
From Local to Systemic: The Journey of Tick Bite Biomarkers in Australian Patients
 ,
,  , , , ,
, , , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
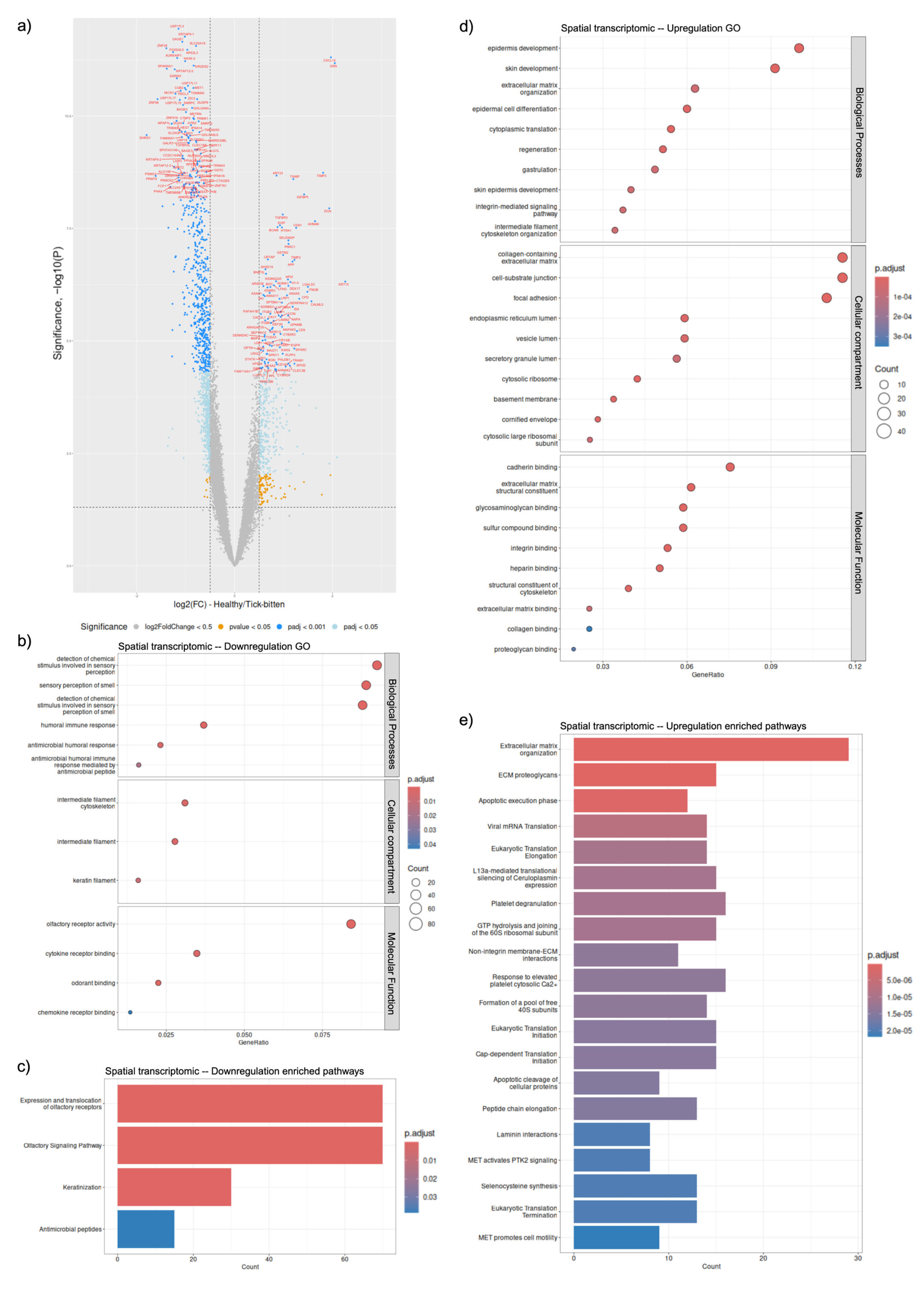
2.1. Local (Solid Biopsy) Signatures with Spatial Transcriptomics
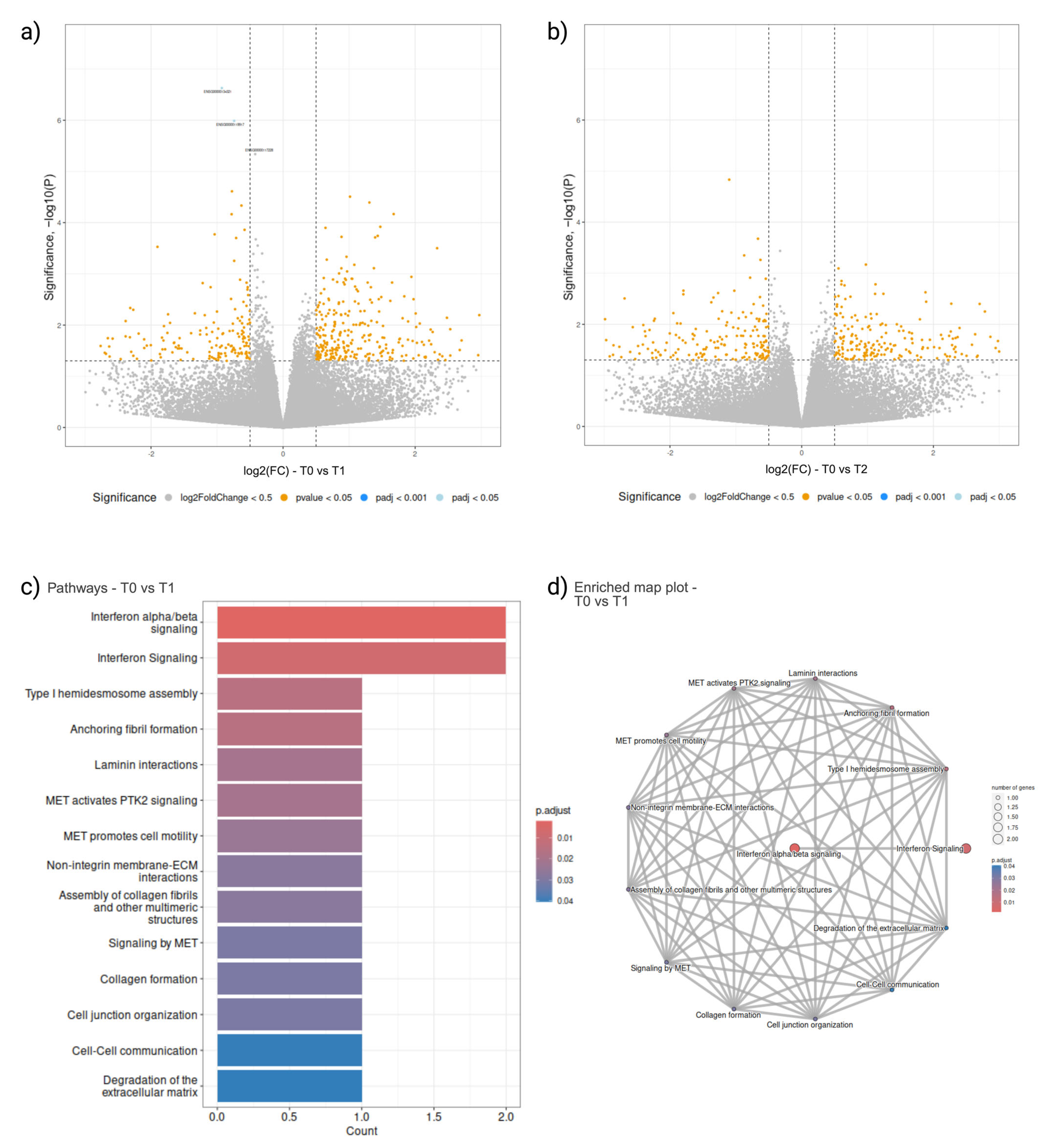
2.2. Peripheral Blood Cell-Free Transcriptomics
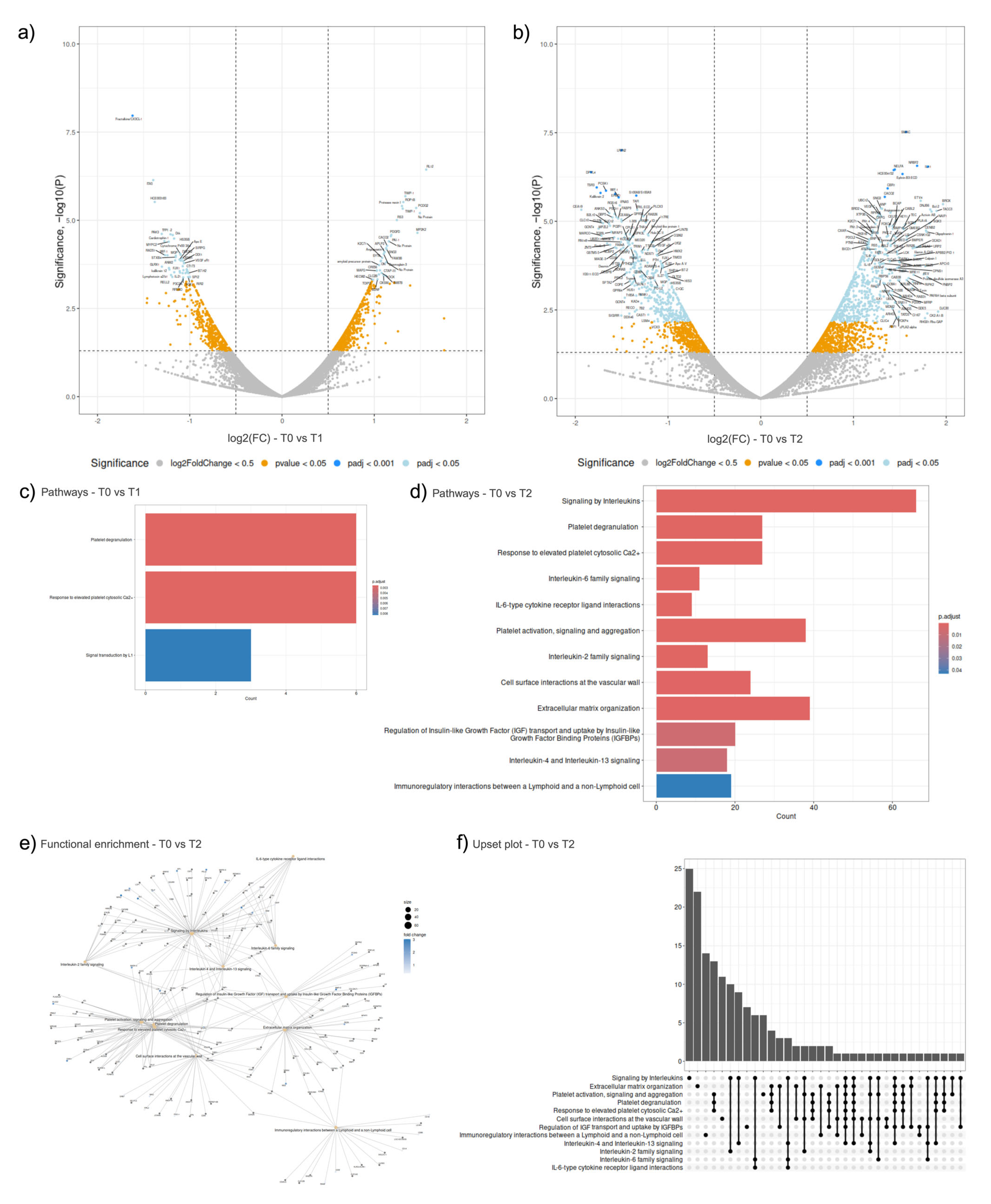
2.3. Peripheral Blood Cellular Transcriptomics
2.4. Peripheral Blood Plasma Proteomics
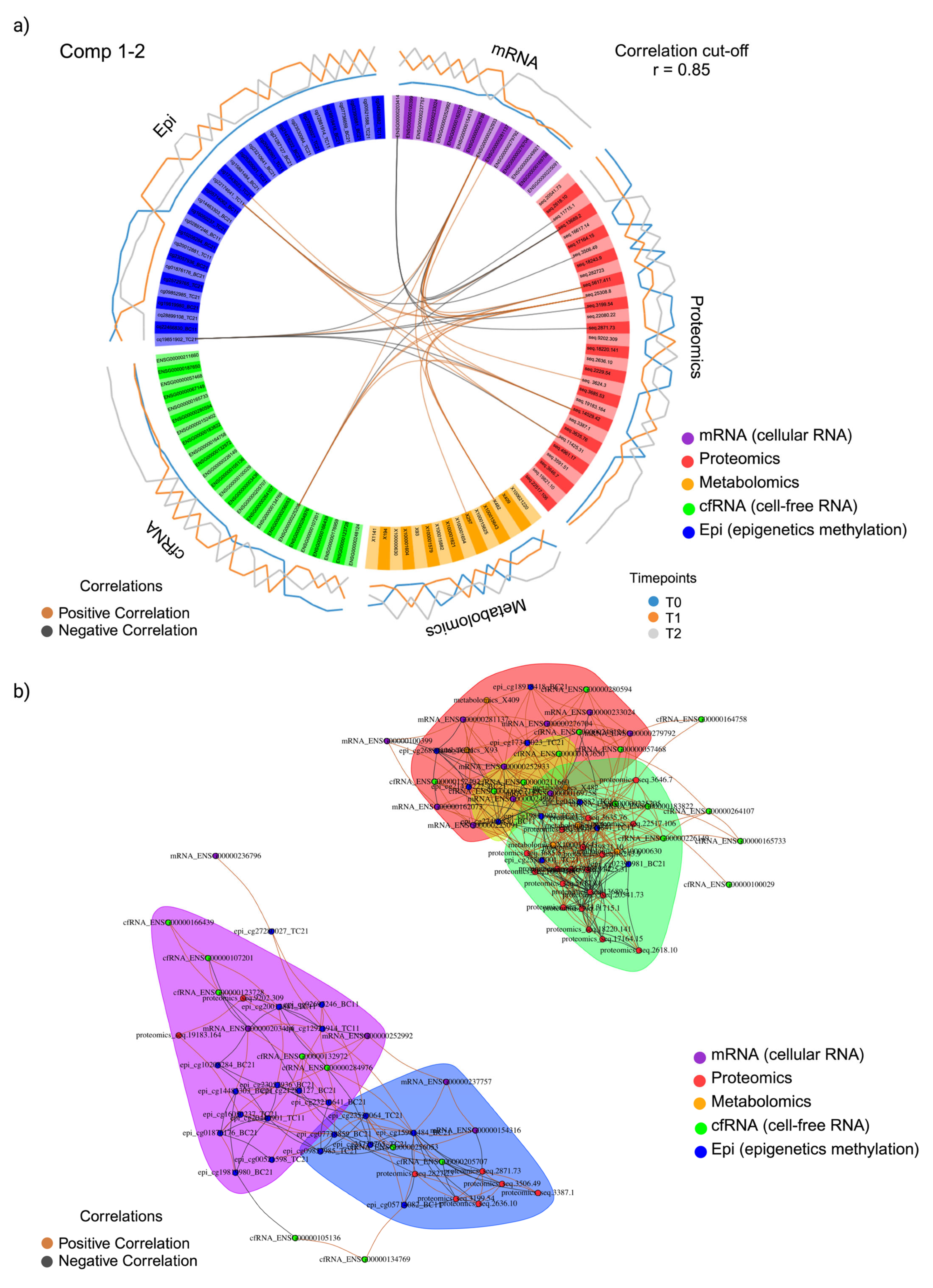
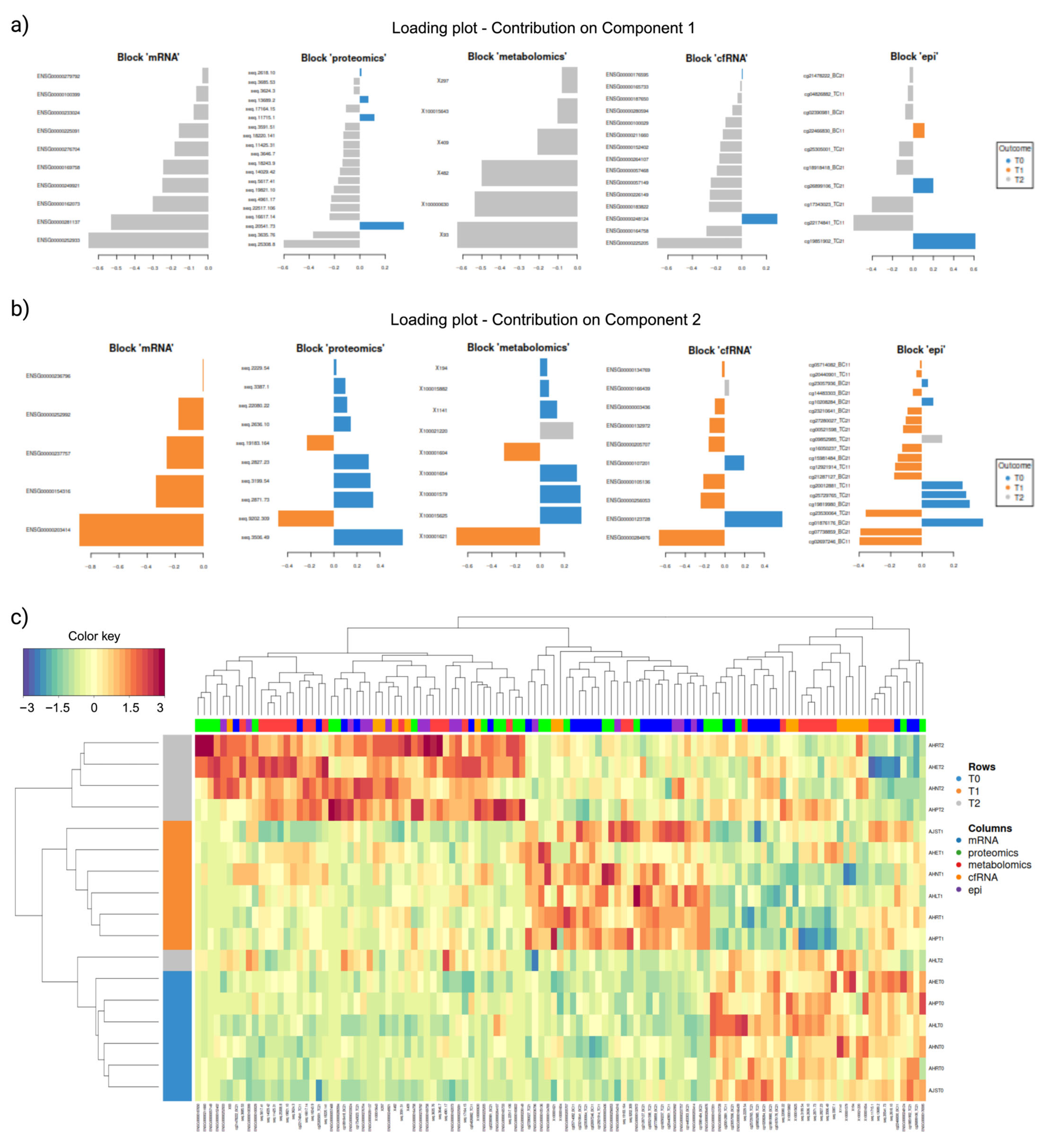
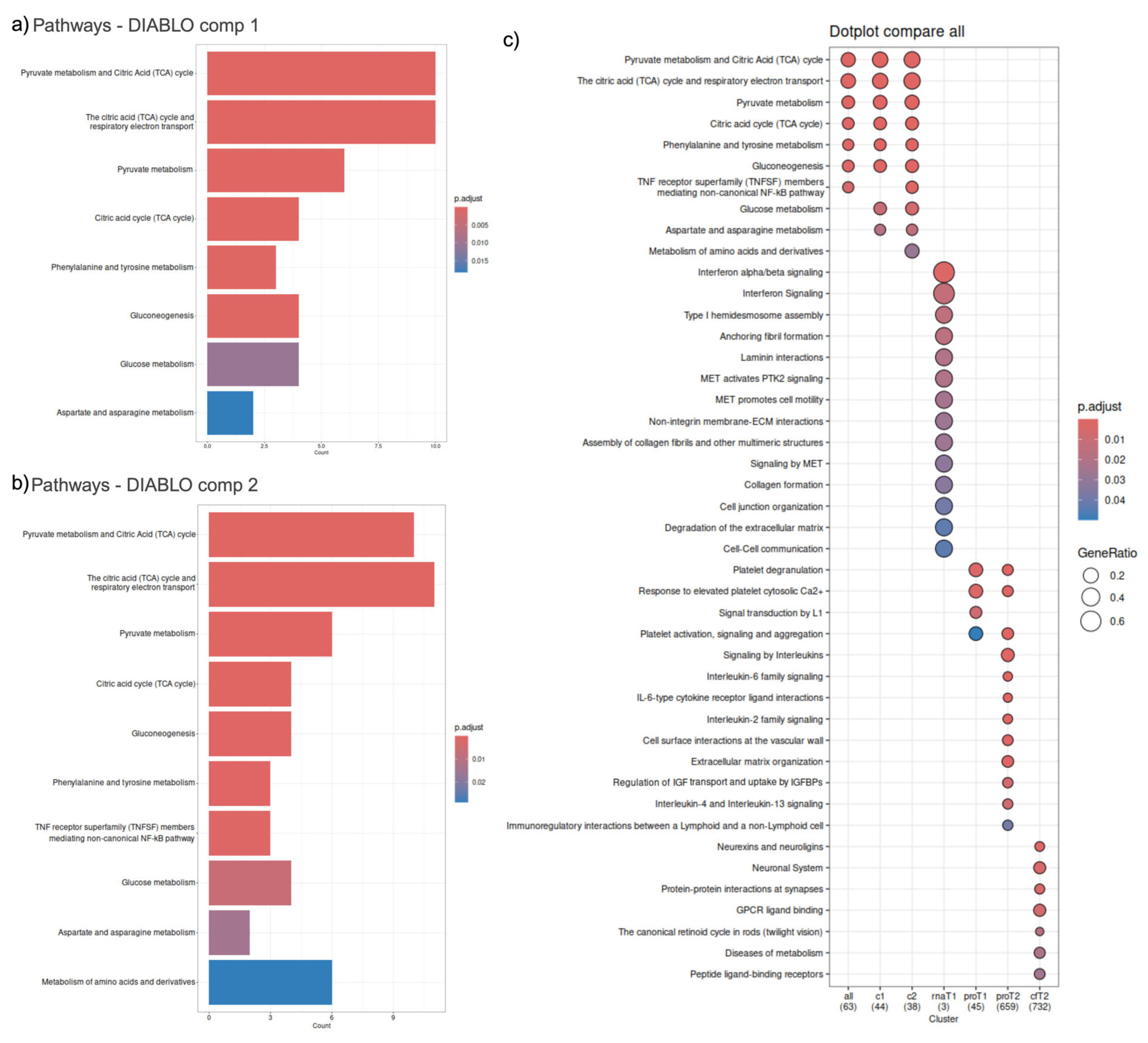
2.5. Integration
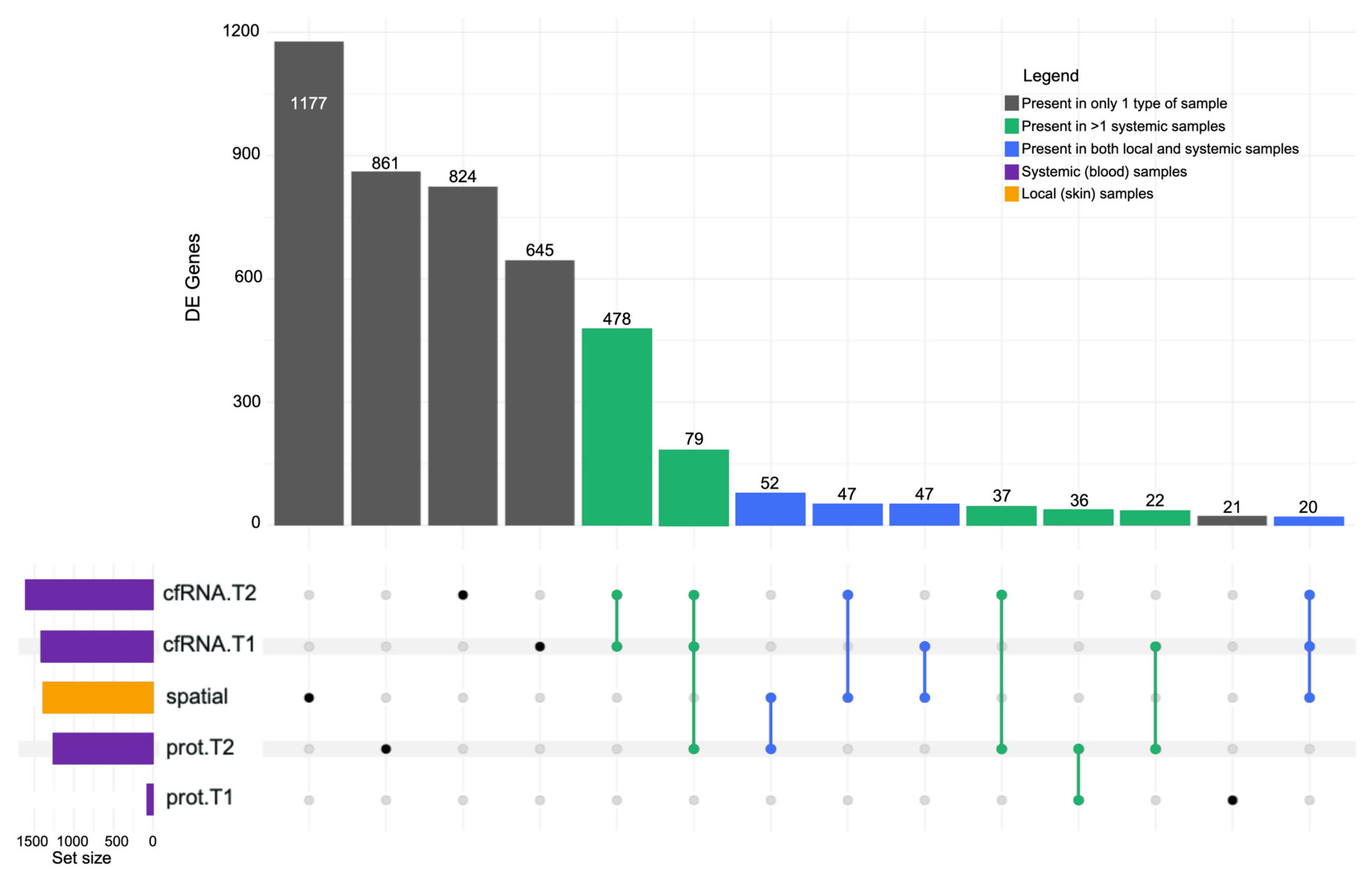
2.6. Correlational Analysis of Local (Solid Biopsy) and Systemic (Liquid Biopsy) Signals
3. Discussion
4. Materials and Methods
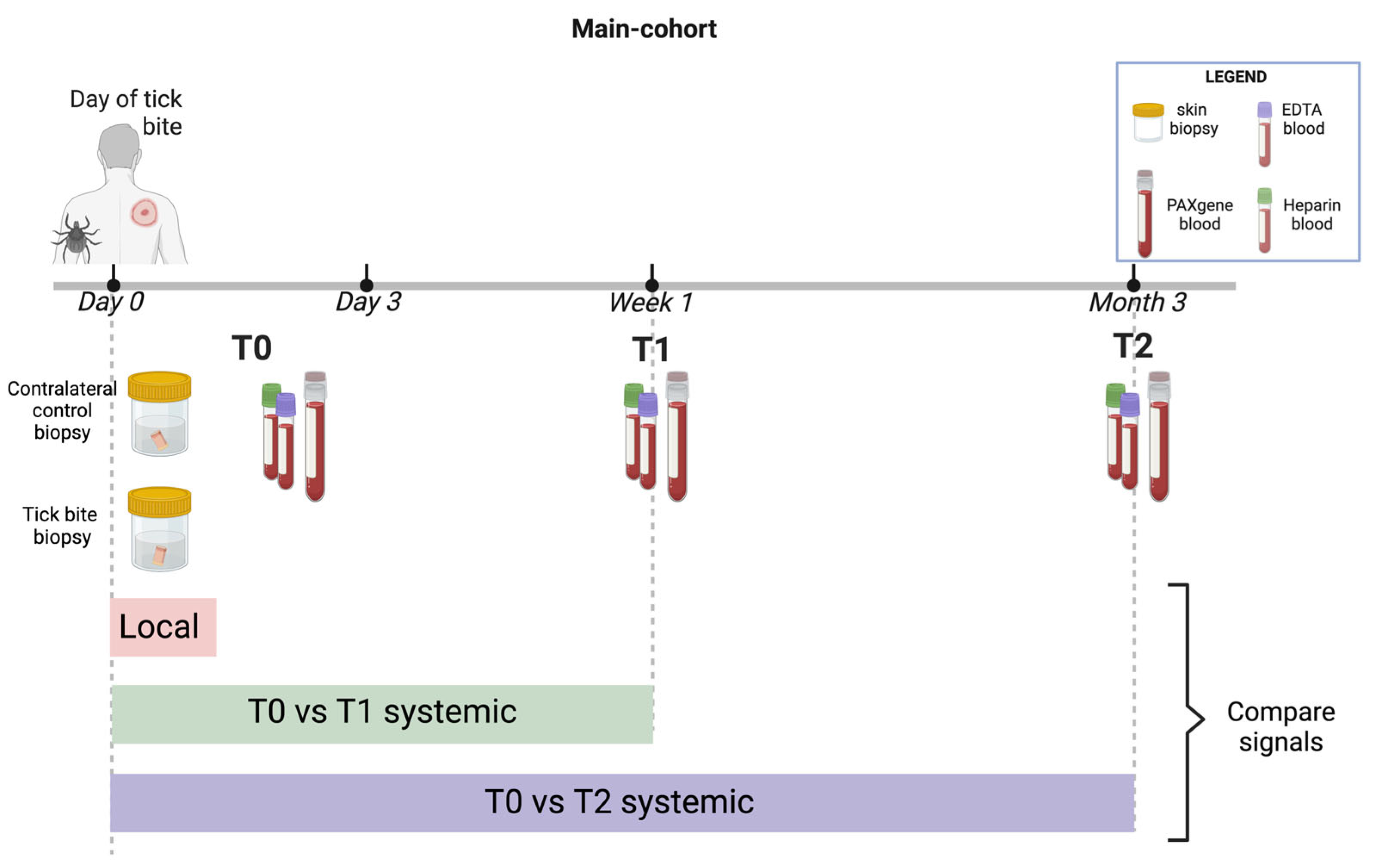
4.1. Overview
4.2. Data Selection Criteria and Extraction
4.3. Participant Details
4.4. Spatial Transcriptomics
4.5. Cell-Free Transcriptomics
4.6. Cellular Transcriptomics
4.7. Plasma Proteomics
4.8. Plasma Metabolomics
4.9. Whole Blood Cell DNA Methylation
4.10. Data Integration
