
Gene Expression Profiling of Pancreatic Ductal Adenocarcinoma Cells in Hypercapnia Identifies SIAH3 as a Novel Prognostic Biomarker

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Chronic Hypercapnia Exposure Induces a Distinct Aggressive Phenotype in Pancreatic Cancer Cells
2.2. Gene Expression Analysis Shows SIAH3 as the Most Differentially Regulated Transcript in Chronic Hypercapnia
2.3. Molecular Pathway Analysis Shows Activation of EMT and TME Regulatory Pathways
2.4. Chronic Hypercapnia Downregulates SIAH3 Gene mRNA and Protein Expression
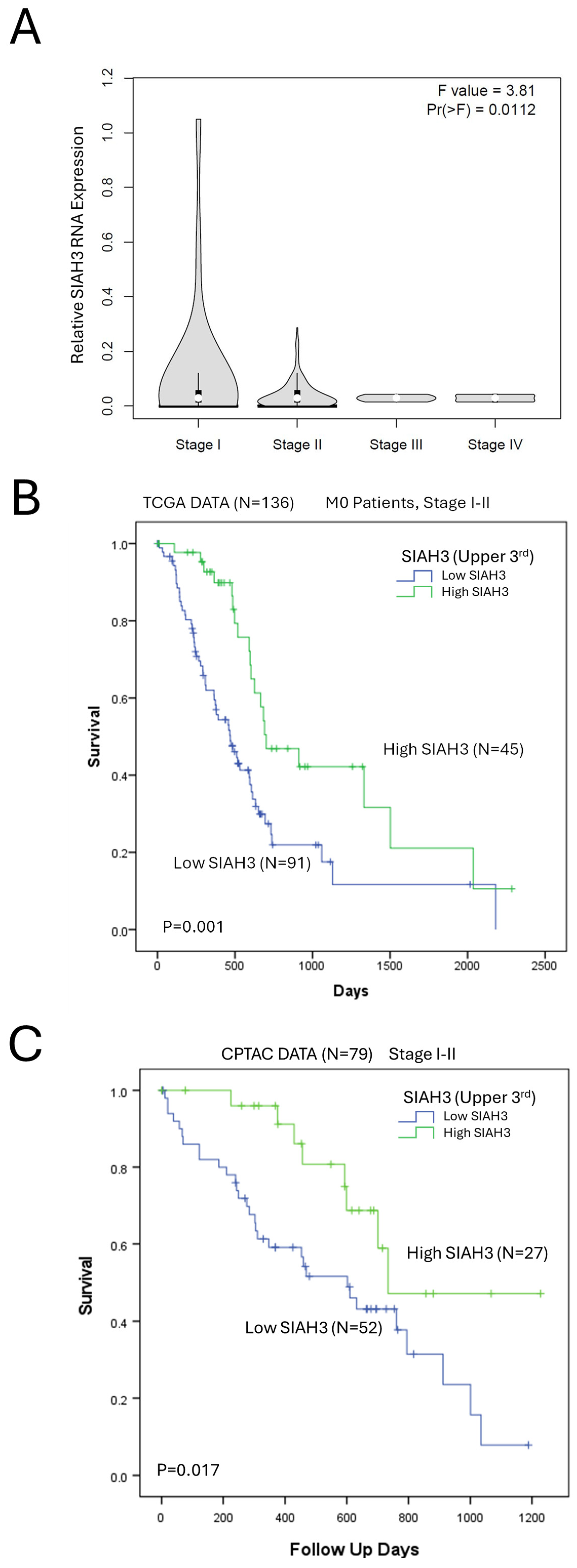
2.5. SIAH3 Expression Is a Positive Prognostic Marker of Pancreatic Adenocarcinoma Survival
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Culturing with CO2 Exposure
4.3. Colony Formation Assays
4.4. Cell Migration (Scratch Wound) Assay
4.5. Cell Transwell Invasion Assay
4.6. RT-qPCR and RNA Quantification
4.7. Whole Exome Sequencing and Gene Expression Analysis
4.8. Reactome Pathway Analysis
4.9. Gene Set Enrichment Analysis (GSEA)
4.10. Immunoblot Analysis
4.11. Normal and Tumor Tissue Expression Data
4.12. RNAi Viability Effect Analysis
4.13. CCLE Analysis of Cell Line Growth Rates
4.14. Clinical Data Collection
4.15. Statistical and Survival Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ECM | Extracellular matrix |
| EMT | Epithelial-to-mesenchymal transition |
| GIST | Gastrointestinal stromal tumor |
| GSEA | Gene set enrichment analysis |
| HIF | Hypoxia-inducible factor |
| HUVEC | Human umbilical vein endothelial cells |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDGF | Platelet-derived growth factor |
| SINA | Seven in absentia |
| SIAH | Seven-in-absentia homolog |
| TME | Tumor microenvironment |
| VEGF | Vascular endothelial growth factor |
References
- Helmlinger, G.; Sckell, A.; Dellian, M.; Forbes, N.S.; Jain, R.K. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 2002, 8, 1284–1291. [Google Scholar]
- Kikuchi, R.; Iwai, Y.; Tsuji, T.; Watanabe, Y.; Koyama, N.; Yamaguchi, K.; Nakamura, H.; Aoshiba, K. Hypercapnic tumor microenvironment confers chemoresistance to lung cancer cells by reprogramming mitochondrial metabolism in vitro. Free Radic. Biol. Med. 2019, 134, 200–214. [Google Scholar] [CrossRef]
- Obata, S.; Goi, T.; Nakazawa, T.; Kimura, Y.; Katayama, K.; Yamaguchi, A. Changes in CO2 concentration increase the invasive ability of colon cancer cells. Anticancer Res. 2013, 33, 1881–1885. [Google Scholar]
- Shen, M.Y.; Huang, I.P.; Chen, W.S.; Chang, J.T.; Lin, J.K. Influence of pneumoperitoneum on tumor growth and pattern of intra-abdominal tumor spreading: In vivo study of a murine model. Hepatogastroenterology 2008, 55, 947–951. [Google Scholar]
