Abstract
In this study, PTA&PMA/NiMoO4@NF was synthesized on nickel foam through wet chemical etching to promote the kinetics of the oxygen evolution reaction (OER) effectively. OER benefits from two cationic (Ni and Mo) defects and the optimized electronic configuration of PTA&PMA/NiMoO4@NF. Thus, it only needs 200 mV to reach the current density of 10 mA cm−2 in 1.0 mol/L of KOH. This value is nearly 100 mV lower than the value needed by pure NiMoO4. After being used as an anode for water splitting in an alkaline solution, the as-obtained catalyst can operate at a current density of 10 mA cm−2 for 24 h of good stability. The synthesis strategy adopted in this study can provide an effective, low-cost, simple, and convenient strategy for improving the OER electrocatalytic performance of other transition metal oxides.
1. Introduction
Electrocatalytic water splitting (2H2O → O2 + 2H2), which is an efficient and sustainable method for hydrogen production, holds promise for addressing the energy crisis and environmental issues caused by traditional fossil fuels [1]. Water electrolysis typically consists of the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). HER is a two-electron proton coupling reaction, whereas OER is a four-electron proton coupling reaction, requiring high energy to overcome the OER barrier. It can significantly reduce the efficiency of the electrochemical water splitting reaction. Therefore, OER is generally considered the decisive step in the water splitting reaction. Selecting an appropriate catalyst is crucial to overcome the impact of the OER process on the electrochemical water splitting process [2]. Hence, developing high-performance electrocatalysts that accelerate OER kinetics is urgently needed.
Currently, the most commonly used anode catalysts are noble metal oxides, such as RuO2 and IrO2. However, their high cost and low reserves hinder their practical application [3,4]. Transition metal oxides are low-cost, have high reserves, and exhibit good OER catalytic activity [5,6,7,8]. Among the transition metal-based compounds, binary nickel-based materials, particularly NiMoO4 (NMO), have been extensively developed as efficient water-splitting electrocatalysts; at present, NMO is the most studied nickel-based electrocatalyst. Although single-metal oxides such as Co3O4 [9], Fe2O3 [10], and MnO2 [11] have been widely explored for similar applications, their performance is often constrained by limited active sites, poor electrical conductivity, or instability under operational conditions. In contrast, the dual-metal composition of NiMoO4 addresses these limitations through its complementary functionalities: Ni atoms contribute high intrinsic redox activity and facilitate charge transfer, and Mo atoms enhance structural stability and modulate the electronic states to optimize intermediate adsorption/desorption kinetics [12,13]. However, NMO electrodes are still affected by poor electronic conductivity, insufficient ion transport and diffusion, and structural instability during long-term cycling. Therefore, fabricating NMO with controlled nanostructures and designing reasonable structural engineering are important but challenging. In particular, various low-dimensional NMO nanostructures directly grown on conductive current collector substrates (e.g., Ni/Cu foam [14], graphene [15], and carbon substrates [16]) reduce charge carrier scattering at grain boundaries and are easily integrated into flexible devices with certain specific applications. These nanostructures are particularly preferred for directional electron transport. Various NMO/carbon composites have been synthesized to overcome the poor conductivity of pure NMO by combining NMO nanostructures with graphene [17], carbon nanotubes [18], conductive polymers [19], and porous carbon structures [20,21]. In addition to carbon composites, several heteroatoms (e.g., Mn [22,23], P [24], Zn [25], Ce [26]) have been doped into NMO, or oxygen vacancies have been generated in the NMO lattice [27]. Furthermore, NMO has been combined with other metal oxides [28,29,30,31,32,33] or sulfides [34,35,36] to form heterostructure electrodes and improve electrochemical performance. However, the reported composite or doping methods of NMO involve multiple complex chemical and physical processes, which have high economic costs or are environmentally unfriendly in the synthesis path.
Research shows that chemical acid etching has become a research hotspot. A multifunctional polyoxometalate (POM; also called heteropoly acid) etching method could clearly reconstruct NiFe layered double hydroxide (LDH), including three-dimensional morphology nanocutting, Fe3+ and α-Ni(OH)2 active substance reconstruction, and the generation of multiple Ni, Fe, and O vacancies, which subtly coordinate local environments and electronic structures of iron and nickel cations, creating additional active sites for reduction reactions. Moreover, the embedding of POM polyanion clusters can tune the electronic configuration of NiFe LDH to facilitate the formation and transformation of intermediate states, thereby accelerating the electrochemical process [37]. As a result, the high catalytic activity (η10 = 206 mV) and excellent stability (negligible η500 change over 24 h) of NiFe LDH-PMo12 can be achieved [38]. An in-depth understanding of the intrinsic activity origin of POM etching has been gained by combining the theoretical and experimental results, thereby proving the feasibility of POM etching as a promising posttreatment technology. POMs are a class of well-defined transition metal oxide clusters with tunable redox properties and strong acidity. They have been widely explored as molecular catalysts for acid and oxidation reactions.
On the basis of the above statement, highly active NMO@NF was prepared via a low-cost, simple, and convenient wet chemical etching method that used phosphotungstic acid (PTA) and phosphomolybdic acid (PMA). It also offered potential avenues for designing new materials. Experimental characterizations revealed two cationic (Ni and Mo) defects and the optimized electronic configuration of NMO after PTA and PMA etching, which promoted the OER process.
2. Results and Discussion
2.1. Material Synthesis and Characterization
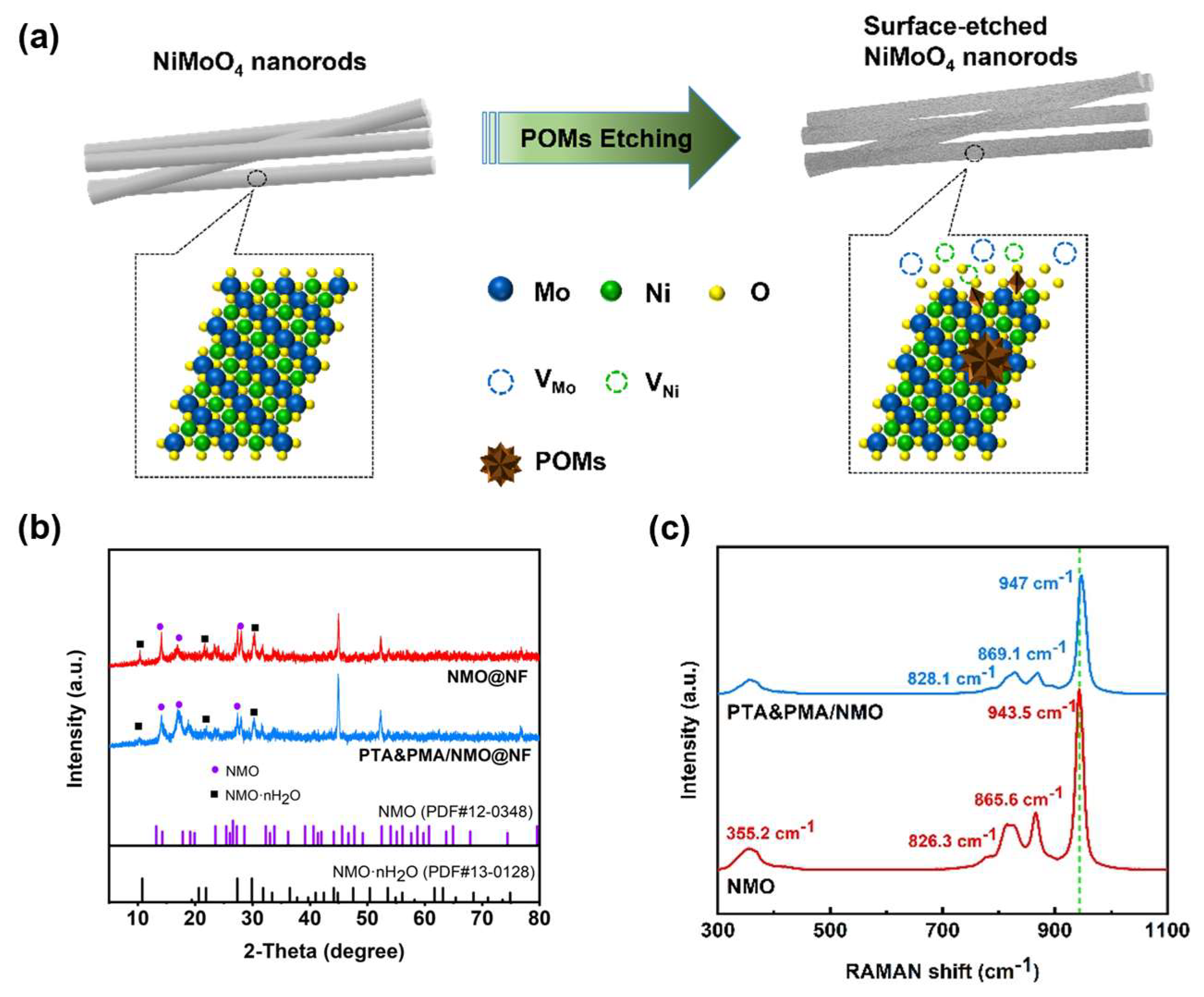
The electrocatalytic samples of PTA&PMA/NMO@NF are prepared using a simple hydrothermal synthesis method and the wet chemical etching method. A mixed solution of ammonium molybdate and nickel molybdate is prepared, and NMO@NF is synthesized in situ on the NF using the hydrothermal method. Subsequently, a weakly acidic mixed solution of PTA and PMA is prepared. This solution easily etches the surface of NMO@NF, thereby obtaining a PTA&PMA/NMO@NF electrocatalyst. The etching schematic diagram is shown in Figure 1a.
