
Chalcogen-Bonded [Se–N]2 Cyclic Supramolecular Synthons Enhanced by Halogen Bonds: Studies in the Gas Phase and Crystalline Phase

Abstract

1. Introduction
2. Results and Discussion
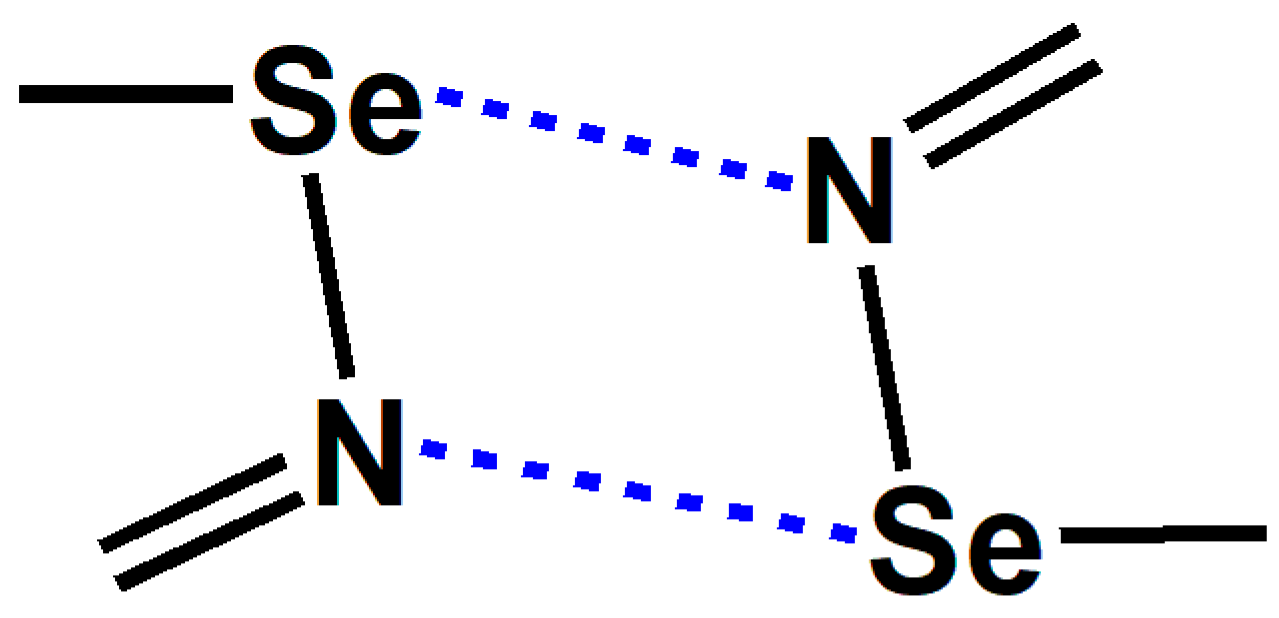
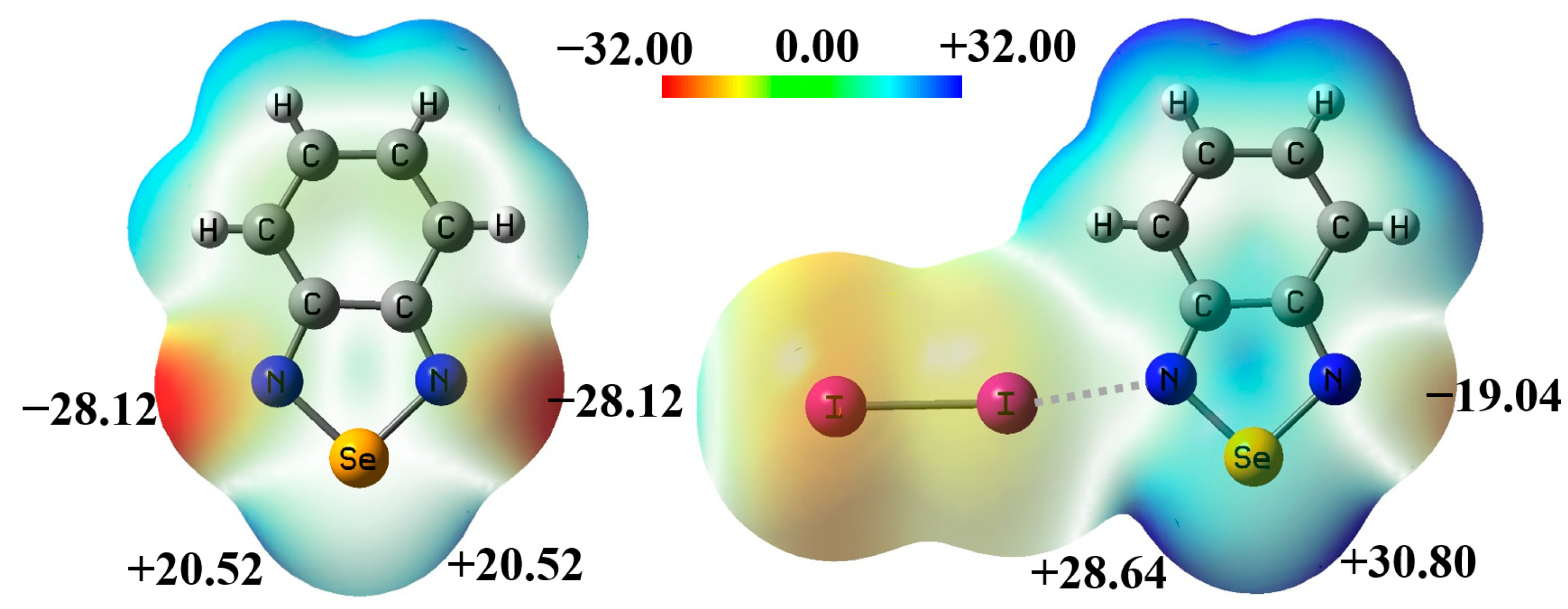
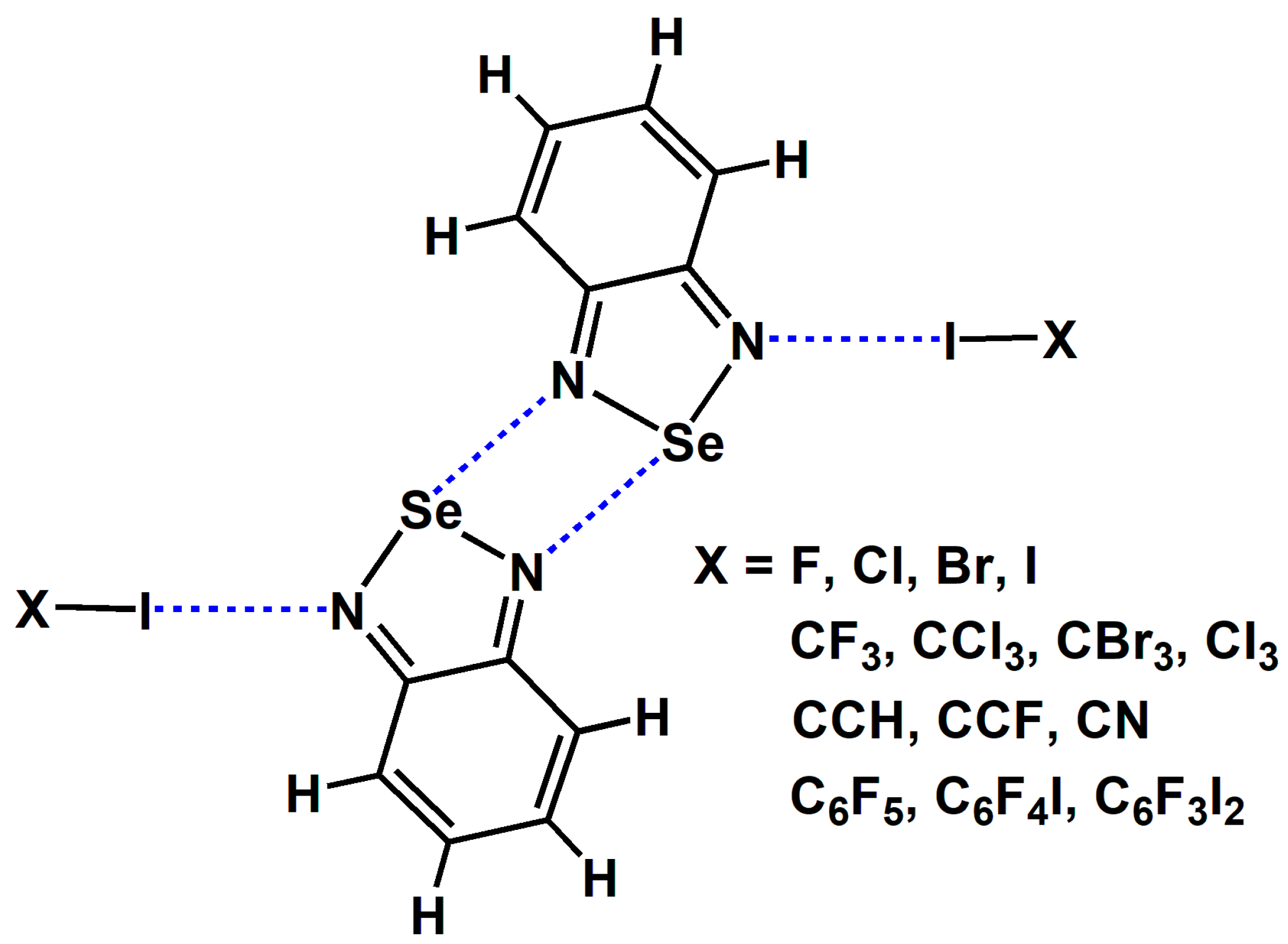
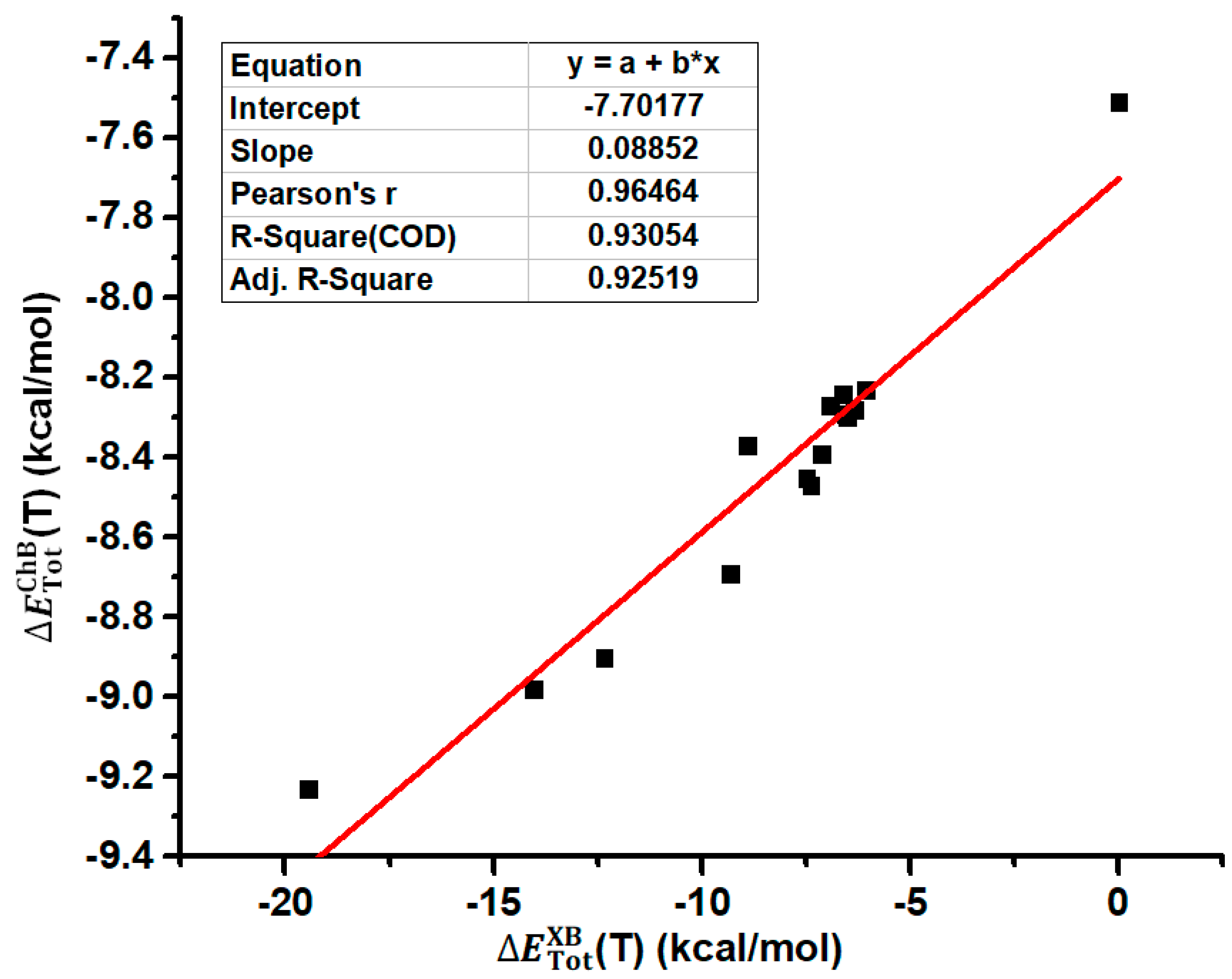
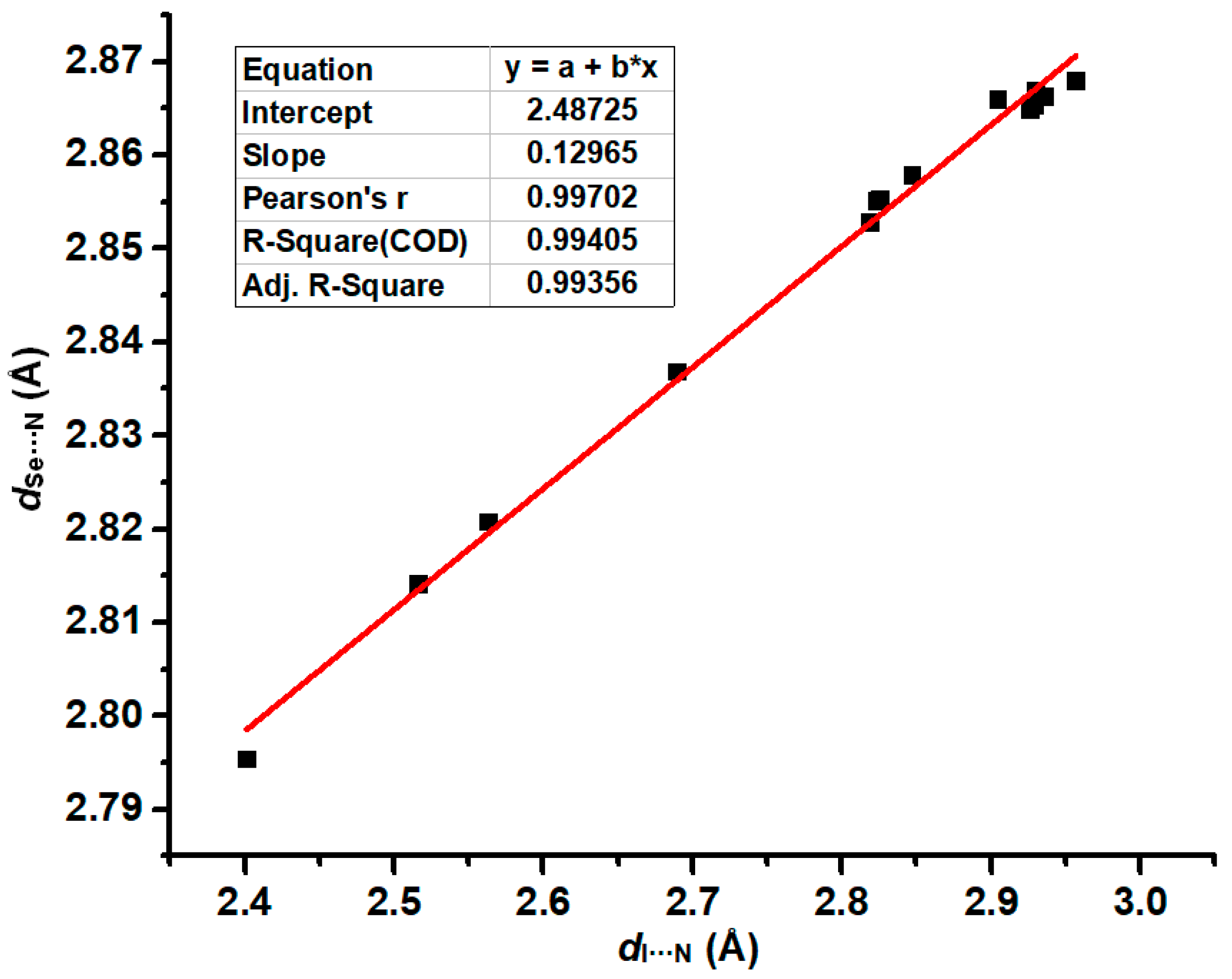
2.1. The Effect of Halogen Bonds on the [Se–N]2 Supramolecular Synthons in the Gas Phase
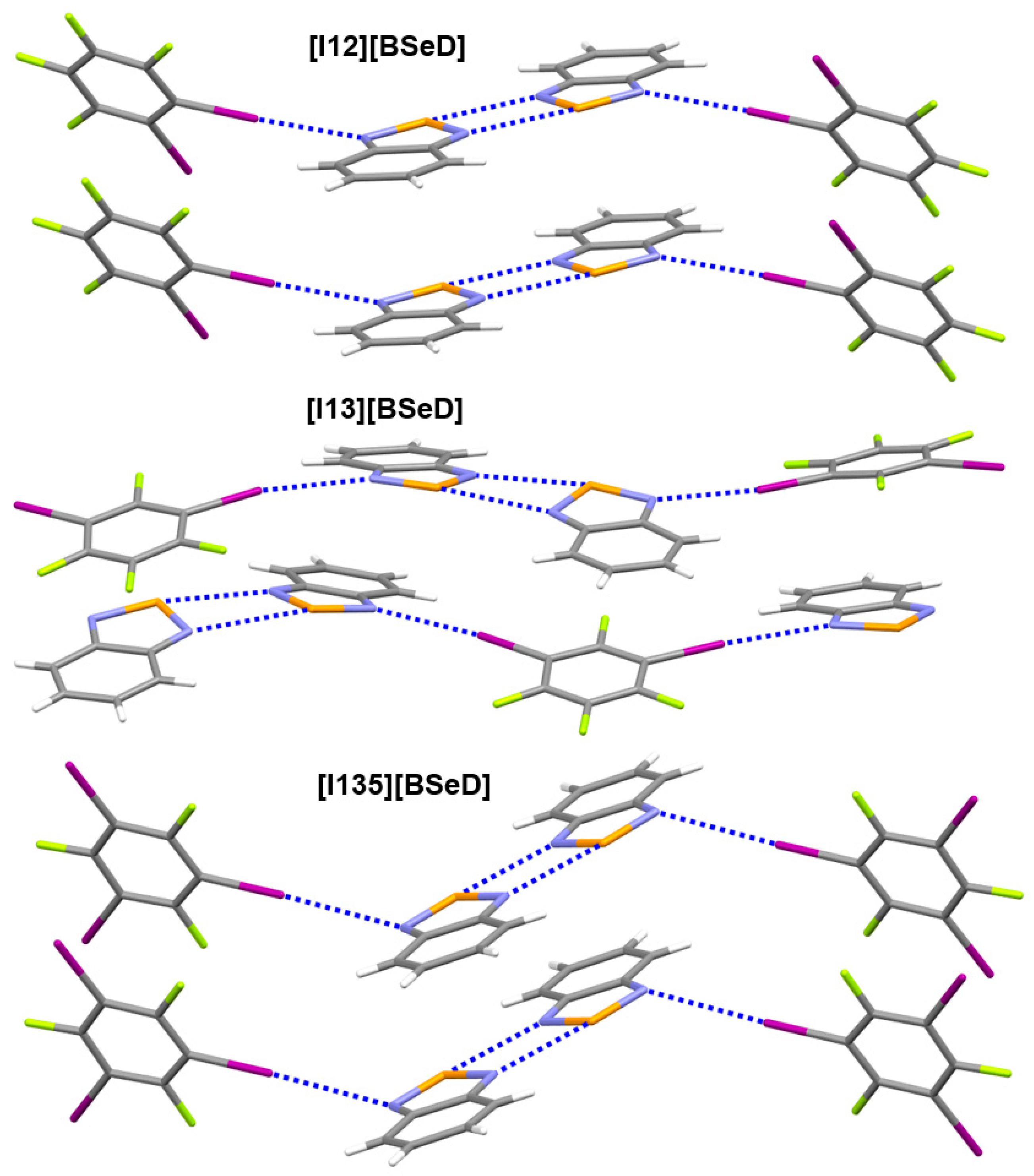
2.2. The Effect of Halogen Bonds on [Se–N]2 Supramolecular Synthons in the Crystalline Phase
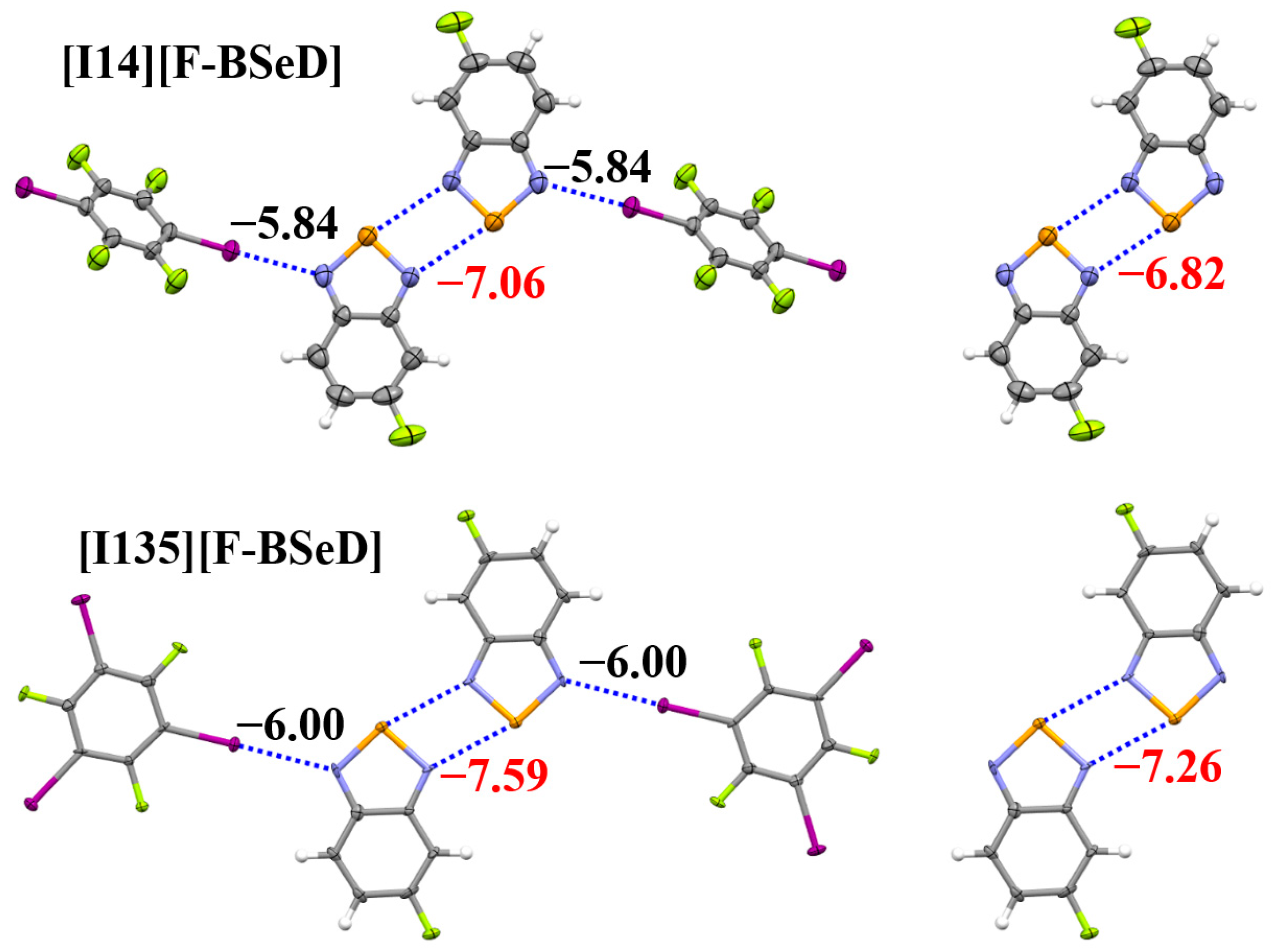
2.3. Expansion of the [Se–N]2 Supramolecular Synthon
3. Materials and Methods
3.1. Computational Details
3.2. Syntheses of Cocrystals
3.3. X-Ray Structure Determinations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond. Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-Hole Concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-Hole Revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Breugst, M.; Koenig, J.J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 2020, 5473–5487. [Google Scholar] [CrossRef]
- Pizzi, A.; Dhaka, A.; Beccaria, R.; Resnati, G. Anion⋯Anion Self-Assembly under the Control of σ- and π-Hole Bonds. Chem. Soc. Rev. 2024, 53, 6654–6674. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Konda, M.; Kauffmann, B.; Manna, M.K.; Das, A.K. Effects of Donor and Acceptor Units Attached with Benzoselenadiazole: Optoelectronic and Self-Assembling Patterns. Cryst. Growth Des. 2015, 15, 5548–5554. [Google Scholar] [CrossRef]
- Lee, L.M.; Corless, V.B.; Tran, M.; Jenkins, H.; Britten, J.F.; Vargas-Baca, I. Synthetic, Structural, and Computational Investigations of N-alkyl Benzo-2,1,3-selenadiazolium Iodides and Their Supramolecular Aggregates. Dalton Trans. 2016, 45, 3285–3293. [Google Scholar] [CrossRef] [PubMed]