- Nevler, A.; Brown, S.Z.; Nauheim, D.; Portocarrero, C.; Rodeck, U.; Bassig, J.; Schultz, C.W.; McCarthy, G.A.; Lavu, H.; Yeo, T.P.; et al. Effect of Hypercapnia, an Element of Obstructive Respiratory Disorder, on Pancreatic Cancer Chemoresistance and Progression. J. Am. Coll. Surg. 2020, 230, 659–667. [Google Scholar] [CrossRef]
- Nevler, A.; Khalilieh, S.; Lavu, H.; Bowne, W.; Yeo, C.J. Hypercapnic Tissue Gene Expression and Survival in Early-Stage Pancreatic Ductal Adenocarcinoma. J. Am. Coll. Surg. 2023, 236, 913–922. [Google Scholar] [CrossRef]
- Shigemura, M.; Welch, L.C.; Sznajder, J.I. Hypercapnia Regulates Gene Expression and Tissue Function. Front. Physiol. 2020, 11, 598122. [Google Scholar] [CrossRef]
- Pelletier-Galarneau, M.; deKemp, R.A.; Hunter, C.; Klein, R.; Klein, M.; Ironstone, J.; Fisher, J.A.; Ruddy, T.D. Effects of Hypercapnia on Myocardial Blood Flow in Healthy Human Subjects. J. Nucl. Med. 2018, 59, 100–106. [Google Scholar] [CrossRef]
- Hof, S.; Truse, R.; Weber, L.; Herminghaus, A.; Schulz, J.; Weber, A.P.M.; Maleckova, E.; Bauer, I.; Picker, O.; Vollmer, C. Local Mucosal CO2 but Not O2 Insufflation Improves Gastric and Oral Microcirculatory Oxygenation in a Canine Model of Mild Hemorrhagic Shock. Front. Med. 2022, 9, 867298. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Itoh, M.; Nakamura, H.; Yamaguchi, K. Hypercapnia accelerates wound healing in endothelial cell monolayers exposed to hypoxia. Open Respir. Med. J. 2013, 7, 6–12. [Google Scholar] [CrossRef]
- Reivich, M. Arterial Pco2 and Cerebral Hemodynamics. Am. J. Physiol. 1964, 206, 25–35. [Google Scholar] [CrossRef]
- Kety, S.S.; Schmidt, C.F. The Effects of Altered Arterial Tensions of Carbon Dioxide and Oxygen on Cerebral Blood Flow and Cerebral Oxygen Consumption of Normal Young Men. J. Clin. Investig. 1948, 27, 484–492. [Google Scholar] [CrossRef]
- Vohwinkel, C.U.; Lecuona, E.; Sun, H.; Sommer, N.; Vadasz, I.; Chandel, N.S.; Sznajder, J.I. Elevated CO2 levels cause mitochondrial dysfunction and impair cell proliferation. J. Biol. Chem. 2011, 286, 37067–37076. [Google Scholar] [CrossRef]
- Phelan, D.E.; Mota, C.; Strowitzki, M.J.; Shigemura, M.; Sznajder, J.I.; Crowe, L.; Masterson, J.C.; Hayes, S.E.; Reddan, B.; Yin, X.; et al. Hypercapnia alters mitochondrial gene expression and acylcarnitine production in monocytes. Immunol. Cell Biol. 2023, 101, 556–577. [Google Scholar] [CrossRef]
- Ceco, E.; Celli, D.; Weinberg, S.; Shigemura, M.; Welch, L.C.; Volpe, L.; Chandel, N.S.; Bharat, A.; Lecuona, E.; Sznajder, J.I. Elevated CO2 Levels Delay Skeletal Muscle Repair by Increasing Fatty Acid Oxidation. Front. Physiol. 2020, 11, 630910. [Google Scholar] [CrossRef]
- Bolshette, N.; Ezagouri, S.; Dandavate, V.; Karavaeva, I.; Golik, M.; Wang, H.; Espenshade, P.J.; Osborne, T.F.; Han, X.; Asher, G. Carbon dioxide regulates cholesterol levels through SREBP2. PLoS Biol. 2023, 21, e3002367. [Google Scholar] [CrossRef]