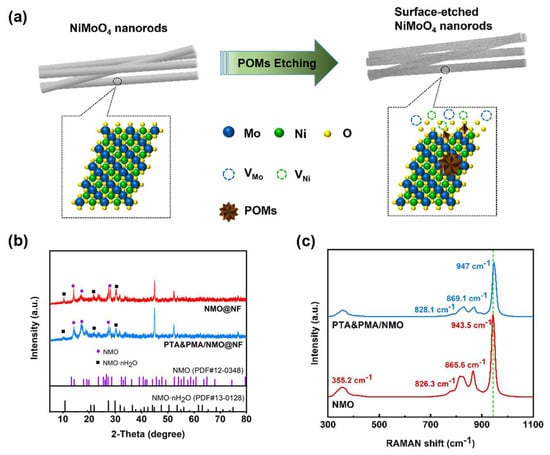
Figure 1.
Structure and phase composition of the catalysts. (a) Diagram of the synthesis process for PTA&PMA/NMO@NF; (b) XRD pattern Raman spectra of the as-prepared NMO@NF and PTA&PMA/NMO@NF; and (c) Raman spectra of the as-prepared NMO and PTA&PMA/NMO.
The atomic ratio is obtained via the ICP test, during which the contents of Ni, Mo, P, and W cations are detected in the NMO and PTA&PMA/NMO electrocatalysts. After POM etching, two cationic (Ni and Mo) defects are produced in NMO, and a small number of P and W atoms are attached to PTA&PMA/NMO during the etching process (Table S1). Additionally, the Ni/Mo element ratio in NMO is approximately 5:8, whereas that in PTA&PMA/NMO is approximately 5:9. In particular, the P and W element contents in PTA&PMA/NMO are approximately 280.7 μg/g and 67.3 μg/g, respectively. This finding confirms the insertion of POM multianion clusters. The partial decomposition of POMs retains residual P substances by combining the TEM/EDX analysis results, and some n(PO4)3− structural building unitsare preserved at the surface of the nanosheets [34].
The XRD analysis results show that for NMO@NF and PTA&PMA/NMO@NF electrodes (Figure 1b), the NF substrate had three strong characteristic peaks at 44.5°, 51.8°, and 76.3°. Figure 1b shows that the crystal phases of NMO·nH2O (JCPDS card number: 13-0128) and NMO (JCPDS card number: 12-0348) coexist in NMO@NF after wet chemical etching. These crystals are retained in PTA&PMA/NMO@NF [39]. The peak widths of PTA&PMA/NMO@NF at 14.2°, 16.8°, and 18.9° significantly increase because of the lack of cations and changes in the lattice structure after etching. The lattice constants and volume slightly increase. This phenomenon can be attributed to POMs exhibiting tunable redox properties, superacidic characteristics, and exceptional stability in both solution and solid states. These attributes enable them to replace traditional liquid acids (e.g., HF, HCl, H2SO4) and molecular catalysts for oxidation reactions [38,40]. Concurrently, H⁺ ions can etch and corrode the catalyst surface to generate abundant defect sites. Additionally, interlayer anion exchange between the POM clusters of heteropoly acids and the intrinsic anions (such as MoO42−) contributes to this phenomenon [38]. This result is consistent with the findings from the SEM, TEM, and XPS analysis.
The Raman spectra are displayed in Figure 1c. The Raman spectrum of NMO shows a strong peak at 943.5 cm−1 and some low-intensity peaks at 865.6, 826.3, and 355.2 cm−1, all of which are Raman characteristic peaks of NMO [41]. The Raman spectrum of NMO/PT-A&PMA shows a strong peak at 947 cm−1 and some low-intensity peaks at 869, 828.1, and 355.2 cm−1 [42,43,44]. The Raman spectrum of PTA&PMA/NMO shifts to high frequencies and the peak intensity of PTA&PMA/NMO weakens because the lack of cations causes neighboring anions to contract or expand to maintain charge balance, resulting in a blue shift in Raman peaks after POM etching. Additionally, interlayer anion exchange between the POM clusters of heteropoly acids and the intrinsic anions alters the interlayer interactions and spacing, increasing the frequency of intra- and interlayer vibrations. This observation is consistent with the XRD results.
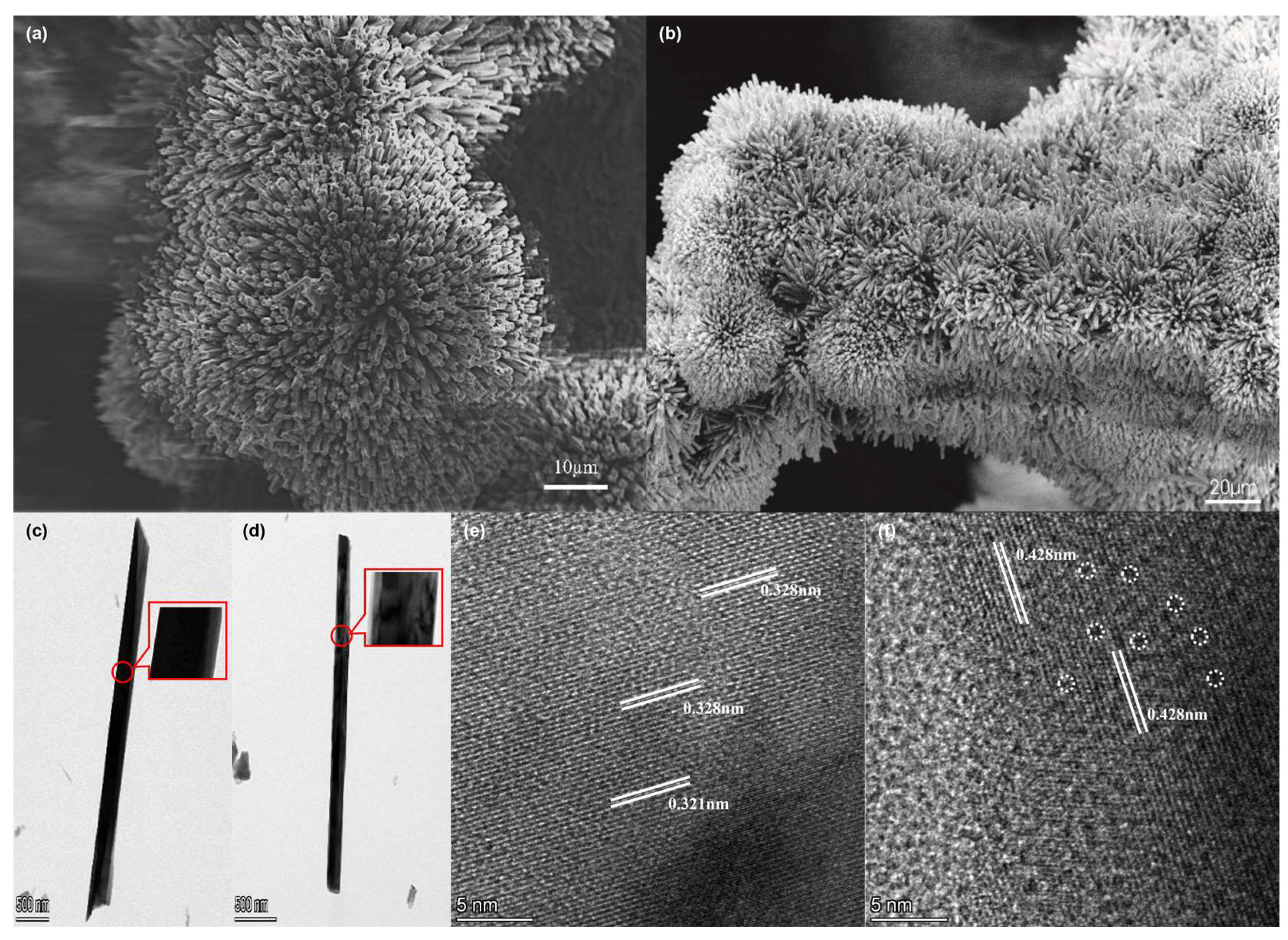
The morphology and the lattice fringes of samples were characterized by SEM, TEM, and HRTEM. Figure 2a shows that after the hydrothermal synthesis of NMO, NF is almost completely covered by flowerlike clusters composed of NMO nanorods. Although the electrode was platinum-sputtered, the SEM images of NMO@NF showed significant discharging. This finding indicates that NMO has poor conductivity. After chemical etching, the conductivity of PTA&PMA/NMO@NF is significantly improved. This phenomenon may be related to the structural changes caused by the etching [39]. As shown in Figure 2b, the flowerlike clusters are well retained in the PTA&PMA/NMO@NF. In Figure 2c, the surface of NMO is smooth with an average diameter of ~300 nm. After etching, nanorods exhibit a diameter that does not change significantly, but PTA&PMA/NMO (Figure 2d) possesses a surface area that loosens and shows defects because of metal exsolution during the etching process. The HRTEM image in Figure 2e shows that the marked lattice spacing of NMO is 0.321 and 0.328 nm. By contrast, the marked lattice spacing of PTA&PMA/NMO is 0.428 nm. Moreover, some lattice distortion can be found at the interface because of the structure termination due to metal precipitation during etching, as shown in Figure 2f. The element distribution in NMO and PTA&PMA/NMO is shown by EDX elemental mapping (Figure S1), in which all elements are uniformly distributed. The detailed EDX elemental analysis (Table S1) shows that the P/Mo ratio is 1:1.4, which is far higher than the nominal ratio of 1:12 in pure PTA and PMA. This finding indicates that the acid–base reaction between pure PTA and PMA and NMO leads to the partial decomposition of pure PTA and PMA. Moreover, a small number of n(PO4)3− structural building units are anchored [45].