- Eichstaedt, K.; Wasilewska, A.; Wicher, B.; Gdaniec, M.; Połoński, T. Supramolecular Synthesis Based on a Combination of Se···N Secondary Bonding Interactions with Hydrogen and Halogen Bonds. Cryst. Growth Des. 2016, 16, 1282–1293. [Google Scholar] [CrossRef]
- Lekin, K.; Leitch, A.A.; Assoud, A.; Yong, W.; Desmarais, J.; Tse, J.S.; Desgreniers, S.; Secco, R.A.; Oakley, R.T. Benzoquinone-Bridged Heterocyclic Zwitterions as Building Blocks for Molecular Semiconductors and Metals. Inorg. Chem. 2018, 57, 4757–4770. [Google Scholar] [CrossRef]
- Centore, R.; Borbone, F.; Carella, A.; Causà, M.; Fusco, S.; Gentile, F.S.; Parisi, E. Hierarchy of Intermolecular Interactions and Selective Topochemical Reactivity in Different Polymorphs of Fused-Ring Heteroaromatics. Cryst. Growth Des. 2020, 20, 1229–1236. [Google Scholar] [CrossRef]
- Biot, N.; Bonifazi, D. Concurring Chalcogen- and Halogen-Bonding Interactions in Supramolecular Polymers for Crystal Engineering Applications. Chem. Eur. J. 2020, 26, 2904–2913. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular Aggregation Patterns Featuring Se⋯N Secondary-Bonding Interactions in Mono-Nuclear Selenium Compounds: A Comparison with Their Congeners. Coord. Chem. Rev. 2021, 443, 214031. [Google Scholar] [CrossRef]
- Alfuth, J.; Zadykowicz, B.; Wicher, B.; Kazimierczuk, K.; Połoński, T.; Olszewska, T. Cooperativity of Halogen- and Chalcogen-Bonding Interactions in the Self-Assembly of 4-Iodoethynyl- and 4,7-Bis(iodoethynyl)benzo-2,1,3-chalcogenadiazoles: Crystal Structures, Hirshfeld Surface Analyses, and Crystal Lattice Energy Calculations. Cryst. Growth Des. 2022, 22, 1299–1311. [Google Scholar] [CrossRef]
- Rahman, F.-U.; Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ballester, P.; Rebek, J., Jr.; Yu, Y. Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces. J. Am. Chem. Soc. 2020, 142, 5876–5883. [Google Scholar] [CrossRef]
- Yu, Y.; Rebek, J., Jr. Confined Molecules: Experiment Meets Theory in Small Spaces. Q. Rev. Biophys. 2020, 53, e6. [Google Scholar] [CrossRef] [PubMed]