- Li, G.; Zhou, D.; Vicencio, A.G.; Ryu, J.; Xue, J.; Kanaan, A.; Gavrialov, O.; Haddad, G.G. Effect of carbon dioxide on neonatal mouse lung: A genomic approach. J. Appl. Physiol. (1985) 2006, 101, 1556–1564. [Google Scholar] [CrossRef]
- Ryu, J.; Heldt, G.P.; Nguyen, M.; Gavrialov, O.; Haddad, G.G. Chronic hypercapnia alters lung matrix composition in mouse pups. J. Appl. Physiol. (1985) 2010, 109, 203–210. [Google Scholar] [CrossRef]
- Dada, L.A.; Welch, L.C.; Magnani, N.D.; Ren, Z.; Han, H.; Brazee, P.L.; Celli, D.; Flozak, A.S.; Weng, A.; Herrerias, M.M.; et al. Hypercapnia alters stroma-derived Wnt production to limit beta-catenin signaling and proliferation in AT2 cells. JCI Insight 2023, 8, e159331. [Google Scholar] [CrossRef]
- Mizuguchi, S.; Iwata, T.; Izumi, N.; Tsukioka, T.; Hanada, S.; Komatsu, H.; Nishiyama, N. Arterial blood gases predict long-term prognosis in stage I non-small cell lung cancer patients. BMC Surg. 2016, 16, 3. [Google Scholar] [CrossRef]
- Dehne, S.; Kirschner, L.; Strowitzki, M.J.; Kilian, S.; Kummer, L.C.; Schneider, M.A.; Michalski, C.W.; Buchler, M.W.; Weigand, M.A.; Larmann, J. Low intraoperative end-tidal carbon dioxide levels are associated with improved recurrence-free survival after elective colorectal cancer surgery. J. Clin. Anesth. 2024, 96, 111495. [Google Scholar] [CrossRef] [PubMed]
- Benderro, G.F.; Tsipis, C.P.; Sun, X.; Kuang, Y.; LaManna, J.C. Increased HIF-1alpha and HIF-2alpha accumulation, but decreased microvascular density, in chronic hyperoxia and hypercapnia in the mouse cerebral cortex. Adv. Exp. Med. Biol. 2013, 789, 29–35. [Google Scholar] [PubMed]
- Shigemura, M.; Lecuona, E.; Angulo, M.; Dada, L.A.; Edwards, M.B.; Welch, L.C.; Casalino-Matsuda, S.M.; Sporn, P.H.S.; Vadasz, I.; Helenius, I.T.; et al. Elevated CO2 regulates the Wnt signaling pathway in mammals, Drosophila melanogaster and Caenorhabditis elegans. Sci. Rep. 2019, 9, 18251. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sessions, E.H.; Zhang, F.; Ban, F.; Placencio-Hickok, V.; Ma, C.T.; Zeng, F.Y.; Pass, I.; Terry, D.B.; Cadwell, G.; et al. Identification and characterization of small molecule inhibitors of the ubiquitin ligases Siah1/2 in melanoma and prostate cancer cells. Cancer Lett. 2019, 449, 145–162. [Google Scholar] [CrossRef]
- Nakayama, K.; Frew, I.J.; Hagensen, M.; Skals, M.; Habelhah, H.; Bhoumik, A.; Kadoya, T.; Erdjument-Bromage, H.; Tempst, P.; Frappell, P.B.; et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell 2004, 117, 941–952. [Google Scholar] [CrossRef]
- Pepper, I.J.; Van Sciver, R.E.; Tang, A.H. Phylogenetic analysis of the SINA/SIAH ubiquitin E3 ligase family in Metazoa. BMC Evol. Biol. 2017, 17, 182. [Google Scholar] [CrossRef]