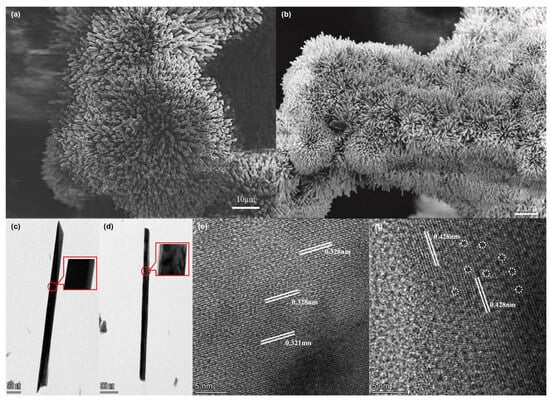
Figure 2.
Morphology of catalysts. The SEM images of (a) NMO@NF and (b) PTA&PMA/NMO@NF. The TEM images of (c) NMO and (d) PTA&PMA/NMO. The HRTEM images of (e) NMO and (f) PTA&PMA/NMO.
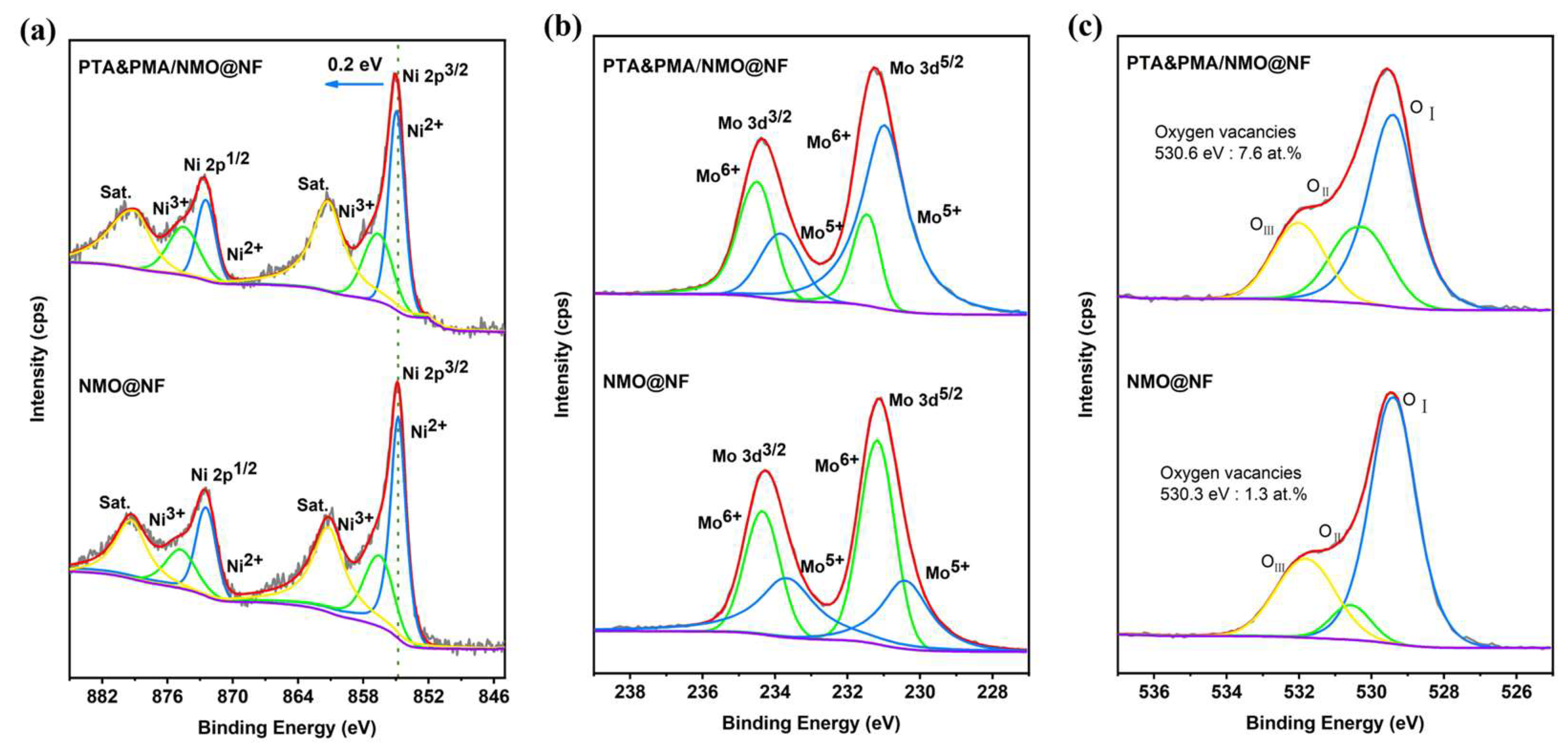
XPS is used to determine the elemental composition and chemical state of NMO@NF and PTA&PMA/NMO@NF electrocatalytic materials and to understand the changes in the oxidation state of atoms in the NMO catalyst caused by POM etching. The full spectrum shows that Ni, Mo, and O elements are present in NMO@NF and PTA&PMA/NMO@NF (Figure S2). As shown in Figure 3a, the high-resolution Ni 2p spectrum of NMO@NF shows that Ni 2P1/2 and Ni 2P3/2 doublets are identified at 872.5 and 874.8 eV and at 854.8 and 856.5 eV, respectively. They are also accompanied by two satellite peaks at 861.2 and 879.3 eV [37]. The high-resolution Ni 2p spectrum of PTA&PMA/NMO@NF shows that Ni 2P1/2 and Ni 2P3/2 doublets are identified at 872.5 and 874.8 eV and at 855 and 856.7eV, respectively. They are also accompanied by two satellite peaks at 861.3 and 879 eV. Unlike the peak position of Ni 2p in NMO@NF, that in PTA&PMA/NMO@NF shifts by 0.2 eV toward a high binding energy. This finding indicates that the valence state of Ni increases and numerous Ni3+ exist in PTA&PMA/NMO@NF [46]. The Ni3+/Ni2+ ratio of PTA&PMA/NMO@NF is 0.77, which is higher than that of NMO@NF in Table S2. This finding suggests that this phenomenon is beneficial for the formation of NiOOH during the OER process [47].
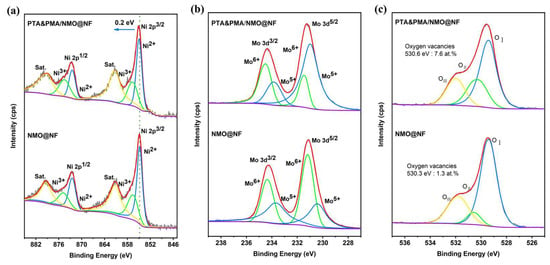
Figure 3.
High-resolution XPS spectra of (a) Ni 2p, (b) Mo 3d, and (c) O 1s for NMO@NF and PTA&PMA/NMO@NF.
The high-resolution Mo 3d of NMO@NF is shown in Figure 3b, and Mo 3d3/2 and Mo 3d5/2 doublets are identified at 233.7 and 234.4 eV and at 230.4 and 231.2 eV, respectively. The high-resolution Mo 3d spectrum of PTA&PMA/NMO@NF shows that Mo 3d3/2 and Mo 3d5/2 doublets are identified at 233.8 and 234.5 eV and 231 and 231.5 eV, respectively. Compared with the intensity of Mo5+ NMO@NF, that in PTA&PMA/NMO@NF, which corresponds to the cations (Ni and Mo) in the ICP-OES results, is significantly enhanced. This finding indicates that the oxygen vacancy defect content increases significantly during the etching process. In addition, the O 1s spectrum of NMO@NF (Figure 3c) shows three characteristic peaks at 529.4, 530.6, and 531.8 eV, and the O 1s spectrum of PTA&PMA/NMO@NF shows three characteristic peaks at 529.4, 530.3, and 532 eV [48,49,50]. The characteristic peaks are attributed to metal oxygen (OI), relative oxygen vacancies (OII), and surface adsorbed oxygen (OIII). For PTA&PMA/NMO@NF, OIII is slightly increased by 0.2 eV; the metal oxygen concentration measured by XPS is 23.7 at.%, which is much higher than that for NMO@NF (17.1 at.%); and the oxygen vacancy concentration measured by XPS is 7.6 at.%, which is much higher than that for NMO@NF (3.1 at.%). The peak density of oxygen vacancies increases significantly; with the introduction of oxygen vacancies, the electronic structure at catalytic sites is effectively modulated to facilitate the formation and transformation of intermediate states, thereby accelerating the electrochemical process [51].