- Allen, F. The Cambridge Structure Database: A Quarter of a Million Crystal Structures and Rising. Acta Crystallogr. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Allen, F.; Motherwell, W.D.S. Applications of the Cambridge Structural Database in Organic and Crystal Chemistry. Acta Crystallogr. 2002, B58, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, W.; Yang, W.; Zhu, Z.; Xu, Z.; Liu, H. 2Ch–2N Square and Hexagon Interactions: A Combined Crystallographic Data Analysis and Computational Study. Phys. Chem. Chem. Phys. 2019, 21, 21568–21576. [Google Scholar] [CrossRef]
- Michalczyk, M.; Malik, M.; Zierkiewicz, W.; Scheiner, S. Experimental and Theoretical Studies of Dimers Stabilized by Two Chalcogen Bonds in the Presence of a N···N Pnicogen Bond. J. Phys. Chem. A 2021, 125, 657–668. [Google Scholar] [CrossRef]
- Scheiner, S. Principles Guiding the Square Bonding Motif Containing a Pair of Chalcogen Bonds between Chalcogenadiazoles. J. Phys. Chem. A 2022, 126, 1194–1203. [Google Scholar] [CrossRef]
- Wang, H.; Li, B.; Wang, X.; Yin, F.; Wei, Q.; Wang, X.; Ni, Y.; Wang, H. First-Principles Study of Square Chalcogen Bond Interactions and its Adsorption Behavior on Silver Surface. Phys. Chem. Chem. Phys. 2023, 25, 10836–10844. [Google Scholar] [CrossRef] [PubMed]
- Radiush, E.A.; Wang, H.; Chulanova, E.A.; Ponomareva, Y.A.; Li, B.; Wei, Q.Y.; Salnikov, G.E.; Petrakova, S.Y.; Semenov, N.A.; Zibarev, A.V. Halide Complexes of 5,6-Dicyano-2,1,3-Benzoselenadiazole with 1:4 Stoichiometry: Cooperativity between Chalcogen and Hydrogen Bonding. ChemPlusChem 2023, 88, e202300523. [Google Scholar] [CrossRef] [PubMed]
- Estarellas, C.; Frontera, A.; Quiñonero, D.; Deyà, P.M. Theoretical Study on Cooperativity Effects between Anion-π and Halogen-Bonding Interactions. ChemPhysChem 2011, 12, 2742–2750. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Li, H.; Zhu, X.; Liu, H.; Zhu, W. Mutual Influence between Halogen Bonds and Cation-π Interactions: A Theoretical Study. ChemPhysChem 2012, 13, 2154–2161. [Google Scholar] [CrossRef]
