
Cell Lineage Affiliation During Hematopoiesis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HSCs Are Rare and Quiescent Cells That Self-Renew
3. Are HSCs Homogenous or a Mixture of Cells?
3.1. The HSC Compartment Is a Homogeneous Population of Multipotent Cells
3.2. HSCs Are a Complex Mixture of Cells
4. What Are the Lineage Options That Are Available to HSCs?
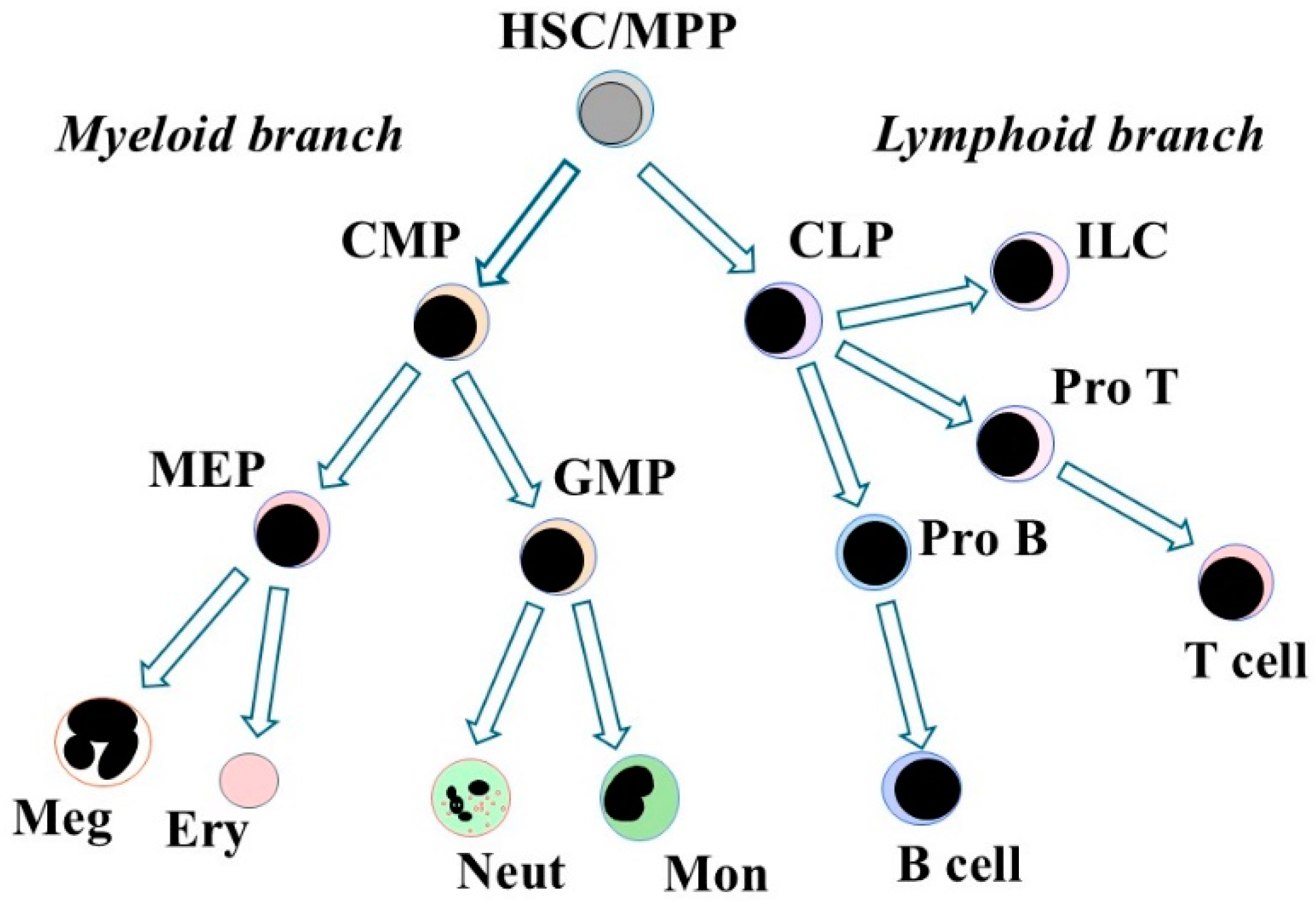
4.1. HSCs Choose to Develop Towards Either Myeloid or Lymphoid Cells
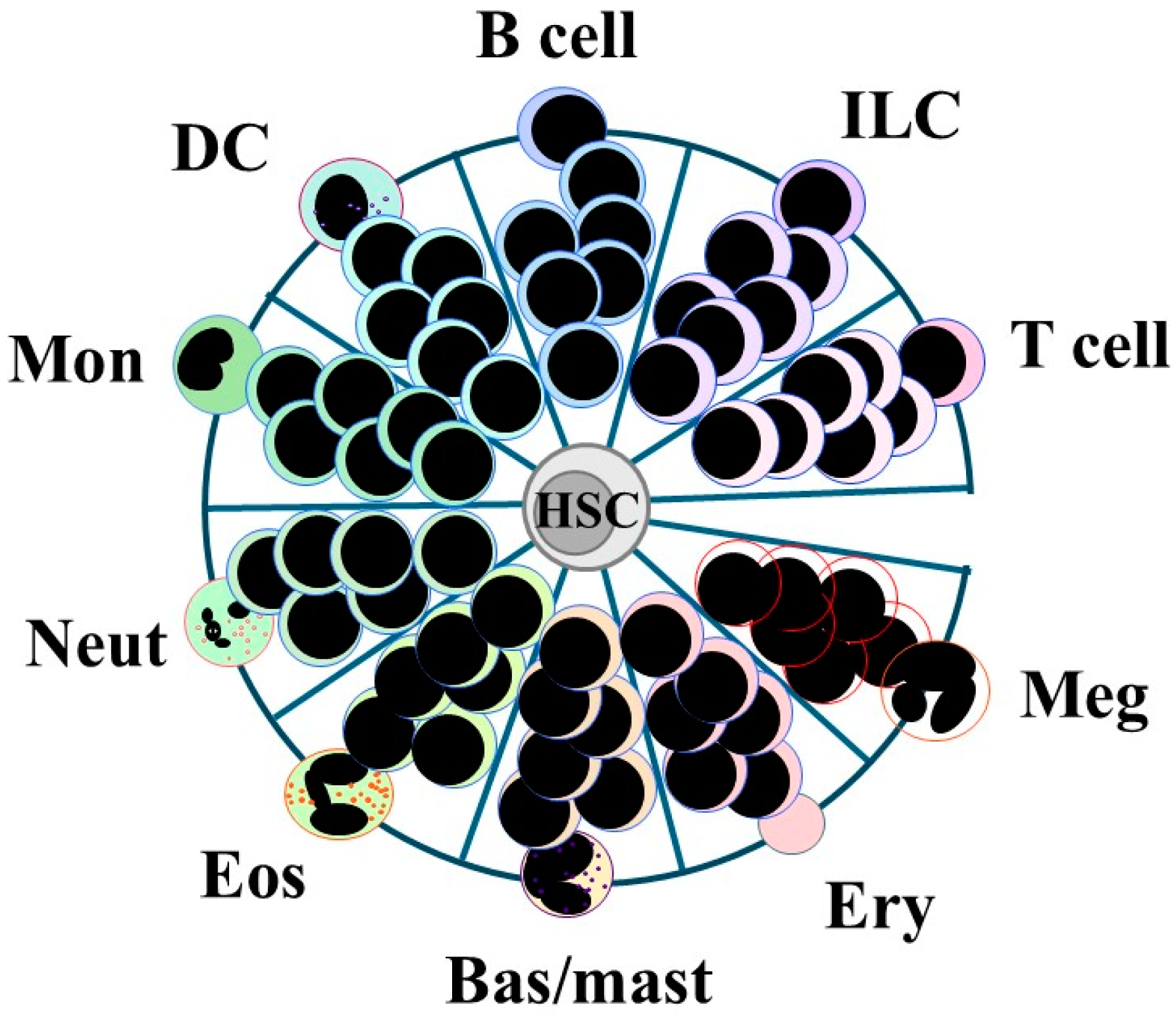
4.2. A Continuum View of the Options That Are Available to HSCs
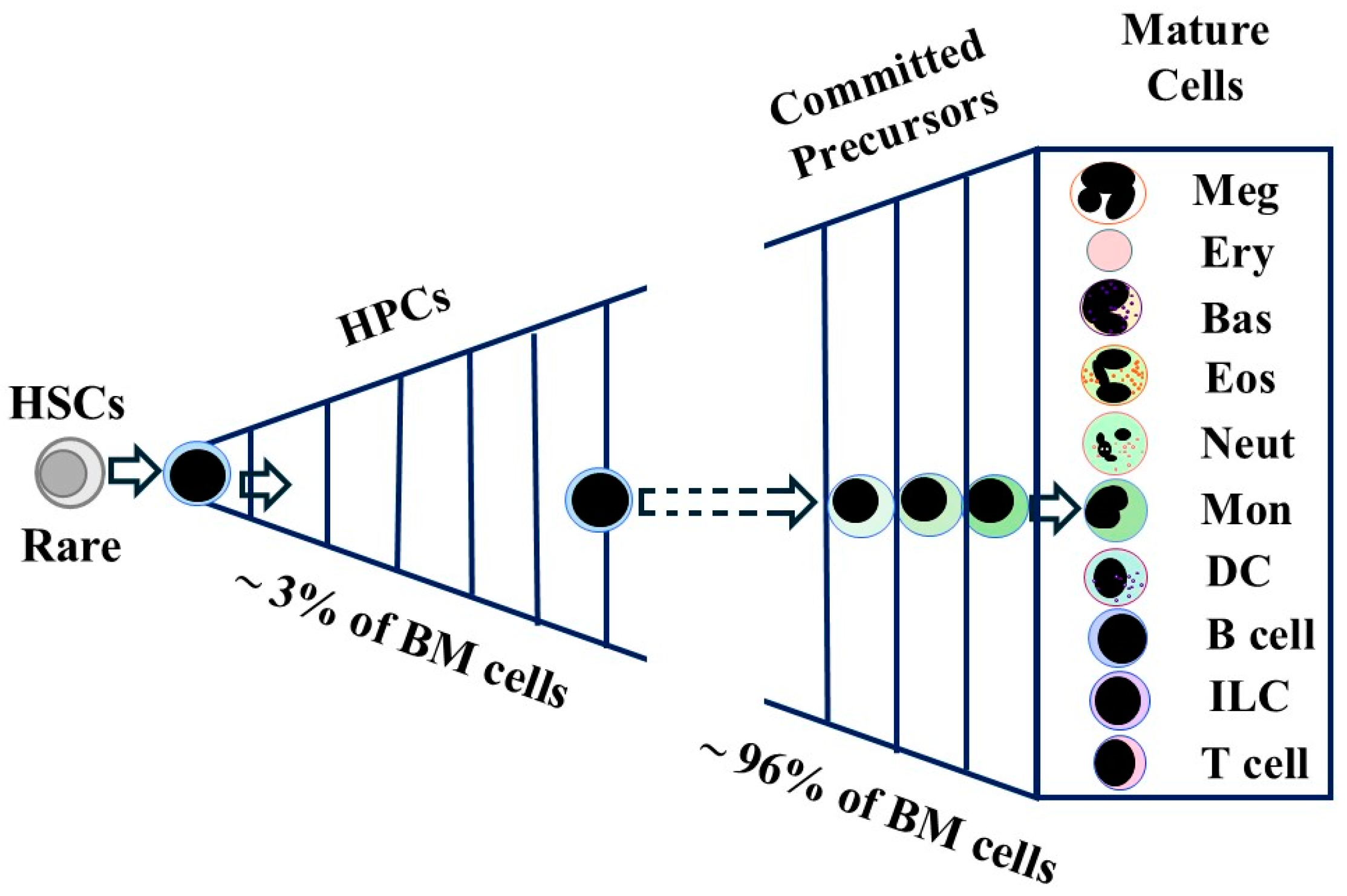
5. HSC/HPC Developmental Progression
5.1. A Canonical Cell Lineage Tree for Developmental Progression
5.2. HPC Progression Is Gradual and Versatile
6. Can Findings Be Reconciled to a Consensus Model?
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar]
- Macedo, A.; Orfao, A.; Ciudad, J.; Gonzalez, M.; Vidriales, B.; Lopez-Berges, C.; Martinez, A.; Landolf, C.; Canizo, C.; San Miguel, J.F. Phenotypic analysis of CD34 subpopulations in normal human bone marrow and its application for the detection of minimal residual disease. Leukemia 1995, 9, 1896–1901. [Google Scholar]
- Pang, W.W.; Price, E.A.; School, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef] [PubMed]
- Catlin, S.N.; Busque, L.; Gale, R.E.; Guttorp, P.; Abkowitz, J.L. The replication rate of human hematopoietic stem cells in vivo. Blood 2011, 117, 4460–4466. [Google Scholar] [PubMed]
- Szilvassy, S.J.; Humphries, R.K.; Landsdorp, P.M.; Eaves, A.C.; Eaves, C.J. Quantitative assay for totipotent reconstitution hematopoietic stem cells by a competitive repopulation strategy. Proc. Natl. Acad. Sci. USA 1990, 87, 8736–8740. [Google Scholar] [PubMed]
- Harrison, D.E.; Jordon, C.T.; Zhang, R.H.; Astle, C.M. Primitive hematopoietic stem cells: Direct assay of most productive populations by competitive repopulation with simple binomial correlation and covariance calculations. Exp. Hematol. 1993, 21, 206–219. [Google Scholar]
- Kent, D.G.; Copley, M.R.; Benz, C.; Wohner, S.; Dykstra, B.J.; Ma, E.; Cheyne, J.; Zhao, Y.; Bowie, M.B.; Zhao, Y.; et al. Prospective isolation and molecular characterisation of haematopoietic stem cells with durable self-renewal potential. Blood 2009, 113, 6342–6350. [Google Scholar]