2.2. Electrochemical OER Performance
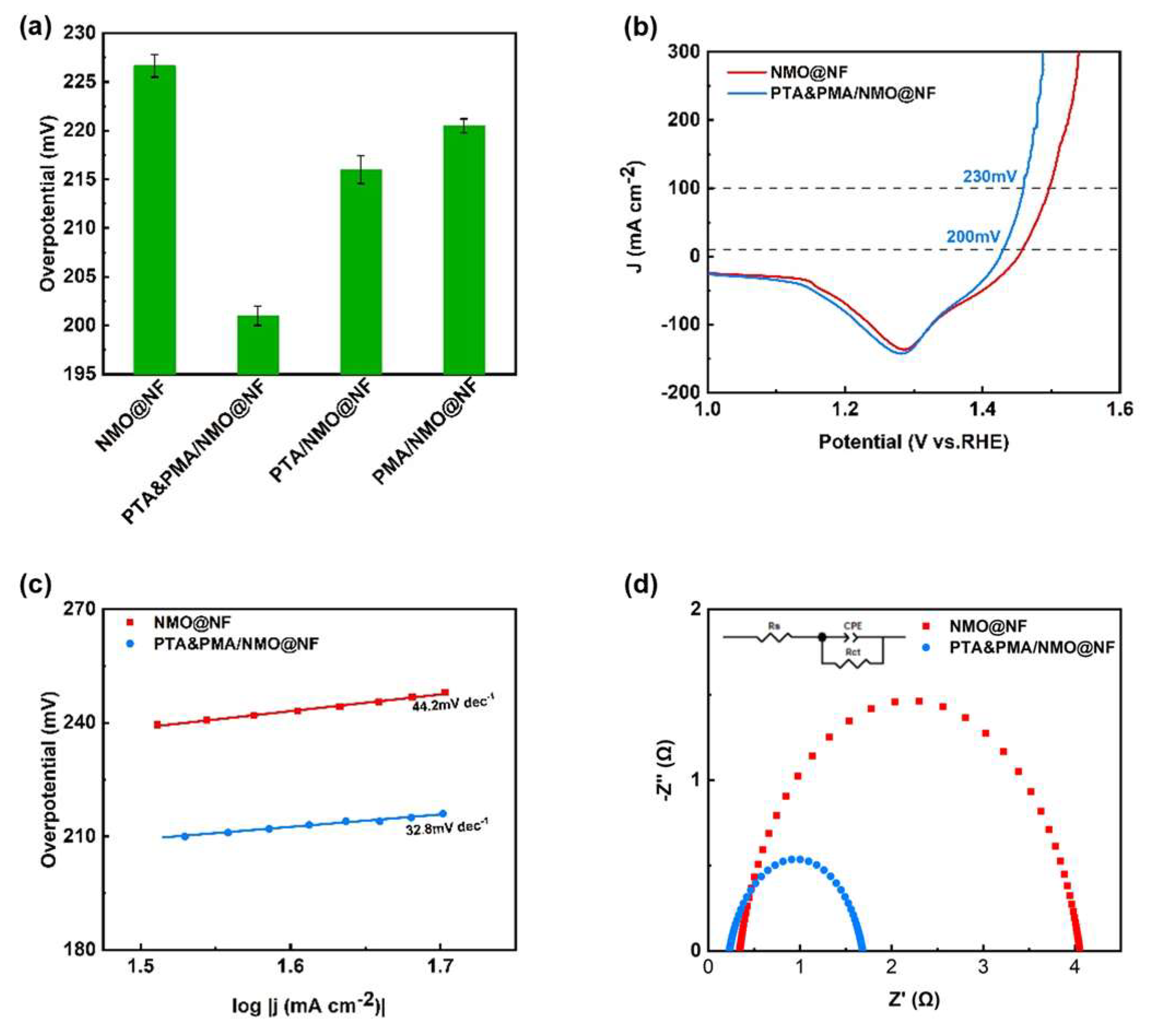
OER testing was conducted at room temperature in a 1 M KOH electrolyte solution using a three-electrode system, the electrocatalytic activity of four OER catalysts (NMO@NF, PTA&PMA/NMO@NF, PTA/NMO@NF, and PMA/NMO@NF) for OER was tested. Figure 4a presents a visual comparison of the overpotential at 10mA cm−2 among the various catalysts, demonstrating that PTA&PMA/NMO@NF outperforms the others by having the least overpotential. The LSV polarization curves were swept from high potential to low potential to avoid the influence of oxidation current. Figure 4b shows that in the potential range of 1.35–1.28 V, Ni3+ was reduced to Ni2+, and the overpotential of PTA&PMA/NMO@NF was significantly reduced. As a result, current densities of 10 and 100 mA cm−2 were achieved at only 200 and 230 mV, respectively. NMO@NF required overpotentials of 227 and 267 mV to achieve current densities of 10 and 100 mA cm−2, while RuO2@NF needed an overpotential of 314 mV to afford 100 mA cm−2 [43]. ICP-OES and XPS analysis results show that the generation of oxygen vacancies significantly improved the OER performance of the PTA&PMA/NMO@NF electrocatalyst after POM etching because two cationic (Ni and Mo) defects were produced, and the ion leaching behavior in the pristine material may have triggered favorable OH⁻ adsorption on the catalyst surface, thereby facilitating electron transfer during the OER process [52,53,54,55]. As shown in the Tafel slope in Figure 4c, the Tafel slope of PTA&PMA/NMO@NF (32.8 mV dec−1) was significantly smaller than that of NMO@NF (44.2 mV dec−1) and RuO2@NF (73 mV dec−1) [14]. This finding indicates that the reaction kinetics of PTA&PMA/NMO@NF were better and more favorable for the OER than those of NMO@NF. The kinetics of the OER were studied using EIS. As shown in Figure 4d, the Rct of PTA&PMA/NMO@NF was the lowest (1.5 Ω), which was lower than that of NMO@NF (3.7 Ω). This phenomenon can be attributed to the generation of abundant cationic defect sites and oxygen vacancies on the catalyst surface through chemical etching, which enhances favorable OH⁻ adsorption. Consequently, this promotes faster electron transfer and reduces charge transfer resistance [53,54,55]; the PTA&PMA/NMO@NF electrode has better conductivity than NMO@NF, leading to fast electron transfer rates and superior OER performance.
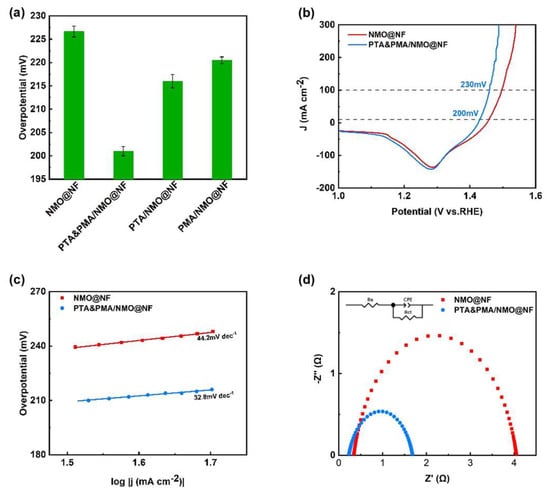
Figure 4.
Electrochemical performance tests of the catalysts. (a) The overpotential at 10mA cm−2 of NMO@NF, PTA&PMA/NMO@NF, PTA/NMO@NF, and PMA/NMO@NF; (b) The iR-compensated polarization curves and (c) the corresponding Tafel plots of NMO@NF and PTA&PMA/NMO@NF for OER at a scan rate of 5 mV s−1. (d) Nyquist plots of the electrodes at an overpotential of 10 mV.
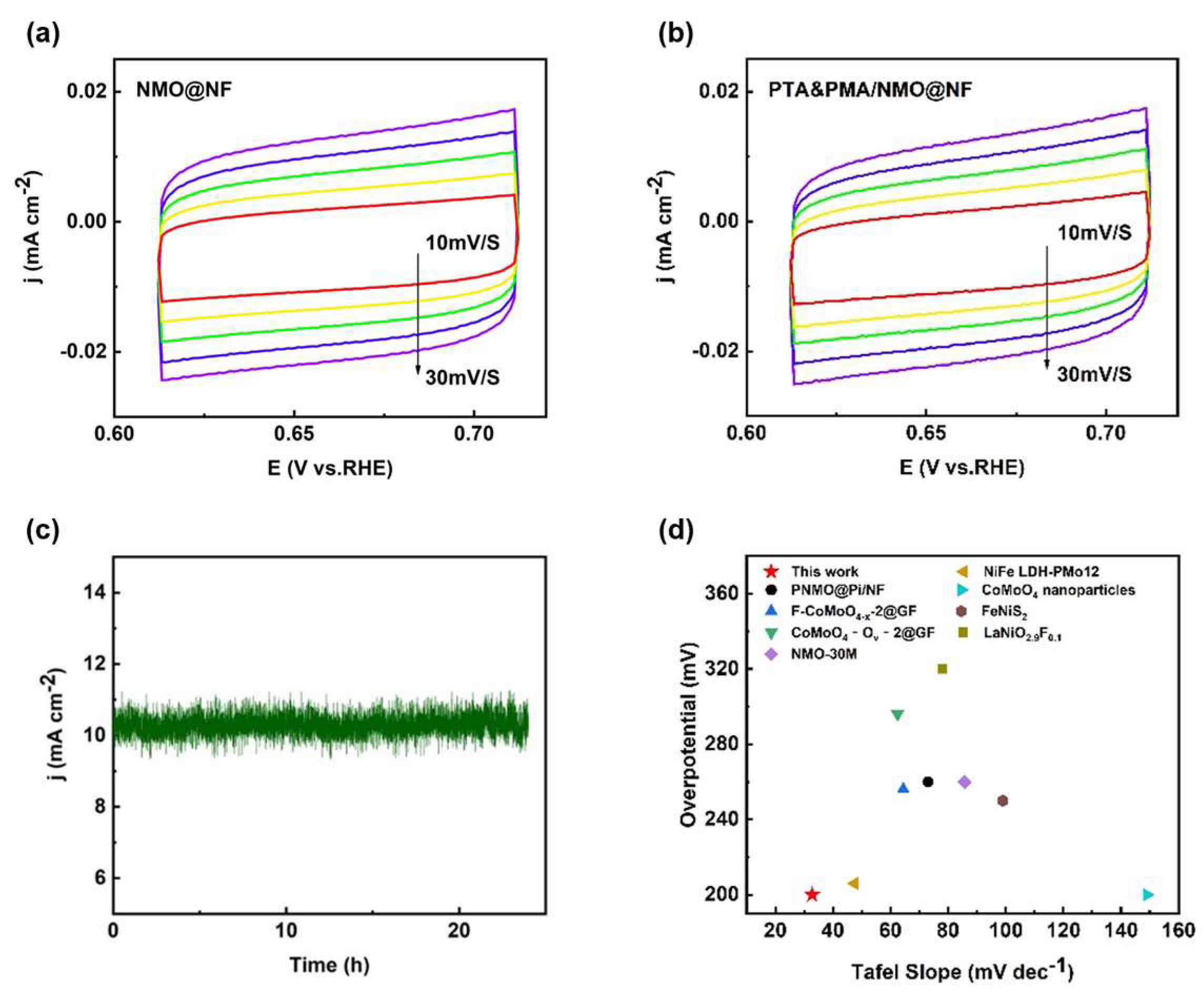
The Cdl of the catalyst was determined by integrating the CV curves and was found to be proportional to the electrochemically active surface area (ECSA). The capacitance values associated with the double layer (Cdl) were derived from the CV curves of NMO@NF (Figure 5a) and PTA&PMA/NMO@NF (Figure 5b), which were 1.07 and 1.10 mF cm−2, respectively (Figure S3). The Cdl of NMO@NF/PTA&PMA did not show noticeable changes, indicating that its catalytic active surface underwent no significant changes. Catalyst stability had practical significance for its application. The stability of NMO@NF/PTA&PMA was evaluated by the current time method. Figure 5c shows that when the reversible hydrogen potential of NMO@NF/PTA&PMA was 1.43 V and the current density was 10 mA cm−2, the curve remained stable after 24 h of the time current stability test, and the change in overpotential was negligible. This finding highlights that NMO@NF/PTA&PMA has outstanding electrochemical stability in the electrochemical process of OER.