- Behling, K.C.; Tang, A.; Freydin, B.; Chervoneva, I.; Kadakia, S.; Schwartz, G.F.; Rui, H.; Witkiewicz, A.K. Increased SIAH expression predicts ductal carcinoma in situ (DCIS) progression to invasive carcinoma. Breast Cancer Res. Treat. 2011, 129, 717–724. [Google Scholar] [CrossRef]
- Moller, A.; House, C.M.; Wong, C.S.; Scanlon, D.B.; Liu, M.C.; Ronai, Z.; Bowtell, D.D. Inhibition of Siah ubiquitin ligase function. Oncogene 2009, 28, 289–296. [Google Scholar] [CrossRef]
- Abd Elghani, F.; Safory, H.; Hamza, H.; Savyon, M.; Farhoud, M.; Toren-Hershoviz, M.; Vitic, Z.; Ebanks, K.; Shani, V.; Bisharat, S.; et al. SIAH proteins regulate the degradation and intra-mitochondrial aggregation of PINK1: Implications for mitochondrial pathology in Parkinson’s disease. Aging Cell 2022, 21, e13731. [Google Scholar] [CrossRef]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef]
- Watzlawik, J.O.; Hou, X.; Richardson, T.; Lewicki, S.L.; Siuda, J.; Wszolek, Z.K.; Cook, C.N.; Petrucelli, L.; DeTure, M.; Dickson, D.W.; et al. Development and characterization of phospho-ubiquitin antibodies to monitor PINK1-PRKN signaling in cells and tissue. Autophagy 2024, 20, 2076–2091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Schetter, A.; He, P.; Funamizu, N.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Hassan, R.; Yfantis, H.G.; Lee, D.H.; et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e31507. [Google Scholar] [CrossRef]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.R.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009, 16, 259–266. [Google Scholar] [CrossRef]
- Yang, S.; He, P.; Wang, J.; Schetter, A.; Tang, W.; Funamizu, N.; Yanaga, K.; Uwagawa, T.; Satoskar, A.R.; Gaedcke, J.; et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res. 2016, 76, 3838–3850. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.H.; Hoefer, R.A.; Guye, M.L.; Bear, H.D. Persistent EGFR/K-RAS/SIAH pathway activation drives chemo-resistance and early tumor relapse in triple-negative breast cancer. Cancer Drug Resist. 2022, 5, 691–702. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Almanza-Hurtado, A.; Polanco Guerra, C.; Martinez-Avila, M.C.; Borre-Naranjo, D.; Rodriguez-Yanez, T.; Duenas-Castell, C. Hypercapnia from Physiology to Practice. Int. J. Clin. Pract. 2022, 2022, 2635616. [Google Scholar] [CrossRef]
- Cherniack, N.S.; Edelman, N.H.; Lahiri, S. Hypoxia and hypercapnia as respiratory stimulants and depressants. Respir. Physiol. 1970, 11, 113–126. [Google Scholar] [CrossRef]
- Stefanik, E.; Drewnowska, O.; Lisowska, B.; Turek, B. Causes, Effects and Methods of Monitoring Gas Exchange Disturbances during Equine General Anaesthesia. Animals 2021, 11, 2049. [Google Scholar] [CrossRef]