4.11. Correlation Analysis and Visualisation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, W.; Barbosa, A.D.; Irwin, P.J.; Currie, A.; Kollmann, T.R.; Beaman, M.; Lee, A.H.; Oskam, C.L. A Systems Biology Approach to Better Understand Human Tick-Borne Diseases. Trends Parasitol. 2022, 39, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Shor, S.; Green, C.; Szantyr, B.; Phillips, S.; Liegner, K.; Burrascano, J.; Bransfield, R.; Maloney, E.L. Chronic Lyme Disease: An Evidence-Based Definition by the ILADS Working Group. Antibiotics 2019, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, M.; Pös, O.; Márton, É.; Buglyó, G.; Soltész, B.; Keserű, J.; Penyige, A.; Szemes, T.; Nagy, B. Circulating Cell-Free Nucleic Acids: Main Characteristics and Clinical Application. Int. J. Mol. Sci. 2020, 21, 6827. [Google Scholar] [CrossRef] [PubMed]
- Cabús, L.; Lagarde, J.; Curado, J.; Lizano, E.; Pérez-Boza, J. Current Challenges and Best Practices for Cell-Free Long RNA Biomarker Discovery. Biomark. Res. 2022, 10, 62. [Google Scholar] [CrossRef]
- Alexandrou, G.; Mantikas, K.-T.; Allsopp, R.; Yapeter, C.A.; Jahin, M.; Melnick, T.; Ali, S.; Coombes, R.C.; Toumazou, C.; Shaw, J.A.; et al. The Evolution of Affordable Technologies in Liquid Biopsy Diagnostics: The Key to Clinical Implementation. Cancers 2023, 15, 5434. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Marchetti, D.; Lang, J.E. Liquid Biopsy: From Concept to Clinical Application. Sci. Rep. 2023, 13, 21685. [Google Scholar] [CrossRef]