- Li, H.; Lu, Y.; Liu, Y.; Zhu, X.; Liu, H.; Zhu, W. Interplay between Halogen Bonds and π-π Stacking Interactions: CSD Search and Theoretical Study. Phys. Chem. Chem. Phys. 2012, 14, 9948–9955. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; van der Lee, A.; Velde, C.V.; Wintjens, R.; Dubois, P.; Clément, S.; Meyer, F. Interplay between Halogen Bonding and Lone Pair-π Interactions: A Computational and Crystal Packing Study. ChemPlusChem 2014, 79, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Robeyns, K.; Soubhye, J.; Wintjens, R.; Meyer, F. Halogen Bonding in Multi-Connected 1,2,2-Triiodo-Alkene Involving Geminal and/or Vicinal Iodines: A Crystallographic and DFT Study. CrystEngComm 2016, 18, 683–690. [Google Scholar] [CrossRef]
- Scheiner, S. Competition between a Tetrel and Halogen Bond to a Common Lewis Acid. J. Phys. Chem. A 2021, 125, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. σ-Hole Bonding: Molecules Containing Group VI Atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Rahm, M.; Hoffmann, R. Distinguishing Bonds. J. Am. Chem. Soc. 2016, 138, 3731–3744. [Google Scholar] [CrossRef]
- Su, P.; Tang, Z.; Wu, W. Generalized Kohn-Sham Energy Decomposition Analysis and its Applications. WIREs Comput. Mol. Sci. 2020, 10, e1460. [Google Scholar] [CrossRef]
- Tang, Z.; Song, Y.; Zhang, S.; Wang, W.; Xu, Y.; Wu, D.; Wu, W.; Su, P. XEDA, A Fast and Multipurpose Energy Decomposition Analysis Program. J. Comput. Chem. 2021, 42, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Wu, W.; Su, P. Assessments of DFT-Based Energy Decomposition Analysis Methods for Intermolecular Interactions. J. Chem. Phys. 2023, 158, 124116. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.B. The Benzene⋯Naphthalene Complex: A More Challenging System Than the Benzene Dimer for Newly Developed Computational Methods. J. Chem. Phys. 2015, 143, 114312. [Google Scholar] [CrossRef]