- Petersson, J.; Glenny, R.W. Gas exchange and ventilation-perfusion relationships in the lung. Eur. Respir. J. 2014, 44, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Graf, N.; Mullen, A.; Kanter, J.; Andrei, A.C.; Sporn, P.H.; DeCamp, M.M.; Sznajder, J.I. Pleural Hypercarbia After Lung Surgery Is Associated With Persistent Alveolopleural Fistulae. Chest 2016, 149, 220–227. [Google Scholar] [CrossRef]
- O’Toole, D.; Hassett, P.; Contreras, M.; Higgins, B.D.; McKeown, S.T.; McAuley, D.F.; O’Brien, T.; Laffey, J.G. Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-kappaB dependent mechanism. Thorax 2009, 64, 976–982. [Google Scholar] [CrossRef]
- Zhao, J.; Cai, Y.; Yin, C.; Lv, Y.; Wei, W.; Wang, X.; Hao, Z.; Shen, C.; Wang, H.; Chen, J. Study on the inhibition of hyperthermic CO2 pneumoperitoneum on the proliferation and migration of colon cancer cells and its mechanism. Oncol. Rep. 2016, 35, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, D.; Lu, L.; Liu, Z.; Fang, L. Effects of CO2 pneumoperitoneum on proliferation, apoptosis, and migration of gastrointestinal stromal tumor cells. Surg. Endosc. 2019, 33, 3384–3395. [Google Scholar] [CrossRef]
- Yu, Y.; Kuebler, J.; Groos, S.; Metzelder, M.; Kurpanik, S.; Ure, B.M.; Vieten, G. Carbon dioxide modifies the morphology and function of mesothelial cells and facilitates transepithelial neuroblastoma cell migration. Pediatr. Surg. Int. 2010, 26, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Selfridge, A.C.; Cavadas, M.A.; Scholz, C.C.; Campbell, E.L.; Welch, L.C.; Lecuona, E.; Colgan, S.P.; Barrett, K.E.; Sporn, P.H.; Sznajder, J.I.; et al. Hypercapnia Suppresses the HIF-dependent Adaptive Response to Hypoxia. J. Biol. Chem. 2016, 291, 11800–11808. [Google Scholar] [CrossRef]
- Molinas, C.R.; Campo, R.; Elkelani, O.A.; Binda, M.M.; Carmeliet, P.; Koninckx, P.R. Role of hypoxia inducible factors 1alpha and 2alpha in basal adhesion formation and in carbon dioxide pneumoperitoneum-enhanced adhesion formation after laparoscopic surgery in transgenic mice. Fertil. Steril. 2003, 80 (Suppl. S2), 795–802. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Xu, Y.F.; Zheng, J.H. Effect of carbon dioxide pneumoperitoneum on human renal cell carcinoma proliferation and metastasis in an orthotropic xenograft nude mouse model. Arch. Med. Sci. 2014, 10, 1041–1046. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Semenza, G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am. J. Physiol. Cell Physiol. 2015, 309, C775–C782. [Google Scholar] [CrossRef]