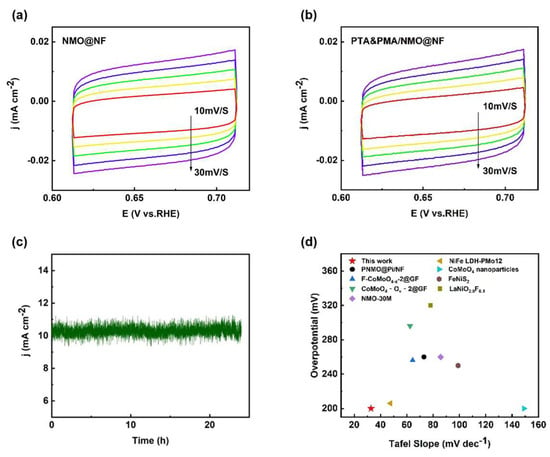
Figure 5.
Electrochemical performance tests of the catalysts. (a) Comparison of the Rct values for NMO@NF and PTA&PMA/NMO@NF; (b) CV curves of NMO@NF/PTA&PMA at different scan rates from 10 mV s−1 to 30 mV s−1; (c) I-t curve of PTA&PMA/NMO@NF at a potential of 1.43 V versus RHE; and (d) a comparison chart of different material properties.
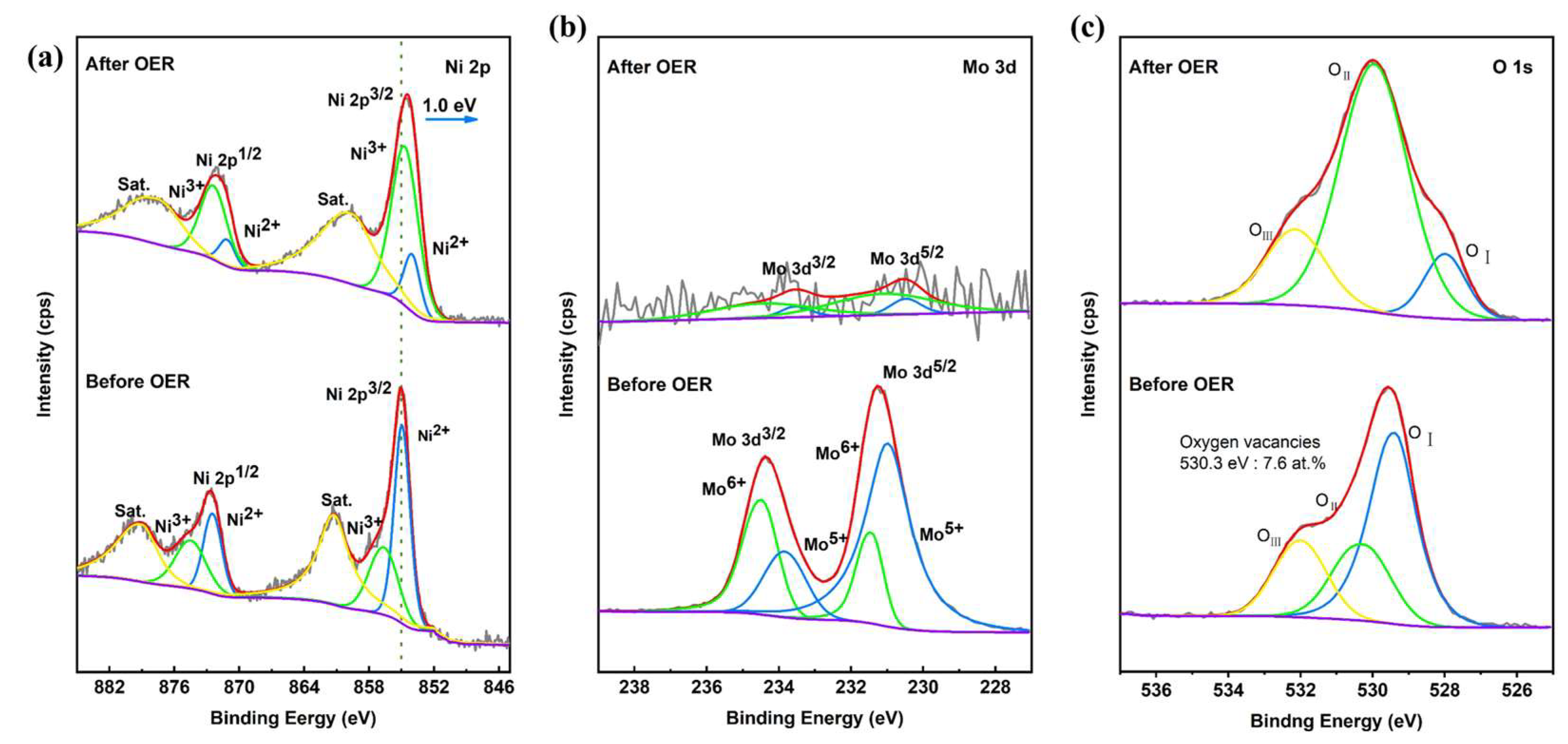
As shown in Figure S4, the SEM image of PTA&PMA/NMO@NF after 24 h of the stability test (PTA&PMA/NMO@NF Sat.) shows that the nanorods covered on the NF did not disappear and were still well preserved. However, the diameter of the nanorods was reduced to approximately 200 nm. The reduction in the nanorod diameter of the PTA&PMA/NMO@NF catalyst after 24 h of stability testing was attributed to the decrease in Mo atoms, as revealed by the ICP-OES and EDS results. Notably, Mo atoms in the catalyst were almost entirely depleted after 24 h of stability testing. Therefore, no significant further changes in nanorod diameter would occur in catalysts subjected to stability tests exceeding 24 h. The transmission electron microscopy results (Figure S5) also confirmed this result. The HRTEM image in Figure S6 shows that the lattice fringes of PTA&PMA/NMO@NF Sat. were not obvious, possibly because of the significant reduction in the content of the Mo element. This observation is in good agreement with the results of ICP-OES (Table S1) and TEM-EDX (Table S3). The element distribution in PTA&PMA/NMO@NF Sat. was shown by the EDX element mapping (Figure S7), in which all elements were evenly distributed. Compared with the XRD spectra of PTA&PMA/NMO@NF Sat. and PTA&PMA/NMO@NF (Figure S8), the peak positions were unchanged. The peak widths of the diffraction peaks of PTA&PMA/NMO@NF Sat. at 14.2°, 16.8°, and 18.9° increased significantly because of the lack of cations and the change in lattice structure after etching. The lattice constant and volume increased slightly. This phenomenon can be attributed to the substantial reduction in the Mo element [47]. These results further demonstrate that the PTA&PMA/NMO@NF electrode has excellent long-term stability. In the Raman spectra of PTA&PMA/NMO@NF Sat. (Figure S9), the peaks belonging to Mo-O and Mo-O-Ni disappearred because of the reduction in Mo atoms. As shown in Figure 6a, the XPS results of PTA&PMA/NMO@NF before and after OER show that after OER, the peak position of Ni 2p moved 1.0 eV to the direction with low binding energy. This observation can be attributed to the decrease in the concentration of Mo atoms [47]. The Ni3+/Ni2+ ratio was 4.5, indicating that the valence state of Ni increased. This finding suggests that this phenomenon is beneficial to the formation of NiOOH during the OER process. The high-resolution Mo 3d of PTA&PMA/NMO@NF Sat. is shown in Figure 6b. Given the surface reconstruction, the intensity of Mo after OER was weaker than that before OER. The O 1s spectrum of PTA&PMA/NMO@NF Sat. (Figure 6c) showed three characteristic peaks at 528, 530, and 532.1 eV, which were attributed to OI, OII, and OIII, respectively. The oxygen vacancy concentration was 18.4 at.%, and the peak density of oxygen vacancy density increased significantly. The metal oxygen concentration was 2.95 at.%, and the peak density of metal oxygen density decreased significantly. The peak position of metallic oxygen moved 1.4 eV toward the low binding energy direction. This phenomenon can be attributed to the reduction in Mo atoms [47].