- Challen, G.A.; Boles, N.; Kuan-Yin, K.; Goodall, M.A. Mouse hematopoietic stem cell identification and analysis. Cytometry A 2009, 75, 14–24. [Google Scholar]
- Harrison, D.E.; Astle, C.M.; Lerner, C. Number and continuous proliferative pattern of transplanted immune hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1988, 89, 822–826. [Google Scholar]
- Harrison, D.E.; Astle, C.M. Loss of stem cell repopulating ability upon transplantation. Effects of donor age, cell number, and transplantation procedure. J. Exp. Med. 1982, 156, 1767–1779. [Google Scholar]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar]
- Sieburg, H.B.; Cattarossi, G.; Muller-Sieburg, C.E. Lifespan differences in hematopoietic stem cells are due to imperfect repair and unstable mean reversion. PLoS Comput. Biol. 2013, 9, e1003006. [Google Scholar]
- Till, J.E.; McCulloch, E.A. A direct measurement of the radiosensitivity of normal bone marrow cells. Radiat. Res. 1961, 14, 213–222. [Google Scholar] [PubMed]
- Wu, A.M.; Till, J.E.; Simmonovitch, L.; McCulloch, E.A. A cytological study of the capacity for differentiation of normal colony-forming cells. J. Cell Physiol. 1967, 69, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.M.; Till, J.E.; Simmonovitch, L.; McCulloch, E.A. Cytological evidence for a relationship between normal hematopoietic colony-forming cells and cells of the lymphoid system. J. Exp. Med. 1968, 127, 455–464. [Google Scholar]
- Osawa, M.; Hanada, K.-I.; Hamada, H.; Nakauchi, H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 1996, 273, 242–245. [Google Scholar]
- Abe, T.; Masuya, M.; Ogawa, M. An efficient method for single hematopoietic stem cell (HSC) engraftment in mice based on cell cycle dormancy of HSCs. Exp. Hematol. 2010, 38, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 2013, 154, 1112–1126. [Google Scholar]
- Harrison, D.E.; Zhang, R.K. The same exhaustible multilineage precursor produces both myeloid and lymphoid cells as early as 3-4 weeks after marrow transplantation. Proc. Natl. Acad. Sci. USA 1992, 89, 10134–10138. [Google Scholar]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar]