- Lee, W.; Ben-Othman, R.; Skut, P.; Lee, A.H.-Y.; Barbosa, A.D.; Beaman, M.; Currie, A.; Harvey, N.T.; Kumarasinghe, P.; Hall, R.A.; et al. Molecular Analysis of Human Tick-Bitten Skin Yields Signatures Associated with Distinct Spatial and Temporal Trajectories—A Proof-of-Concept Study. Heliyon 2024, 10, e33600. [Google Scholar] [CrossRef]
- Chen, Z.; Li, C.; Zhou, Y.; Yao, Y.; Liu, J.; Wu, M.; Su, J. Liquid Biopsies for Cancer: From Bench to Clinic. MedComm 2023, 4, e329. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 50, D20–D26. [Google Scholar] [CrossRef]
- Barker, S.C.; Barker, D. Ticks in Australia: Endemics; Exotics; Which Ticks Bite Humans? Microbiol. Aust. 2018, 39, 194–199. [Google Scholar] [CrossRef]
- Lantos, P.M.; Rumbaugh, J.; Bockenstedt, L.K.; Falck-Ytter, Y.T.; Aguero-Rosenfeld, M.E.; Auwaerter, P.G.; Baldwin, K.; Bannuru, R.R.; Belani, K.K.; Bowie, W.R.; et al. Clinical Practice Guidelines by the Infectious Diseases Society of America (IDSA), American Academy of Neurology (AAN), and American College of Rheumatology (ACR): 2020 Guidelines for the Prevention, Diagnosis and Treatment of Lyme Disease. Clin. Infect. Dis. 2021, 72, e1–e48. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.J.; Auwaerter, P.G.; Bannuru, R.R.; Branda, J.A.; Falck-Ytter, Y.T.; Lantos, P.M.; Lavergne, V.; Meissner, H.C.; Osani, M.C.; Rips, J.G.; et al. Clinical Practice Guidelines by the Infectious Diseases Society of America (IDSA): 2020 Guideline on Diagnosis and Management of Babesiosis. Clin. Infect. Dis. 2021, 72, e49–e64. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.H.; Allison, D.H.R.; Feng, Y.; Jour, G.; Park, K.; Zhou, F.; Moreira, A.L.; Shen, G.; Feng, X.; Sabari, J.; et al. Comparison of Solid Tissue Sequencing and Liquid Biopsy Accuracy in Identification of Clinically Relevant Gene Mutations and Rearrangements in Lung Adenocarcinomas. Mod. Pathol. 2021, 34, 2168–2174. [Google Scholar] [CrossRef]
- Liu, M.C.; MacKay, M.; Kase, M.; Piwowarczyk, A.; Lo, C.; Schaeffer, J.; Finkle, J.D.; Mason, C.E.; Beaubier, N.; Blackwell, K.L.; et al. Longitudinal Shifts of Solid Tumor and Liquid Biopsy Sequencing Concordance in Metastatic Breast Cancer. JCO Precis. Oncol. 2022, e2100321. [Google Scholar] [CrossRef]
- Potekaev, N.N.; Borzykh, O.B.; Medvedev, G.V.; Pushkin, D.V.; Petrova, M.M.; Petrov, A.V.; Dmitrenko, D.V.; Karpova, E.I.; Demina, O.M.; Shnayder, N.A. The Role of Extracellular Matrix in Skin Wound Healing. J. Clin. Med. 2021, 10, 5947. [Google Scholar] [CrossRef]