- Jacquemin, C.; El Orch, W.; Diaz, O.; Lalande, A.; Aublin-Gex, A.; Jacolin, F.; Toesca, J.; Si-Tahar, M.; Mathieu, C.; Lotteau, V.; et al. Pharmacological induction of the hypoxia response pathway in Huh7 hepatoma cells limits proliferation but increases resilience under metabolic stress. Cell. Mol. Life Sci. 2024, 81, 320. [Google Scholar] [CrossRef]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Fotia, C.; Massa, A.; Boriani, F.; Baldini, N.; Granchi, D. Hypoxia enhances proliferation and stemness of human adipose-derived mesenchymal stem cells. Cytotechnology 2015, 67, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Gaelzer, M.M.; Santos, M.S.D.; Coelho, B.P.; de Quadros, A.H.; Simao, F.; Usach, V.; Guma, F.C.R.; Setton-Avruj, P.; Lenz, G.; Salbego, C.G. Hypoxic and Reoxygenated Microenvironment: Stemness and Differentiation State in Glioblastoma. Mol. Neurobiol. 2017, 54, 6261–6272. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.K.; Park, G.Y.; Bae, M.J.; Kim, J.S.; Jo, W.S.; Lee, C.G. Hypoxia induces immunogenic cell death of cancer cells by enhancing the exposure of cell surface calreticulin in an endoplasmic reticulum stress-dependent manner. Oncol. Lett. 2019, 18, 6269–6274. [Google Scholar] [CrossRef]
- Taylor, C.T. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J. Physiol. 2008, 586, 4055–4059. [Google Scholar] [CrossRef]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef]
- Ma, S.; Zhao, Y.; Lee, W.C.; Ong, L.T.; Lee, P.L.; Jiang, Z.; Oguz, G.; Niu, Z.; Liu, M.; Goh, J.Y.; et al. Hypoxia induces HIF1alpha-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat. Commun. 2022, 13, 4118. [Google Scholar] [CrossRef]
- Horsman, M.R.; Vaupel, P. Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Hammelef, E.; Sabitsky, M.; Ream, C.; Khalilieh, S.; Zohar, N.; Lavu, H.; Bowne, W.B.; Yeo, C.J.; Nevler, A. Chronic Obstructive Pulmonary Disease Is Associated with Worse Oncologic Outcomes in Early-Stage Resected Pancreatic and Periampullary Cancers. Biomedicines 2023, 11, 1684. [Google Scholar] [CrossRef]
- Schmidt, R.L.; Park, C.H.; Ahmed, A.U.; Gundelach, J.H.; Reed, N.R.; Cheng, S.; Knudsen, B.E.; Tang, A.H. Inhibition of RAS-mediated transformation and tumorigenesis by targeting the downstream E3 ubiquitin ligase seven in absentia homologue. Cancer Res. 2007, 67, 11798–11810. [Google Scholar] [CrossRef]
- van Reesema, L.L.S.; Zheleva, V.; Winston, J.S.; Jansen, R.J.; O’Connor, C.F.; Isbell, A.J.; Bian, M.; Qin, R.; Bassett, P.T.; Hinson, V.J.; et al. SIAH and EGFR, Two RAS Pathway Biomarkers, are Highly Prognostic in Locally Advanced and Metastatic Breast Cancer. EBioMedicine 2016, 11, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.S.; Sceneay, J.; House, C.M.; Halse, H.M.; Liu, M.C.; George, J.; Hunnam, T.C.; Parker, B.S.; Haviv, I.; Ronai, Z.; et al. Vascular normalization by loss of Siah2 results in increased chemotherapeutic efficacy. Cancer Res. 2012, 72, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- Brauckhoff, A.; Malz, M.; Tschaharganeh, D.; Malek, N.; Weber, A.; Riener, M.O.; Soll, C.; Samarin, J.; Bissinger, M.; Schmidt, J.; et al. Nuclear expression of the ubiquitin ligase seven in absentia homolog (SIAH)-1 induces proliferation and migration of liver cancer cells. J. Hepatol. 2011, 55, 1049–1057. [Google Scholar] [CrossRef]