- Sakamaki, T.; Kao, K.S.; Nishi, K.; Chen, J.Y.; Sadaoke, M.; Takaori-Kondo, A.; Weissman, I.L.; Miyanishi, M. Hoxb5 defines the heterogeneity of self-renewal capacity in the haematopoietic stem cell compartment. Biochem. Biophys. Res. Commun. 2021, 539, 34–41. [Google Scholar] [PubMed]
- England, S.J.; McGrath, K.E.; Frame, J.M.; Palis, J. Immature erythroblasts with extensive ex vivo self-renewal capacity emerge from the early mammalian fetus. Blood 2010, 117, 2708–2717. [Google Scholar] [PubMed]
- Hu, M.; Krause, D.; Greaves, M.; Sharkis, S.; Dexter, M.; Heyworth, C.; Enver, T. Multilineage gene expression precedes commitment in the hematopoietic system. Genes Dev. 1997, 11, 774–785. [Google Scholar] [PubMed]
- Mooney, C.J.; Cunningham, A.; Tsapogas, P.; Toellner, K.-M.; Brown, G. Selective expression of Flt3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017, 18, 1037. [Google Scholar] [CrossRef]
- Grover, R.; Mancini, I.C.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussan, K.D.; O’Carrol, D.D.; Jacobsen, S.E.W.; Nerlov, C. Erythropoietin guides multipotent progenitors towards an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar]
- Rieger, M.A.; Hoppe, P.S.; Smeijkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Haematopoietic cytokines can instruct lineage choice. Science 2009, 325, 217–218. [Google Scholar]
- Brown, G.; Ceredig, R. Modelling the hematopoietic landscape. Front. Cell Dev. Biol. 2019, 7, 104. [Google Scholar]
- Adolfsson, J.; Mansson, R.; Buza-Vidas, N.; Huitquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Berge, O.-J.; Thoren, L.A.M.; et al. Identification of Flt3+ lymphoid-myeloid stem cell lacking erythro-megakaryocytic potential: A revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306. [Google Scholar]
- Muller-Sieburg, C.E.; Cho, R.H.; Karlsson, L.; Huang, J.-F.; Sieburg, H.B. Myeloid biased hematopoietic stem cells have extensive self-renewal capacity but generate diminished lymphoid progeny with impaired IL-7 responsiveness. Blood 2004, 104, 4111–4118. [Google Scholar]