- Moniuszko-Malinowska, A.; Czupryna, P.; Dunaj, J.; Pancewicz, S.; Garkowski, A.; Kondrusik, M.; Grygorczuk, S.; Zajkowska, J. Acrodermatitis Chronica Atrophicans: Various Faces of the Late Form of Lyme Borreliosis. Postep. Dermatol. Alergol. 2018, 35, 490–494. [Google Scholar] [CrossRef]
- Gindl, A.; Schötta, A.-M.; Berent, S.; Markowicz, M.; Stockinger, H.; Thalhammer, F.; Stary, G.; Strobl, J. Persistent Lyme Disease with Cutaneous Borrelia Biofilm Formation. Br. J. Dermatol. 2022, 186, 1041–1043. [Google Scholar] [CrossRef]
- Talbot, N.C.; Spillers, N.J.; Luther, P.; Flanagan, C.; Soileau, L.G.; Ahmadzadeh, S.; Viswanath, O.; Varrassi, G.; Shekoohi, S.; Cornett, E.M.; et al. Lyme Disease and Post-Treatment Lyme Disease Syndrome: Current and Developing Treatment Options. Cureus 2023, 15, e43112. [Google Scholar] [CrossRef]
- Mans, B.J.; Neitz, A.W.H. Adaptation of Ticks to a Blood-Feeding Environment: Evolution from a Functional Perspective. Insect Biochem. Mol. Biol. 2004, 34, 1–17. [Google Scholar] [CrossRef]