- Qi, J.; Nakayama, K.; Gaitonde, S.; Goydos, J.S.; Krajewski, S.; Eroshkin, A.; Bar-Sagi, D.; Bowtell, D.; Ronai, Z. The ubiquitin ligase Siah2 regulates tumorigenesis and metastasis by HIF-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 16713–16718. [Google Scholar] [CrossRef]
- Qin, R.; Smyrk, T.C.; Reed, N.R.; Schmidt, R.L.; Schnelldorfer, T.; Chari, S.T.; Petersen, G.M.; Tang, A.H. Combining clinicopathological predictors and molecular biomarkers in the oncogenic K-RAS/Ki67/HIF-1alpha pathway to predict survival in resectable pancreatic cancer. Br. J. Cancer 2015, 112, 514–522. [Google Scholar] [CrossRef]
- Varghese, A.M.; Perry, M.A.; Chou, J.F.; Nandakumar, S.; Muldoon, D.; Erakky, A.; Zucker, A.; Fong, C.; Mehine, M.; Nguyen, B.; et al. Clinicogenomic landscape of pancreatic adenocarcinoma identifies KRAS mutant dosage as prognostic of overall survival. Nat. Med. 2025, 31, 466–477. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, Y.; Gong, Y.; Zhao, S.; Li, M.; Li, X.; Song, G.; Zhai, B.; Liu, J.; Shao, Y.; et al. The genetic landscape of pancreatic head ductal adenocarcinoma in China and prognosis stratification. BMC Cancer 2022, 22, 186. [Google Scholar] [CrossRef]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically Detected Expression of 3 Major Genes (CDKN2A/p16, TP53, and SMAD4/DPC4) Strongly Predicts Survival in Patients With Resectable Pancreatic Cancer. Ann. Surg. 2013, 258, 336–346. [Google Scholar] [CrossRef]
- Khan, S.; Zafar, N.; Khan, S.S.; Setua, S.; Behrman, S.W.; Stiles, Z.E.; Yallapu, M.M.; Sahay, P.; Ghimire, H.; Ise, T.; et al. Clinical significance of MUC13 in pancreatic ductal adenocarcinoma. HPB 2018, 20, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.M.; Cannon, A.; West, S.; Ghersi, D.; Atri, P.; Bhatia, R.; Smith, L.; Rachagani, S.; Wichman, C.; Kumar, S.; et al. Mucin Expression and Splicing Determine Novel Subtypes and Patient Mortality in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 6787–6799. [Google Scholar] [CrossRef]
- Stubbe, B.E.; Larsen, A.C.; Madsen, P.H.; Krarup, H.B.; Pedersen, I.S.; Lundbye-Christensen, S.; Hansen, C.P.; Hasselby, J.P.; Johansen, A.Z.; Thorlacius-Ussing, O.; et al. Promoter hypermethylation of SFRP1 as a prognostic and potentially predictive blood-based biomarker in patients with localized pancreatic ductal adenocarcinoma. Front. Oncol. 2023, 13, 1211292. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.W.; McCarthy, G.A.; Nerwal, T.; Nevler, A.; DuHadaway, J.B.; McCoy, M.D.; Jiang, W.; Brown, S.Z.; Goetz, A.; Jain, A.; et al. The FDA-Approved Anthelmintic Pyrvinium Pamoate Inhibits Pancreatic Cancer Cells in Nutrient-Depleted Conditions by Targeting the Mitochondria. Mol. Cancer Ther. 2021, 20, 2166–2176. [Google Scholar] [CrossRef]