- Gieger, H.; de Hann, G.; Florian, M. The aging haematopoietic stem cell compartment. Nat. Rev. Immunol. 2013, 13, 376–389. [Google Scholar]
- Dorshkind, K.; Hofer, T.; Montecino-Rodriguez, E.; Poili, D.D.; Rodewald, H.-R. Do haematopoietic stem cells age? Nat. Rev. Immunol. 2020, 20, 196–202. [Google Scholar]
- Montecino-Rodriguez, E.; Kong, Y.; Casero, D.; Rouoult, A.; Dorshkind, K.; Poili, P.D. Lymphoid-biased HSC are maintained with age and efficiently generate lymphoid progeny. Stem Cell Rep. 2019, 12, 584–598. [Google Scholar]
- Vanickova, K.; Milosevic, M.; Bas, I.R.; Burocziove, M.; Yokota, A.; Danek, P.; Grusanovic, S.; Chilinski, M.; Plewczynski, D.; Rohlena, J.; et al. Hematopoietic stem cells undergo a lymphoid to myeloid switch in early stages of emergency granulopoiesis. EMBO J. 2023, 42, e113527. [Google Scholar] [PubMed]
- Sanjuan-Pla, A.; Macaulay, I.C.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Corella, C.; Matsuoka, S.; Jones, T.B.; et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [PubMed]
- Ninos, J.M.; Jefferis, L.C.; Cogle, C.R.; Kerr, W.G. The thrombopoietin receptor, c-Mpl, is a selective surface marker for human hematopoietic stem cells. J. Trans. Med. 2006, 4, 9. [Google Scholar]
- Knapp, D.J.H.F.; Mammond, C.A.; Hul, T.; van Loenhout, M.T.J.; Wang, F.; Aghaeepour, N.; Miller, P.H.; Moksa, M.; Rabu, G.M.; Beer, P.A.; et al. Single-cell analysis identifies a CD33C subset of human cord blood cells with high regenerative potential. Nat. Cell Biol. 2018, 20, 710–720. [Google Scholar]
- Kondo, M.; Weissman, I.L.; Akashi, K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997, 91, 661–672. [Google Scholar]
- Akashi, K.; Trover, D.; Miyamoto, Y.; Weissman, I.L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404, 193–197. [Google Scholar]
- Ceredig, R.; Rolink, A.G.; Brown, G. Models of haematopoiesis: Seeing the wood for the trees. Nat. Rev. Immunol. 2009, 9, 293–299. [Google Scholar]
- Wong, A.K.-Y.; Bunce, C.M.; Lord, J.M.; Salt, J.; Brown, G. Evidence that precursor cells of monocytes and B lymphocytes are closely related. Exp. Hematol. 1989, 17, 968–973. [Google Scholar]
- Boyd, A.W.; Schrader, J.W. Derivation of macrophage-like lines from the pre-B lymphoma ABLS 8.1 using 5-azacytidine. Nature 1982, 297, 691–693. [Google Scholar] [PubMed]