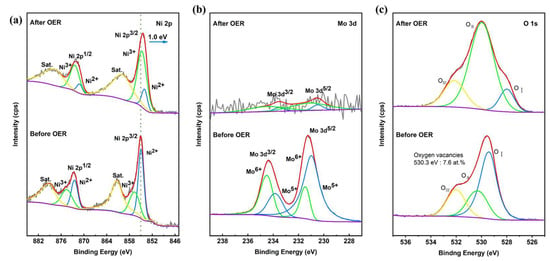
Figure 6.
XPS of NMO@NF/PTA&PMA before and after OER. (a) Ni 2p, (b) Mo 3d, and (c) O 1s.
Additionally, the OER performance of our catalyst was comprehensively compared with the OER performance in the existing literature [1,38,39,42,56,57,58,59] and that of commercial catalysts. The results showed that the OER efficiency of PTA&PMA/NMO@NF surpassed many previously described transition metal electrocatalysts (Figure 5d and Table S4), thereby making it a strong competitor with noble metal-based OER catalysts. The slight change in the overpotential of PTA&PMA/NMO@NF indicated good electrocatalytic performance. By contrast, the reduced Tafel slope indicated that the rate-limiting step was close to the end of the multielectron transfer process. This observation emphasizes the adaptability of the electrocatalyst.
3. Materials and Methods
3.1. Material
Nickel foam (NF; size of 20 × 30 cm, thickness of 1 mm) was purchased from Kunshan XZH Electronic Materials Co., Ltd. (Suzhou, China) Ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) and hydrochloric acid were purchased from China Shanghai Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) Potassium hydroxide (KOH; purity: ≥85%) and nickel nitrate (Ni(NO3)2·6H2O) were purchased from China Tianjin Damao Chemical Reagent Factory. Anhydrous ethanol (C2H5OH) was purchased from China Chengdu Cologne Chemicals Co., Ltd. (Chengdu, China) PTA hydrate and PMA hydrate were purchased from China Shanghai Macklin Biochemical Co., Ltd. All the above chemicals were of an analytical grade and were used directly without any further treatment.
3.2. Synthesis of NMO@NF
Preparation of NMO@NF using a hydrothermal method: Before use, the commercial bulk NF was cut into small pieces (1.0 cm × 1.5 cm) and ultrasonically treated for 10 min in a mixed solution of hydrochloric acid (12.0 mol/L), ethanol, and water (hydrochloric acid/ethanol/water = 1:1:1). After treatment, the pieces were washed three times with ethanol and pure water, and the surface moisture was absorbed with filter paper. A piece of pretreated NF was placed in a 25.0 mL polytetrafluoroethylene reaction kettle containing 7.5 mL water. Subsequently, ammonium molybdate tetrahydrate (0.1125 mmol, 139.0 mg) and nickel nitrate hexahydrate (0.2625 mmol, 76.0 mg) were sequentially dissolved, and were ultrasonically dissolved for 30 min. The reaction kettle was heated in an oven at 150 °C for 6 h. After being cooled naturally to room temperature, the resulting NMO@NF was removed, washed three times with ethanol and deionized water, and finally dried at 60 °C for 10 h [39].
3.3. Synthesis of PMA/NMO@NF, PTA/NMO@NF, and PTA&PMA/NMO@NF
Preparation of PMA/NMO@NF, PTA/NMO@NF, and PTA&PMA/NMO@NF: The PMA/NMO@NF, PTA/NMO@NF, and PTA&PMA/NMO@NF sample was synthesized through a simple wet chemical etching process. Before soaking, PMA (0.04 mmol, 73.0 mg) was dissolved in an 8.0 mL mixture solution of ethanol and deionized water (ethanol/water = 2:1) to prepare solution A; PTA (0.04 mmol, 115.2 mg) was dissolved in an 8.0 mL mixture solution of ethanol and deionized water (ethanol/water = 2:1) to prepare solution B; and PMA (0.008 mmol, 14.6 mg) and PTA (0.032 mmol, 92.2 mg) were dissolved in an 8.0 mL mixture solution of ethanol and deionized water (ethanol/water = 2:1) to prepare solution C. Then, the NMO@NF precursor was separately soaked in the above mixed solution for 15 min; the obtained PMA/NMO@NF, PTA/NMO@NF, and PTA&PMA/NMO@NF were washed three times with ethanol and deionized water. Finally, they were dried in an oven at 60 °C for 1 h.
3.4. Material Characterization
The surface microstructure of the synthesized catalyst was studied using a high-resolution transmission electron microscope (HR-TEM; FEI Talos F200S, Thermo Fisher Scientific, New York, NY, USA) with a field emission gun. Additionally, the surface morphology and microstructure of the catalyst were investigated using a scanning electron microscope (SEM; SUPRA 55 Sapphire, Tokyo JEOL Ltd., Tokyo, Japan), an X-ray diffractometer (XRD; Mini Flex 600, Tokyo Rigaku Corporation, Tokyo, Japan), and a Raman spectrometer. The cation content of the samples was detected using an inductively coupled plasma optical emission spectrometer (ICP-OES; Agilent 5110, Agilent Technologies Inc., Santa Clara, CA, USA). The elemental composition and valence states of the catalyst were studied using X-ray photoelectron spectroscopy (XPS; Thermo Scientific K-Alpha, Thermo Fisher Scientific, New York, NY, USA), with the binding energy of the samples being calibrated using the C-C peak (284.8 eV) of the C 1s orbital. Elemental spectra and mapping were obtained using an energy-dispersive spectrometer (Bruker SuperX, Thermo Fisher Scientific, New York, NY, USA). Furthermore, the FEI Talos F200S transmission electron microscope was used to explore the microscope images, elemental mapping, and linear scanning analysis.
3.5. Electrochemical Characterization
All electrochemical data were obtained using a CHI 660E electrochemical workstation (CH Instruments Inc., Shanghai, China) equipped with a three-electrode system. The prepared NMO-based catalyst was loaded onto NF as the working electrode, with the Hg/HgO electrode and graphite electrode serving as the reference and counter electrodes, respectively. In the electrochemical tests involved in this study, the geometric area of the working electrode immersed in the electrolyte was 1.0 cm × 1.0 cm. According to Equation (1), all potentials tested under the Hg/HgO electrode were calibrated to the potential under the reversible hydrogen electrode (RHE):
E (vs. RHE) = E (vs. Hg/HgO) + 0.098 V + 0.059 pH
In a 1 M of KOH solution, an OER test was conducted using linear sweep voltammetry (LSV) at a scan rate of 5 mV/s within the voltage range of 0 to 1.0 V. The Tafel slope of the samples was linearly fitted according to the Tafel equation (η = b log j + a, where η is the overpotential, b is the Tafel slope, and j is the current density). Electrochemical impedance spectroscopy (EIS) was used to determine the electrolyte resistance (Rs) and charge transfer resistance (Rct) for different catalysts in the frequency range of 10 kHz to 0.01 Hz. The double-layer capacitance (Cdl) was obtained from cyclic voltammograms (CVs) at scan rates of 10, 15, 20, 25, and 30 mV/s within the non-Faradaic current region. The 24 h stability was measured using chronoamperometry. All reported current densities were adjusted to account for ohmic potential drop, and reverse scans were conducted to avoid the effects of redox currents.
4. Conclusions
In summary, PTA&PMA/NMO@NF was successfully prepared via an easy and accessible hydrothermal process, followed by chemical etching with PTA and PMA. The obtained catalyst exhibited superior electrocatalytic effects compared to NMO@NF in OER. For the OER in 1.0 M of KOH, PTA&PMA/NMO@NF achieved a low overpotential of 200 mV at a current density of 10 mA·cm−2. After a 24 h current time test, the electrode maintained good stability, and the overpotential remained unchanged. The excellent performance of PTA&PMA/NMO@NF is attributed to the etching of PTA and PMA. For instance, PTA and PMA can result in the defect of the two cations (Ni and Mo). Moreover, the narrowing of the lattice structure improves the charge transfer efficiency, thereby reducing the charge transfer electrons. These changes are beneficial to electron transfer in the OER process. Moreover, the increase in the valence state of Ni ions is beneficial to the generation of NiOOH in the OER process. In addition, (PO4)3− is attached to the NMO surface, thereby regulating the electronic configuration of the two cations (Ni and Mo). This work presents low-cost, simple, and convenient synthetic strategies to improve the catalytic activity of NMO. It also offers potential avenues for designing new materials.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms26073107/s1.