- Li, M.M.; Wang, Y.B.; Zhang, Y.; Wang, W. The Nature of the Noncovalent Interactions between Benzene and C60 Fullerene. J. Phys. Chem. A 2016, 120, 5766–5772. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Highly Accurate Benchmark Calculations of the Interaction Energies in the Complexes C6H6⋯C6X6 (X = F, Cl, Br, and I). Int. J. Quantum Chem. 2017, 117, e25345. [Google Scholar] [CrossRef]
- Kolari, K.; Sahamies, J.; Kalenius, E.; Novikov, A.S.; Kukushkin, V.Y.; Haukka, M. Metallophilic Interactions in Polymeric Group 11 Thiols. Solid State Sci. 2016, 60, 92–98. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Novikov, A.S.; Kinzhalov, M.A.; Zolotarev, A.A.; Boyarskiy, V.P. Intra-/Intermolecular Bifurcated Chalcogen Bonding in Crystal Structure of Thiazole/Thiadiazole Derived Binuclear (Diaminocarbene)PdII Complexes. Crystals 2018, 8, 112. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Novikov, A.S.; Kukushkin, V.Y. Recognition of the π-Hole Donor Ability of Iodopentafluorobenzene—A Conventional σ-Hole Donor for Crystal Engineering Involving Halogen Bonding. CrystEngComm 2019, 21, 616–628. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Zolotarev, A.A.; Boyarskiy, V.P.; Kukushkin, V.Y. (Isocyano Group π-Hole)⋯[dz2-MII] Interactions of (Isocyanide)[MII] Complexes, in which Positively Charged Metal Centers (d8-M=Pt, Pd) Act as Nucleophiles. Chem. Eur. J. 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Shikhaliyev, N.G.; Maharramov, A.M.; Bagirova, K.N.; Suleymanova, G.T.; Novikov, A.S.; Khrustalev, V.N.; Tskhovrebov, A.G. Halogenated Diazabutadiene Dyes: Synthesis, Structures, Supramolecular Features, and Theoretical Studies. Molecules 2020, 25, 5013. [Google Scholar] [CrossRef]
- Baykov, S.V.; Katlenok, E.A.; Baykova, S.O.; Semenov, A.V.; Bokach, N.A.; Boyarskiy, V.P. Conformation-Associated C···dz2-PtII Tetrel Bonding: The Case of Cyclometallated Platinum(II) Complex with 4-Cyanopyridyl Urea Ligand. Int. J. Mol. Sci. 2024, 25, 4052. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Rikagu Oxford Diffraction. CrysAlisPro Revision 1.171.40.53; Rikagu Oxford Diffraction Inc.: Yarnton, UK, 2019. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tetramer | dI···N | dSe···N | (T) | (T) | (D) | (D) |