- Borzillo, G.V.; Ashmun, R.A.; Sherr, C.J. Macrophage lineage switching of murine early pre-B lymphoid cells expressing transduced fms genes. Mol. Cell. Biol. 1990, 10, 2703–2714. [Google Scholar]
- Fisher, A.G.; Burdet, C.; Bunce, C.; Merkenschlager, M.; Ceredig, R. Lymphoproliferative disorders in IL-7 transgenic mice: Expansion of immature B cells which retain macrophage potential. Int. Immunol. 1995, 7, 415–423. [Google Scholar]
- Cumano, A.; Paige, C.J.; Iscove, N.N.; Brady, G. Bipotential precursors of B cells and macrophages in murine fetal liver. Nature 1992, 356, 612–615. [Google Scholar] [PubMed]
- Montecino-Rodriguez, E.; Leathers, H.; Dorshkind, K. Bipotential B-macrophage progenitors are present in adult bone marrow. Nat. Immunol. 2001, 2, 83–88. [Google Scholar] [PubMed]
- Paul, F.; Arkin, Y.; Giladi, A.; Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Winter, D.; Lara-Astiaso, D.; Gury, M.; Weiner, A.; et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 2015, 163, 1663–1677. [Google Scholar]
- Kucinski, I.; Campos, J.; Barile, M.; Severi, F.; Bohin, N.; Mareira, P.N.; Allen, L.; Lawson, H.; Haltalli, M.L.R.; Kinston, S.J.; et al. A time and single-cell resolved model of murine bone marrow hematopoiesis. Cell Stem Cell 2024, 31, 244–259. [Google Scholar]
- Patel, S.H.; Christodoulou, C.; Weinreb, C.; Yu, Q.; Lummertz da Rocha, E.; Pepe-Mooney, B.J.; Bowling, S.; Li, L.; Osario, F.G.; Doley, G.Q.; et al. Lifelong multilineage contribution by embryonic-born blood progenitors. Nature 2021, 606, 747–753. [Google Scholar]
- Kapadia, C.D.; Willams, N.; Dawson, K.J.; Watson, C.; Yousefzadeh, M.J.; Le, D.; Nyamondo, K.; Kodavali, S.; Cogan, A.; Waldvogel, S.; et al. Clonal dynamics and somatic evolution of haematopoiesis in mouse. bioRxiv 2025. [Google Scholar] [CrossRef]
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar]