- Papamanoli, A.; Kaplun, O.; Lobo, Z.; LeMaitre, B.; Romano, C.L.; Campbell, S.R.; Psevdos, G. Tick-Borne Disease–Associated Thrombocytopenia: Epidemiological Description Among United States Veterans and Tick Surveillance Data From Suffolk County, Long Island, New York. Infect. Dis. Clin. Pract. 2022, 30, e1090. [Google Scholar] [CrossRef]
- Lin, Y.-P.; Yu, Y.; Marcinkiewicz, A.L.; Lederman, P.; Hart, T.M.; Zhang, F.; Linhardt, R.J. Non-Anticoagulant Heparin as a Pre-Exposure Prophylaxis Prevents Lyme Disease Infection. ACS Infect. Dis. 2020, 6, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Torina, A.; Villari, S.; Blanda, V.; Vullo, S.; La Manna, M.P.; Shekarkar Azgomi, M.; Di Liberto, D.; de la Fuente, J.; Sireci, G. Innate Immune Response to Tick-Borne Pathogens: Cellular and Molecular Mechanisms Induced in the Hosts. Int. J. Mol. Sci. 2020, 21, 5437. [Google Scholar] [CrossRef]
- Baquer, F.; Jaulhac, B.; Barthel, C.; Paz, M.; Wolfgramm, J.; Müller, A.; Boulanger, N.; Grillon, A. Skin Microbiota Secretomes Modulate Cutaneous Innate Immunity against Borrelia Burgdorferi s.s. Sci. Rep. 2023, 13, 16393. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; Pan, W.; Kim, H.J.; Mauntz, R.E.; Stuart, S.M.; Pimentel, M.; Zhou, Y.; Knudsgaard, P.; Demas, V.; Aravanis, A.M.; et al. A Comprehensive Characterization of the Cell-Free Transcriptome Reveals Tissue- and Subtype-Specific Biomarkers for Cancer Detection. Nat. Commun. 