|---|---|---|---|---|---|---|
| FI···BSeD···BSeD···FI | 2.401 | 2.795 | −19.44 | −9.23 | −8.51 | −0.72 |
| ClI···BSeD···BSeD···ClI | 2.516 | 2.814 | −14.05 | −8.98 | −8.35 | −0.63 |
| BrI···BSeD···BSeD···BrI | 2.563 | 2.821 | −12.35 | −8.90 | −8.29 | −0.61 |
| I2···BSeD···BSeD···I2 | 2.690 | 2.837 | −9.32 | −8.69 | −8.12 | −0.57 |
| CF3I···BSeD···BSeD···CF3I | 2.957 | 2.868 | −6.06 | −8.23 | −7.81 | −0.42 |
| CCl3I···BSeD···BSeD···CCl3I | 2.847 | 2.858 | −7.13 | −8.39 | −7.91 | −0.48 |
| CBr3I···BSeD···BSeD···CBr3I | 2.823 | 2.855 | −7.49 | −8.45 | −7.93 | −0.52 |
| CI4···BSeD···BSeD···CI4 | 2.825 | 2.855 | −7.39 | −8.47 | −7.93 | −0.54 |
| HCCI···BSeD···BSeD···HCCI | 2.930 | 2.867 | −6.62 | −8.24 | −7.81 | −0.43 |
| FCCI···BSeD···BSeD···FCCI | 2.904 | 2.866 | −6.92 | −8.27 | −7.83 | −0.44 |
| NCI···BSeD···BSeD···NCI | 2.819 | 2.853 | −8.91 | −8.37 | −7.94 | −0.43 |
| C6F3I3···BSeD···BSeD···C6F3I3 | 2.935 | 2.866 | −6.34 | −8.28 | −7.82 | −0.46 |
| C6F4I2···BSeD···BSeD···C6F4I2 | 2.928 | 2.865 | −6.53 | −8.30 | −7.83 | −0.47 |
| C6F5I···BSeD···BSeD···C6F5I | 2.926 | 2.865 | −6.55 | −8.29 | −7.83 | −0.46 |
| Dimer | ∆Eele | ∆Eexrep | ∆Epol | ∆Ecorr | ∆Edisp |
|---|---|---|---|---|---|
| BSeD···BSeD(FI···BSeD···BSeD···FI) | −21.40 | 30.89 | −9.40 | −7.23 | −3.38 |
| BSeD···BSeD(ClI···BSeD···BSeD···ClI) | −20.33 | 31.00 | −8.80 | −6.90 | −3.34 |
| BSeD···BSeD(BrI···BSeD···BSeD···BrI) | −19.95 | 30.36 | −8.60 | −6.78 | −3.32 |
| BSeD···BSeD(I2···BSeD···BSeD···I2) | −19.06 | 28.84 | −8.12 | −6.51 | −3.29 |
| BSeD···BSeD(CF3I···BSeD···BSeD···CF3I) | −17.44 | 26.13 | −7.26 | −6.04 | −3.21 |
| BSeD···BSeD(CCl3I···BSeD···BSeD···CCl3I) | −17.95 | 26.99 | −7.52 | −6.19 | −3.24 |
| BSeD···BSeD(CBr3I···BSeD···BSeD···CBr3I) | −18.10 | 27.23 | −7.60 | −6.23 | −3.24 |
| BSeD···BSeD(CI4···BSeD···BSeD···CI4) | −18.09 | 27.21 | −7.60 | −6.22 | −3.24 |