- Bradley, T.R.; Metcalf, D. The growth of mouse bone marrow cells in vitro. Aust. J. Exp. Biol. Med. Sci. 1966, 44, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Niiro, H.; Iino, T.; Yashida, S.; Saito, N.; Onohara, S.; Miyamoto, T.; Minagawa, H.; Fujii, S.-I.; Shultz, L.D.; et al. The developmental program of human dendritic cells is operated independently of conventional myeloid and lymphoid pathways. Blood 2007, 110, 3591–3660. [Google Scholar] [CrossRef]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- Westin, J.A. Motile and social behaviour of neural crest cells. In Cell Behavior; Bellairs, R., Curtis, A., Dunn, G., Eds.; Cambridge University Press: London, UK, 1981; pp. 429–470. [Google Scholar]
- Paczko, M.; Vordos, D.; Szabo, P.; Jekely, G.; Szathmary, E.; Szilagyi, A. A neural network-based framework for cell-fate decisions and development. Commun. Biol. 2024, 7, 323. [Google Scholar] [CrossRef]
- Angeli, D.; Ferrell, J.E.; Sontag, E.D. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive feedback systems. Proc. Natl. Acad. Sci. USA 2004, 101, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Harrington, H.A.; Feliu, E.; Wiuf, C.; Stumpf, M.P. Cellular compartments cause multistability and allow cells to process more information. Biophys. J. 2013, 104, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Craciun, G.; Tang, Y.; Feinberg, M. Understanding bistability in complex enzyme-driven reaction networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8697–8702. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, T.; Tian, T. A robust method for designing multistable systems by embedding bistable subsystems. Syst. Biol. Appl. 2022, 8, 10. [Google Scholar] [CrossRef]
- Nestorowa, S.; Hamey, F.K.; Pijuan Sala, B.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Gottgens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef]
- Brown, G.; Hughes, P.J.; Michell, R.H.; Rolink, A.G.; Ceredig, R. The sequential determination model of hematopoiesis. Trends Immunol. 2007, 28, 442–448. [Google Scholar] [CrossRef]
- Grishechkin, A.; Mulcherjee, A.; Karin, O. Hierarchical control of state transitions in dense associated memories. arXiv 2025, arXiv:2412.111336V2. [Google Scholar]
- Novershtern, N.; Subramanian, A.; Lawton, L.N.; Mak, R.H.; Haining, W.N.; McConkey, M.E.; Habib, N.; Yosef, N.; Chang, C.Y.; Shay, T.; et al. Densely Interconnected Transcriptional Circuits Control Cell States in Human Hematopoiesis. Cell 2011, 144, 296–309. [Google Scholar]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, aaw3381. [Google Scholar]