Author Contributions
Conceptualization, T.C. and X.H.; methodology, X.L.; validation, T.C., X.H. and C.L.; investigation, Y.N. and J.W.; resources, M.L., P.L. and X.L.; data curation, T.C.; writing—original draft preparation, T.C.; writing—review and editing, T.C.; visualization, X.L.; supervision, Z.W.; project administration, X.L.; funding acquisition, M.L., P.L. and X.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the project of Guangxi Natural Science Foundation (2021GXNSFAA075043, 2023GXNSFAA026409, and 2022GXNSFBA035499), and the project of Talent Introduction Program of Guangxi Minzu University (2018KJQD06).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xie, W.; Huang, J.; Huang, L.; Geng, S.; Song, S.; Tsiakaras, P.; Wang, Y. Novel fluorine-doped cobalt molybdate nanosheets with enriched oxygen-vacancies for improved oxygen evolution reaction activity. Appl. Catal. B Environ. 2022, 303, 120871. [Google Scholar]
- Tian, L.; Wang, Q.; Li, Y.; Ren, X.; Wei, Q.; Wu, D. A hierarchical CoMoO4@CoFe-LDH heterostructure as a highly effective catalyst to boost electrocatalytic water oxidation. Dalton Trans. 2022, 51, 10552–10557. [Google Scholar] [PubMed]
- Stoerzinger, K.A.; Diaz-Morales, O.; Kolb, M.; Rao, R.R.; Frydendal, R.; Qiao, L.; Wang, X.R.; Halck, N.B.; Rossmeisl, J.; Hansen, H.A.; et al. Orientation-dependent oxygen evolution on RuO2 without lattice exchange. ACS Energy Lett. 2017, 2, 876–881. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, J.; Zhao, H.; Jiang, R.; Tian, F.; Zhang, R. Tremella-like Ni3S2/MnS with ultrathin nanosheets and abundant oxygen vacancies directly used for high speed overall water splitting. Appl. Catal. B Environ. 2019, 257, 117899. [Google Scholar]
- Liang, Y.; Wang, H.; Zhou, J.; Li, Y.; Wang, J.; Regier, T.; Dai, H. Covalent hybrid of spinel manganese–cobalt oxide and graphene as advanced oxygen reduction electrocatalysts. J. Am. Chem. Soc. 2012, 134, 3517–3523. [Google Scholar]
- Ma, T.Y.; Dai, S.; Jaroniec, M.; Qiao, S.Z. Metal–organic framework derived hybrid Co3O4-carbon porous nanowire arrays as reversible oxygen evolution electrodes. J. Am. Chem. Soc. 2014, 136, 13925–13931. [Google Scholar]
- An, L.; Feng, J.; Zhang, Y.; Wang, R.; Liu, H.; Wang, G.C.; Cheng, F.; Xi, P. Epitaxial heterogeneous interfaces on N-NiMoO4/NiS2 nanowires/nanosheets to boost hydrogen and oxygen production for overall water splitting. Adv. Funct. Mater. 2019, 29, 1805298. [Google Scholar]
- Liu, Z.; Yuan, C.; Teng, F. Crystal facets-predominated oxygen evolution reaction activity of earth abundant CoMoO4 electrocatalyst. J. Alloys Compd. 2019, 781, 460–466. [Google Scholar]
- Xu, J.; Xiao, T.; Tan, X.; Xiang, P.; Jiang, L.; Wu, D.; Li, J.; Wang, S. A new asymmetric aqueous supercapacitor: Co3O4//Co3O4@polypyrrole. J. Alloys Compd. 2017, 706, 351–357. [Google Scholar]
- Wang, Z.; Zheng, K.; Liu, S.; Dai, Z.; Xu, Y.; Li, X.; Wang, H.; Wang, L. Electrocatalytic Nitrogen Reduction to Ammonia by Fe2O3 Nanorod Array on Carbon Cloth. ACS Sustain. Chem. Eng. 2019, 7, 11754–11759. [Google Scholar]
- Salvador, G.M.; Silva, A.L.; Silva, L.P.; Passos, F.B.; Carvalho, N.M. Enhanced activity of Pd/α-MnO2 for electrocatalytic oxygen evolution reaction. Int. J. Hydrogen Energy 2021, 46, 26976–26988. [Google Scholar] [CrossRef]
- Zhang, X.; Su, H.; Du, X. A Nickel molybdenum oxide nanoarray as an efficient and stable electrocatalyst for overall water splitting. New J. Chem. 2020, 44, 8176–8182. [Google Scholar]
- Ratha, S.; Samantara, A.K.; Singha, K.K.; Gangan, A.S.; Chakraborty, B.; Jena, B.K.; Rout, C.S. Urea-assisted room temperature stabilized metastable β-NiMoO4: Experimental and theoretical insights into its unique bifunctional activity toward oxygen evolution and supercapacitor. ACS Appl. Mater. Interfaces 2017, 9, 9640–9653. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xiang, J.; Zhang, W.; Chen, C.; Xu, H.; Huang, Y. 3D interconnected porous NiMoO4 nanoplate arrays on Ni foam as high-performance binder-free electrode for supercapacitors. J. Mater. Chem. A 2015, 3, 22081–22087. [Google Scholar]
- Wang, Z.; Wei, G.; Du, K.; Zhao, X.; Liu, M.; Wang, S.; Zhou, Y.; An, C.; Zhang, J. Ni foam-supported carbon-sheathed NiMoO4 nanowires as integrated electrode for high-performance hybrid supercapacitors. ACS Sustain. Chem. Eng. 2017, 5, 5964–5971. [Google Scholar] [CrossRef]
- Xu, Y.; Xuan, H.; Gao, J.; Liang, T.; Han, X.; Yang, J.; Zhang, Y.; Li, H.; Han, P.; Du, Y. Hierarchical three-dimensional NiMoO4-anchored rGO/Ni foam as advanced electrode material with improved supercapacitor performance. J. Mater. Sci. 2018, 53, 8483–8498. [Google Scholar]
- Murugan, E.; Govindaraju, S.; Santhoshkumar, S. Hydrothermal synthesis, characterization and electrochemical behavior of NiMoO4 nanoflower and NiMoO4/rGO nanocomposite for high-performance supercapacitors. Electrochim. Acta 2021, 392, 138973. [Google Scholar] [CrossRef]
- Yao, P.; Li, C.; Yu, J.; Zhang, S.; Zhang, M.; Liu, H.; Ji, M.; Cong, G.; Zhang, T.; Zhu, C.; et al. High performance flexible energy storage device based on copper foam supported NiMoO4 nanosheets-CNTs-CuO nanowires composites with core–shell holey nanostructure. J. Mater. Sci. Technol. 2021, 85, 87–94. [Google Scholar]
- Zhu, D.; Sun, X.; Yu, J.; Liu, Q.; Liu, J.; Chen, R.; Zhang, H.; Song, D.; Li, R.; Wang, J. Three-dimensional heterostructured polypyrrole/nickel molybdate anchored on carbon cloth for high-performance flexible supercapacitors. J. Colloid Interface Sci. 2020, 574, 355–363. [Google Scholar]
- Hao, Y.; Huang, H.; Wang, Q.; Wang, Q.; Zhou, G. Nitrogen-doped carbon/NiMoO4 nanospheres assembled by nanosheets and ultrasmall nanoparticles for supercapacitors. Chem. Phys. Lett. 2019, 728, 215–223. [Google Scholar]
- Tong, B.; Wei, W.; Chen, X.; Wang, J.; Ye, W.; Cui, S.; Chen, W.; Mi, L. Designed synthesis of porous NiMoO4/C composite nanorods for asymmetric supercapacitors. CrystEngComm 2019, 21, 5492–5499. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Ma, M.; Mu, X.; Zhang, Y.; Du, J.; Hu, Q.; Huang, B.; Hua, X.; Liu, G.; et al. Manganese-doped nickel molybdate nanostructures for high-performance asymmetric supercapacitors. Chem. Eng. J. 2019, 372, 452–461. [Google Scholar] [CrossRef]
- Yuan, J.; Yao, D.; Jiang, L.; Tao, Y.; Che, J.; He, G.; Chen, H. Mn-doped NiMoO4 mesoporous nanorods/reduced graphene oxide composite for high-performance all-solid-state supercapacitor. ACS Appl. Energy Mater. 2020, 3, 1794–1803. [Google Scholar] [CrossRef]
- Wang, F.; Ma, K.; Tian, W.; Dong, J.; Han, H.; Wang, H.; Deng, K.; Yue, H.; Zhang, Y.X.; Jiang, W.; et al. P-Doped NiMoO4 parallel arrays anchored on cobalt carbonate hydroxide with oxygen vacancies and mass transfer channels for supercapacitors and oxygen evolution. J. Mater. Chem. A 2019, 7, 19589–19596. [Google Scholar] [CrossRef]
- Sharma, P.; Minakshi Sundaram, M.; Watcharatharapong, T.; Laird, D.; Euchner, H.; Ahuja, R. Zn metal atom doping on the surface plane of one-dimesional NiMoO4 nanorods with improved redox chemistry. ACS Appl. Mater. Interfaces 2020, 12, 44815–44829. [Google Scholar] [CrossRef]
- Cui, S.; Wang, F.; Sun, K.; Wang, X.; Hu, Q.; Peng, H.; Ma, G.; Lei, Z. High-performance hybrid supercapacitors based on Ce-doped NiMoO4 nanosheets and Fe3O4@Bi2O3 nanoarrays. J. Phys. Chem. C 2021, 125, 18129–18140. [Google Scholar] [CrossRef]