| BSeD···BSeD(HCCI···BSeD···BSeD···HCCI) | −17.49 | 26.22 | −7.29 | −6.04 | −3.22 |
| BSeD···BSeD(FCCI···BSeD···BSeD···FCCI) | −17.55 | 26.31 | −7.31 | −6.06 | −3.22 |
| BSeD···BSeD(NCI···BSeD···BSeD···NCI) | −18.21 | 27.44 | −7.66 | −6.27 | −3.25 |
| BSeD···BSeD(C6F3I3···BSeD···BSeD···C6F3I3) | −17.53 | 26.27 | −7.31 | −6.04 | −3.22 |
| BSeD···BSeD(C6F4I2···BSeD···BSeD···C6F4I2) | −17.58 | 26.36 | −7.33 | −6.06 | −3.22 |
| BSeD···BSeD(C6F5I···BSeD···BSeD···C6F5I) | −17.59 | 26.39 | −7.34 | −6.08 | −3.22 |
| [I12][BSeD] | [I13][BSeD] | [I135][BSeD] | |
|---|---|---|---|
| CCDC deposition number | 2239822 | 2239823 | 2239824 |
| Empirical formula | C12H4F4I2N2Se | C18H8F4I2N4Se2 | C12H4F3I3N2Se |
| Formula weight | 584.93 | 768.00 | 692.83 |
| Temperature/K | 293.00(2) | 290.00(10) | 293.00(2) |
| Crystal system | monoclinic | orthorhombic | monoclinic |
| Space group | P21/n | Pbca | P21/c |
| a/Å | 15.0997(6) | 14.8028(4) | 4.4302(2) |
| b/Å | 4.2279(2) | 15.5137(4) | 29.9768(14) |
| c/Å | 23.6753(9) | 37.2860(14) | 12.2962(7) |
| α/° | 90 | 90 | 90 |
| β/° | 90.124(4) | 90 | 91.505(5) |
| γ/° | 90 | 90 | 90 |
| Volume/Å3 | 1511.43(11) | 8562.6(5) | 1632.41(14) |
| Z | 4 | 16 | 4 |
| ρcalc/g·cm−3 | 2.571 | 2.383 | 2.819 |
| Color | colorless | colorless | colorless |
| Crystal size/mm3 | 0.29 × 0.28 × 0.17 | 0.2 × 0.17 × 0.15 | 0.22 × 0.18 × 0.15 |
| Reflections collected | 16,599 | 57,076 | 19,226 |
| Independent reflections | 3453 | 9660 | 3668 |
| Rint | 0.0371 | 0.0693 | 0.0547 |
| Number of refined parameters | 190 | 541 | 190 |
| Goodness-of-fit on F2 | 1.122 | 1.094 | 1.308 |
| Final R1 index [I ≥ 2σ(I)] | 0.0333 | 0.060 | 0.0770 |
| Final wR2 index [I ≥ 2σ(I)] | 0.0623 | 0.0756 | 0.1240 |
| Final R1 index [all data] | 0.0424 | 0.1217 | 0.0918 |
| Final wR2 index [all data] | 0.0657 | 0.0880 | 0.1286 |
| [I14][F-BSeD] | [I135][F-BSeD] | |
|---|---|---|
| CCDC deposition number | 2417680 | 2417681 |
| Empirical formula | C18H6F6I2N4Se2 | C18H6F5I3N4Se2 |
| Formula weight | 803.99 | 911.89 |