2021, 12, 2357. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, J.; Hu, Y.; Guo, Q.; Zhang, S.; Tian, J.; Niu, Y.; Ji, L.; Xu, Y.; Tang, P.; et al. Terminal Modifications Independent Cell-Free RNA Sequencing Enables Sensitive Early Cancer Detection and Classification. Nat. Commun. 2024, 15, 156. [Google Scholar] [CrossRef]
- Wadi, L.; Meyer, M.; Weiser, J.; Stein, L.D.; Reimand, J. Impact of Outdated Gene Annotations on Pathway Enrichment Analysis. Nat. Methods 2016, 13, 705–706. [Google Scholar] [CrossRef]
- Barbosa, A.D.; Long, M.; Lee, W.; Austen, J.M.; Cunneen, M.; Ratchford, A.; Burns, B.; Kumarasinghe, P.; Ben-Othman, R.; Kollmann, T.R.; et al. The Troublesome Ticks Research Protocol: Developing a Comprehensive, Multidiscipline Research Plan for Investigating Human Tick-Associated Disease in Australia. Pathogens 2022, 11, 1290. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Reeves, J.; Divakar, P.; Ortogero, N.; Griswold, M.; Yang, Z.; Zimmerman, S.; Vitancol, R.; David, H. GeoMxWorkflows: GeoMx Digital Spatial Profiler (DSP) Data Analysis Workflows, R Package version 1.6.0; NanoString Technologies: Seattle, WA, USA, 2023.
