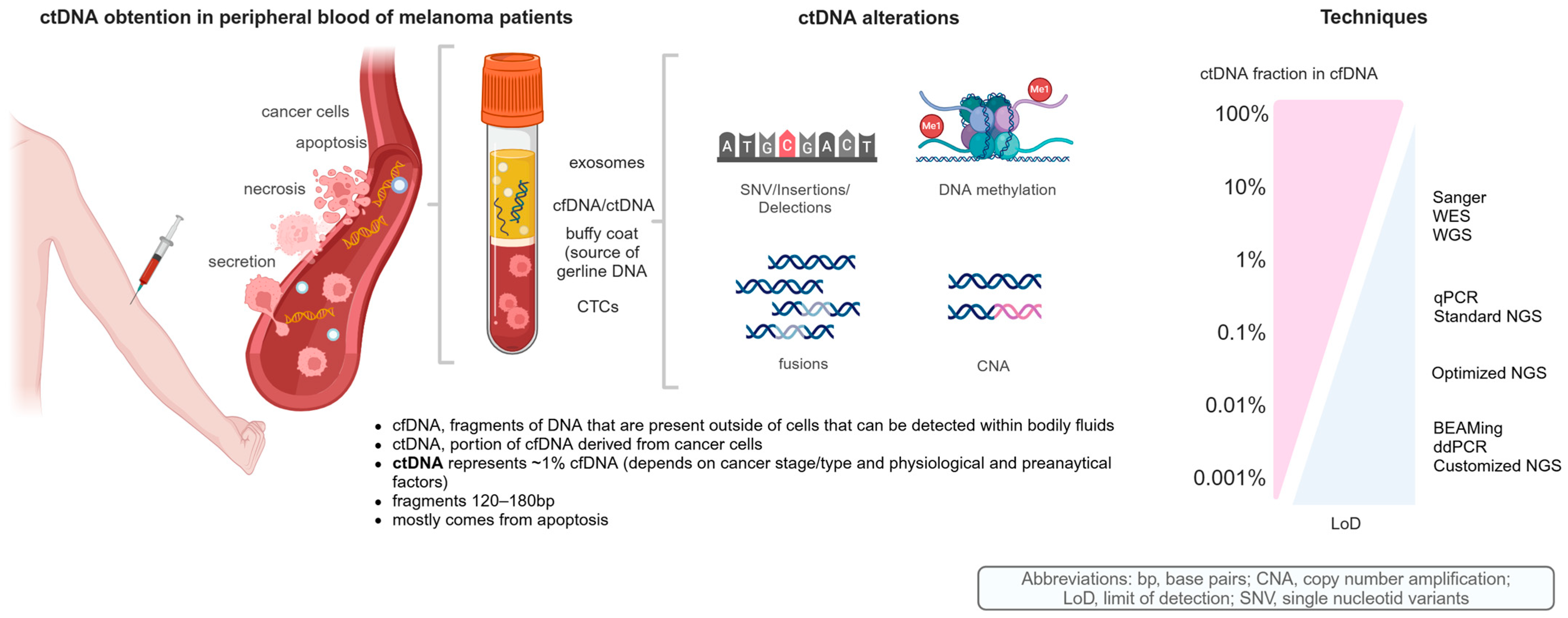

| Strengths | Weaknesses | LoD (Assay Sensitivity) | |
|---|---|---|---|
| Standard PCR-based techniques | Selective amplification of known DNA sequences Cost-efficient and rapid | Particular sequences flanking the sequence of interest must be known, and the process is limited to a single mutation per test Danger of contamination Amplification errors will be further amplified | 0.1% qPCR BRAF 0.005% allele-specific qPCR BRAF |
| ddPCR | Cost-efficient and rapid High sensitivity, accuracy and reproducibility Quantitative: mutant and wild-type copy number | Particular sequences flanking the sequence of interest must be known, and the process is limited to 1–2 mutations per test Danger of contamination Amplification errors will be further amplified | 0.005% BRAF |
| BEAMing | High sensitivity, accuracy and reproducibility | Particular sequences flanking the sequence of interest must be known, and the process is limited to a single mutation per test Danger of contamination Amplification errors will be further amplified | 0.01% BRAF |
| Standard NGS | Several genomic alterations in parallel allow tumor mutational burden analysis Greater mutational landscape information | Semiquantitive: variant allele frequency Higher cost, bioinformatic turn-out time Low sensitivity | 1% targeted NGS 0.1% NGS with molecular barcode |
| Modified NGS Amplicon deep sequencing Hybrid-capture deep sequencing | Higher sensitivity than standard NGS Several genomic alterations in parallel allow tumor mutational burden analysis Greater mutational landscape information Detection of sub-clonal mutations or changes in clonal composition over time | Semiquantitive: variant allele frequency Higher cost, bioinformatic turn-out time | 0.01% modified NGS |
| Bespoke assays (WES)/(WGS) + ddPCR) | High sensitivity and specificity Quantitative: mean tumor molecules (MTM)/mL Overcomes non-tumoral cfDNA contamination CHIP Several genomic alterations in parallel allow tumor mutational burden analysis Greater mutational landscape information Detection of sub-clonal mutations or changes in clonal composition over time | Higher cost, bioinformatic turn-out time Requires large amount of tumor tissue | 0.004% SignateraTM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Vila, C.; Teixido, C.; Aya, F.; Martín, R.; González-Navarro, E.A.; Alos, L.; Castrejon, N.; Arance, A. Detection of Circulating Tumor DNA in Liquid Biopsy: Current Techniques and Potential Applications in Melanoma. Int. J. Mol. Sci. 2025, 26, 861. https://doi.org/10.3390/ijms26020861
Martínez-Vila C, Teixido C, Aya F, Martín R, González-Navarro EA, Alos L, Castrejon N, Arance A. Detection of Circulating Tumor DNA in Liquid Biopsy: Current Techniques and Potential Applications in Melanoma. International Journal of Molecular Sciences. 2025; 26(2):861. https://doi.org/10.3390/ijms26020861
Chicago/Turabian StyleMartínez-Vila, Clara, Cristina Teixido, Francisco Aya, Roberto Martín, Europa Azucena González-Navarro, Llucia Alos, Natalia Castrejon, and Ana Arance. 2025. "Detection of Circulating Tumor DNA in Liquid Biopsy: Current Techniques and Potential Applications in Melanoma" International Journal of Molecular Sciences 26, no. 2: 861. https://doi.org/10.3390/ijms26020861
APA StyleMartínez-Vila, C., Teixido, C., Aya, F., Martín, R., González-Navarro, E. A., Alos, L., Castrejon, N., & Arance, A. (2025). Detection of Circulating Tumor DNA in Liquid Biopsy: Current Techniques and Potential Applications in Melanoma. International Journal of Molecular Sciences, 26(2), 861. https://doi.org/10.3390/ijms26020861