| Temperature/K | 293.00(2) | 100.00(10) |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/n | C2/c |
| a/Å | 13.0258(6) | 14.3599(6) |
| b/Å | 6.3844(3) | 9.2659(4) |
| c/Å | 13.4476(6) | 17.4527(9) |
| α/° | 90 | 90 |
| β/° | 104.852(5) | 101.082(4) |
| γ/° | 90 | 90 |
| Volume/Å3 | 1080.97(9) | 2278.91(18) |
| Z | 2 | 4 |
| ρcalc/g·cm−3 | 2.470 | 2.658 |
| Color | colorless | colorless |
| Crystal size/mm3 | 0.33 × 0.25 × 0.18 | 0.13 × 0.12 × 0.10 |
| Reflections collected | 13343 | 2009 |
| Independent reflections | 2678 | 2009 |
| Rint | 0.0627 | 0.0334 |
| Number of refined parameters | 145 | 142 |
| Goodness-of-fit on F2 | 1.036 | 1.113 |
| Final R1 index [I ≥ 2σ(I)] | 0.0349 | 0.0674 |
| Final wR2 index [I ≥ 2σ(I)] | 0.0555 | 0.1818 |
| Final R1 index [all data] | 0.0579 | 0.0716 |
| Final wR2 index [all data] | 0.0626 | 0.1857 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, S.; Sun, X.; Zhang, Y.; Wang, W. Chalcogen-Bonded [Se–N]2 Cyclic Supramolecular Synthons Enhanced by Halogen Bonds: Studies in the Gas Phase and Crystalline Phase. Int. J. Mol. Sci. 2025, 26, 2324. https://doi.org/10.3390/ijms26052324
Miao S, Sun X, Zhang Y, Wang W. Chalcogen-Bonded [Se–N]2 Cyclic Supramolecular Synthons Enhanced by Halogen Bonds: Studies in the Gas Phase and Crystalline Phase. International Journal of Molecular Sciences. 2025; 26(5):2324. https://doi.org/10.3390/ijms26052324
Chicago/Turabian StyleMiao, Shaobin, Xiaotian Sun, Yu Zhang, and Weizhou Wang. 2025. "Chalcogen-Bonded [Se–N]2 Cyclic Supramolecular Synthons Enhanced by Halogen Bonds: Studies in the Gas Phase and Crystalline Phase" International Journal of Molecular Sciences 26, no. 5: 2324. https://doi.org/10.3390/ijms26052324
APA StyleMiao, S., Sun, X., Zhang, Y., & Wang, W. (2025). Chalcogen-Bonded [Se–N]2 Cyclic Supramolecular Synthons Enhanced by Halogen Bonds: Studies in the Gas Phase and Crystalline Phase. International Journal of Molecular Sciences, 26(5), 2324. https://doi.org/10.3390/ijms26052324