- Aboyoun, P.; Ortogero, N.; Yang, Z. NanoStringNCTools: NanoString nCounter Tools; NanoString Technologies: Seattle, WA, USA, 2023. [Google Scholar]
- Ortogero, N.; Yang, Z.; Vitancol, R.; Griswold, M.; Henderson, D. GeomxTools: NanoString GeoMx Tools; NanoString Technologies: Seattle, WA, USA, 2023. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de Novo Haploid Genome Assemblies Demonstrates the Enduring Quality of the Reference Assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Putri, G.H.; Anders, S.; Pyl, P.T.; Pimanda, J.E.; Zanini, F. Analysing High-Throughput Sequencing Data in Python with HTSeq 2.0. Bioinformatics 2022, 38, 2943–2945. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Lee, A.H.; Shannon, C.P.; Amenyogbe, N.; Bennike, T.B.; Diray-Arce, J.; Idoko, O.T.; Gill, E.E.; Ben-Othman, R.; Pomat, W.S.; van Haren, S.D.; et al. Dynamic Molecular Changes during the First Week of Human Life Follow a Robust Developmental Trajectory. Nat. Commun. 2019, 10, 1092. [Google Scholar] [CrossRef]
- Yu, G.; Petyuk, V. ReactomePA: Reactome Pathway Analysis; Bioconductor: R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Hoffman, G.E.; Roussos, P. Dream: Powerful Differential Expression Analysis for Repeated Measures Designs. Bioinformatics 2021, 37, 192–201. [Google Scholar] [CrossRef]
- Hansen, K.D.; Aryee, M.; Irizarry, R.A.; Jaffe, A.E.; Maksimovic, J.; Houseman, E.A.; Fortin, J.-P.; Triche, T.; Andrews, S.V.; Hickey, P.F. Minfi: Analyze Illumina Infinium DNA Methylation Arrays; Bioconductor: R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Lê Cao, K.-A. DIABLO: An Integrative Approach for Identifying Key Molecular Drivers from Multi-Omics Assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; Boitard, S.; Besse, P. Sparse PLS Discriminant Analysis: Biologically Relevant Feature Selection and Graphical Displays for Multiclass Problems. BMC Bioinform. 2011, 12, 253. [Google Scholar] [CrossRef]
- Qin, S.; Kim, J.; Arafat, D.; Gibson, G. Effect of Normalization on Statistical and Biological Interpretation of Gene Expression Profiles. Front. Genet. 2013, 3, 160. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Durinck, S.; Huber, W.; Davis, S.; Pepin, F.; Buffalo, V.S.; Smith, M. biomaRt: Interface to BioMart Databases (i.e., Ensembl); Bioconductor: R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human; Bioconductor: R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology Knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Hu, E.; Gao, C.-H. Enrichplot: Visualization of Functional Enrichment Result; Bioconductor: R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.; Barbosa, A.D.; Lee, A.H.-Y.; Currie, A.; Martino, D.; Stenos, J.; Long, M.; Beaman, M.; Harvey, N.T.; Kresoje, N.; et al. From Local to Systemic: The Journey of Tick Bite Biomarkers in Australian Patients. Int. J. Mol. Sci. 2025, 26, 1520. https://doi.org/10.3390/ijms26041520
Lee W, Barbosa AD, Lee AH-Y, Currie A, Martino D, Stenos J, Long M, Beaman M, Harvey NT, Kresoje N, et al. From Local to Systemic: The Journey of Tick Bite Biomarkers in Australian Patients. International Journal of Molecular Sciences. 2025; 26(4):1520. https://doi.org/10.3390/ijms26041520
Chicago/Turabian StyleLee, Wenna, Amanda D. Barbosa, Amy Huey-Yi Lee, Andrew Currie, David Martino, John Stenos, Michelle Long, Miles Beaman, Nathan T. Harvey, Nina Kresoje, and et al. 2025. "From Local to Systemic: The Journey of Tick Bite Biomarkers in Australian Patients" International Journal of Molecular Sciences 26, no. 4: 1520. https://doi.org/10.3390/ijms26041520
APA StyleLee, W., Barbosa, A. D., Lee, A. H.-Y., Currie, A., Martino, D., Stenos, J., Long, M., Beaman, M., Harvey, N. T., Kresoje, N., Skut, P., Irwin, P. J., Kumarasinghe, P., Hall, R. A., Ben-Othman, R., Graves, S., Kollmann, T. R., & Oskam, C. L. (2025). From Local to Systemic: The Journey of Tick Bite Biomarkers in Australian Patients. International Journal of Molecular Sciences, 26(4), 1520. https://doi.org/10.3390/ijms26041520