- Chand, S.N.; Zarei, M.; Schiewer, M.J.; Kamath, A.R.; Romeo, C.; Lal, S.; Cozzitorto, J.A.; Nevler, A.; Scolaro, L.; Londin, E.; et al. Posttranscriptional Regulation of PARG mRNA by HuR Facilitates DNA Repair and Resistance to PARP Inhibitors. Cancer Res. 2017, 77, 5011–5025. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- McDonald, E.R., 3rd; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Log2 (Fold Change) MiaPaCa2 | Log2 (Fold Change) Panc1 |
|---|---|---|
| SIAH3/ENSG00000215475 | −4.283 | −3.175 |
| EML5/ENSG00000165521 | −1.892 | −2.393 |
| C3ORF80/ENSG00000180044 | −2.179 | −2.328 |
| TCGA (N = 134) | ||
| Covariate | HR | Sig. |
| T | 1.26 (0.71–2.26) | 0.431 |
| N | 0 (0–0) | 0.047 |
| N1 | 1.47 (0.81–2.65) | 0.205 |
| NX | 12.8 (1.54–106.28) | 0.018 |
| High SIAH3 | 0.4 (0.24–0.67) | 0.001 |
| CPTAC (N = 79) | ||
| Covariate | HR | Sig. |
| T stage | 1.821 (0.98–3.36) | 0.058 |
| N stage | 0.178 | |
| N1 | 0.63 (0.30–1.32) | 0.223 |
| NX | 12.4 (0.26–586.57) | 0.201 |
| R | 0.008 | |
| R1 | 1.07 (0.46–2.46) | 0.877 |
| R2 | 0.23 (0.00–10.83) | 0.457 |
| RX | 2.72 (1.13–6.55) | 0.025 |
| High SIAH3 | 0.36 (0.15–0.85) | 0.020 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zohar, N.; Maguire, R.; Khalilieh, S.; Jain, A.; Bosykh, D.; Bowne, W.B.; Lavu, H.; Yeo, C.J.; Nevler, A. Gene Expression Profiling of Pancreatic Ductal Adenocarcinoma Cells in Hypercapnia Identifies SIAH3 as a Novel Prognostic Biomarker. Int. J. Mol. Sci. 2025, 26, 2848. https://doi.org/10.3390/ijms26072848
Zohar N, Maguire R, Khalilieh S, Jain A, Bosykh D, Bowne WB, Lavu H, Yeo CJ, Nevler A. Gene Expression Profiling of Pancreatic Ductal Adenocarcinoma Cells in Hypercapnia Identifies SIAH3 as a Novel Prognostic Biomarker. International Journal of Molecular Sciences. 2025; 26(7):2848. https://doi.org/10.3390/ijms26072848
Chicago/Turabian StyleZohar, Nitzan, Ryan Maguire, Saed Khalilieh, Aditi Jain, Dmitriy Bosykh, Wilbur B. Bowne, Harish Lavu, Charles J. Yeo, and Avinoam Nevler. 2025. "Gene Expression Profiling of Pancreatic Ductal Adenocarcinoma Cells in Hypercapnia Identifies SIAH3 as a Novel Prognostic Biomarker" International Journal of Molecular Sciences 26, no. 7: 2848. https://doi.org/10.3390/ijms26072848
APA StyleZohar, N., Maguire, R., Khalilieh, S., Jain, A., Bosykh, D., Bowne, W. B., Lavu, H., Yeo, C. J., & Nevler, A. (2025). Gene Expression Profiling of Pancreatic Ductal Adenocarcinoma Cells in Hypercapnia Identifies SIAH3 as a Novel Prognostic Biomarker. International Journal of Molecular Sciences, 26(7), 2848. https://doi.org/10.3390/ijms26072848