- Li, P.; Ruan, C.; Xu, J.; Xie, Y. Supercapacitive performance of CoMoO4 with oxygen vacancy porous nanosheet. Electrochim. Acta 2020, 330, 135334. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, L.; Guo, X. Ultrathin mesoporous NiMoO4-modified MoO3 core/shell nanostructures: Enhanced capacitive storage and cycling performance for supercapacitors. Chem. Eng. J. 2018, 353, 615–625. [Google Scholar] [CrossRef]
- Shen, J.; Wang, Q.; Zhang, K.; Wang, S.; Li, L.; Dong, S.; Zhao, S.; Chen, J.; Sun, R.; Wang, Y.; et al. Flexible carbon cloth based solid-state supercapacitor from hierarchical holothurian-morphological NiCo2O4@NiMoO4/PANI. Electrochim. Acta 2019, 320, 134578. [Google Scholar] [CrossRef]
- Xu, R.; Lin, J.; Wu, J.; Huang, M.; Fan, L.; Xu, Z.; Song, Z. A high-performance pseudocapacitive electrode material for supercapacitors based on the unique NiMoO4/NiO nanoflowers. Appl. Surf. Sci. 2019, 463, 721–731. [Google Scholar]
- Yu, D.; Zhang, Z.; Teng, Y.; Meng, Y.; Wu, Y.; Liu, X.; Hua, Y.; Zhao, X.; Liu, X. Fabrication of CuO@NiMoO4 core-shell nanowire arrays on copper foam and their application in high-performance all-solid-state asymmetric supercapacitors. J. Power Sources 2019, 440, 227164. [Google Scholar]
- Liu, Y.; Ma, Z.; Xin, N.; Ying, Y.; Shi, W. High-performance supercapacitor based on highly active P-doped one-dimension/two-dimension hierarchical NiCo2O4/NiMoO4 for efficient energy storage. J. Colloid Interface Sci. 2021, 601, 793–802. [Google Scholar]
- Zeng, Y.; Liao, J.; Wei, B.; Huang, Z.; Zhu, W.; Zheng, J.; Liang, H.; Zhang, Y.; Wang, Z. Tuning the electronic structure of NiMoO4 by coupling with SnO2 for high-performance hybrid supercapacitors. Chem. Eng. J. 2021, 409, 128297. [Google Scholar] [CrossRef]
- Chen, C.; Yan, D.; Luo, X.; Gao, W.; Huang, G.; Han, Z.; Zeng, Y.; Zhu, Z. Construction of core–shell NiMoO4@Ni-Co-S nanorods as advanced electrodes for high-performance asymmetric supercapacitors. ACS Appl. Mater. Interfaces 2018, 10, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ji, S.; Liu, Q.; Wang, H.; Liu, H.; Brett, D.J.; Wang, G.; Wang, R. Rational design of hierarchically core–shell structured Ni3S2@NiMoO4 nanowires for electrochemical energy storage. Small 2018, 14, 1800791. [Google Scholar] [CrossRef] [PubMed]
- Acharya, J.; Ojha, G.P.; Kim, B.-S.; Pant, B.; Park, M. Modish designation of hollow-tubular rGO–NiMoO4@Ni–Co–S hybrid core–shell electrodes with multichannel superconductive pathways for high-performance asymmetric supercapacitors. ACS Appl. Mater. Interfaces 2021, 13, 17487–17500. [Google Scholar]
- Kong, X.; Peng, H.; Bu, S.; Gao, Q.; Jiao, T.; Cheng, J.; Liu, B.; Hong, G.; Lee, C.; Zhang, W. Defect engineering of nanostructured electrocatalysts for enhancing nitrogen reduction. J. Mater. Chem. A 2020, 8, 7457–7473. [Google Scholar]
- Cai, Z.; Wang, P.; Zhang, J.; Chen, A.; Zhang, J.; Yan, Y.; Wang, X. Reinforced layered double hydroxide oxygen-evolution electrocatalysts: A polyoxometallic acid wet-etching approach and synergistic mechanism. Adv. Mater. 2022, 34, 2110696. [Google Scholar]
- Jiang, R.; Zhao, D.; Fan, H.; Xie, Y.; Li, M.; Lin, H.; Wu, Z.-S. Phosphorus doping and phosphates coating for nickel molybdate/nickel molybdate hydrate enabling efficient overall water splitting. J. Colloid Interface Sci. 2022, 606, 384–392. [Google Scholar]
- Laura, E.B.; Graciela, T.B.; Horacio, J.T. The state of the art on Wells-Dawson heteropoly-compounds A review of their properties and applications. Appl. Catal. A 2003, 256, 37–50. [Google Scholar]
- Ghosh, D.; Giri, S.; Das, C.K. Synthesis, characterization and electrochemical performance of graphene decorated with 1D NiMoO4·nH2O nanorods. Nanoscale 2013, 5, 10428–10437. [Google Scholar] [PubMed]
- Barraclough, C.; Lewis, J.; Nyholm, R. 713. The stretching frequencies of metal–oxygen double bonds. J. Chem. Soc. (Resumed) 1959, 11, 3552–3555. [Google Scholar] [CrossRef]
- Abdel-Dayem, H.M. Dynamic phenomena during reduction of α-NiMoO4 in different atmospheres: In-situ thermo-Raman spectroscopy study. Ind. Eng. Chem. Res. 2007, 46, 2466–2472. [Google Scholar] [CrossRef]
- Zhang, J.; Qian, J.; Ran, J.; Xi, P.; Yang, L.; Gao, D. Engineering lower coordination atoms onto NiO/Co3O4 heterointerfaces for boosting oxygen evolution reactions. ACS Catal. 2020, 10, 12376–12384. [Google Scholar] [CrossRef]
- Guan, X.; Yang, L.; Zhu, G.; Wen, H.; Zhang, J.; Sun, X.; Feng, H.; Tian, W.; Chen, X.; Yao, Y. A hierarchical CoMoO4 nanoparticle decorated nanoplate array as an electrocatalyst toward improved alkaline oxygen evolution reaction. Sustain. Energy Fuels 2020, 4, 1595–1599. [Google Scholar] [CrossRef]
- Shin, H.; Xiao, H.; Goddard, W.A., III. In silico discovery of new dopants for Fe-doped Ni oxyhydroxide (Ni1–xFexOOH) catalysts for oxygen evolution reaction. J. Am. Chem. Soc. 2018, 140, 6745–6748. [Google Scholar]
- Zhu, J.; Qian, J.; Peng, X.; Xia, B.; Gao, D. Etching-induced surface reconstruction of NiMoO4 for oxygen evolution reaction. Nano-Micro Lett. 2023, 15, 30. [Google Scholar]
- Wachs, I.E. Raman and IR studies of surface metal oxide species on oxide supports: Supported metal oxide catalysts. Catal. Today 1996, 27, 437–455. [Google Scholar]
- Zhuang, L.; Ge, L.; Yang, Y.; Li, M.; Jia, Y.; Yao, X.; Zhu, Z. Ultrathin iron-cobalt oxide nanosheets with abundant oxygen vacancies for the oxygen evolution reaction. Adv. Mater. 2017, 29, 1606793. [Google Scholar] [CrossRef]
- Bao, J.; Zhang, X.; Fan, B.; Zhang, J.; Zhou, M.; Yang, W.; Hu, X.; Wang, H.; Pan, B.; Xie, Y. Ultrathin spinel-structured nanosheets rich in oxygen deficiencies for enhanced electrocatalytic water oxidation. Angew. Chem. 2015, 127, 7507–7512. [Google Scholar]
- Tong, Y.; Chen, P.; Zhang, M.; Zhou, T.; Zhang, L.; Chu, W.; Wu, C.; Xie, Y. Oxygen vacancies confined in nickel molybdenum oxide porous nanosheets for promoted electrocatalytic urea oxidation. Acs Catal. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Yan, D.; Li, Y.; Huo, J.; Chen, R.; Dai, L.; Wang, S. Defect chemistry of nonprecious-metal electrocatalysts for oxygen reactions. Adv. Mater. 2017, 29, 1606459. [Google Scholar]
- Emiliana, F.; Maarten, N.; Tobias, B.; Xi, C.; Bae-Jung, K.; Julien, D.; Francesco, B.; Thomas, G.; Robin, S.; Luke, W.; et al. Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting. Nat. Mater. 2017, 16, 925–931. [Google Scholar]
- Marcel, R.; Alexis, G.; Kevin, J.M.; Kelsey, A.S.; Tina, J.C.; Azzam, N.M.; Yang, S. Structural changes of cobalt-based perovskites upon water oxidation investigated by EXAFS. J. Phys. Chem. C 2013, 117, 8628–8635. [Google Scholar]
- Xi, C.; Bae-Jung, K.; Emiliana, F.; Thomas, J.S. Co/Fe oxyhydroxides supported on perovskite oxides as oxygen evolution reaction catalyst systems. ACS Appl. Mater. Interfaces 2019, 11, 34787–34795. [Google Scholar]
- Jiang, T.; Xie, W.; Geng, S.; Li, R.; Song, S.; Wang, Y. Constructing oxygen vacancy-regulated cobalt molybdate nanoflakes for efficient oxygen evolution reaction catalysis. Chin. J. Catal. 2022, 43, 2434–2442. [Google Scholar]
- Ray, S.K.; Bastakoti, B.P. Improved supercapacitor and oxygen evolution reaction performances of morphology-controlled cobalt molybdate. Int. J. Hydrogen Energy 2024, 51, 1109–1118. [Google Scholar] [CrossRef]
- Dalai, N.; Jena, B. Iron nickel sulfide nanorods for oxygen and hydrogen evolution reaction. ChemistrySelect 2023, 8, e202204370. [Google Scholar]
- Choi, S.; Kim, S.-J.; Han, S.; Wang, J.; Kim, J.; Koo, B.; Ryabin, A.A.; Kunze, S.; Hyun, H.; Han, J.; et al. Enhancing Oxygen Evolution Reaction via a Surface Reconstruction-Induced Lattice Oxygen Mechanism. ACS Catal. 2024, 14, 15096–15107. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).