- Gao, S.; Zhang, Y.; Liu, F. Revisiting the lineage contribution of hematopoietic stem and progenitor cells. Development 2023, 150, dev201609. [Google Scholar] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar]
- Waddington, C.H. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology; Allen & Unwin: London, UK, 1957. [Google Scholar]
- Tsapogas, P.; Swee, L.K.; Nusser, A.; Nuber, N.; Kreuzaler, M.; Capoferri, G.; Rolink, H.; Ceredig, R.; Rolink, A. In vivo evidence for an instructive role of fms-like tyrosine kinase-3 (FLT3) ligand in hematopoietic development. Haematologica 2014, 99, 638–646. [Google Scholar] [PubMed]
- Tsapogas, P.; Mooney, C.J.; Brown, G.; Rolink, A. The cytokine Flt3-ligand in normal and malignant hematopoiesis. Int. J. Mol. Sci. 2017, 18, 1115. [Google Scholar] [CrossRef]
- Onai, N.; Obata-Onai, A.; Tussiwand, R.; Lanzavecchia, A.; Manz, M.G. Activation of the FLT3 signal transduction cascade rescues and enhances type 1 interferon-producing and dendritic cell development. J. Exp. Med. 2006, 203, 227–238. [Google Scholar]
- Momenilandi, M.; Levy, R.; Sobrino, S.; Li, J.; Lagresle-Peyrou, C.; Esmaelzadeh, H.; Fayard, A.; Le Floc’h, C.; Guerin, A.; Mina, E.D.; et al. FLT3 governs the development of partially overlapping hematopoietic lineages in humans and mice. Cell 2024, 187, 2817–2837. [Google Scholar]
- Calabria, A.; Spinozzi, G.; Casana, D.; Buscaroli, E.; Benedicenti, F.; Pais, G.; Gazzo, F.; Scala, S.; Lidonnici, M.R.; Scaramuzza, S.; et al. Long-term lineage commitment in haematopoietic stem cell gene therapy. Nature 2024, 636, 162–171. [Google Scholar] [CrossRef]
- Brown, G.; Ceredig, R.; Tsapogas, P. The making of hematopoiesis: Developmental ancestry and environmental nurture. Int. J. Mol. Sci. 2018, 19, 2122. [Google Scholar] [CrossRef] [PubMed]
- Porritt, H.E.; Rumfelt, L.L.; Tabrizifard, S.; Smitt, T.M.; Zuniga-Pflucker, J.C.; Petrie, H.T. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity 2004, 20, 735–745. [Google Scholar]
- Lehar, S.M.; Dooley, J.; Farr, A.G.; Bevan, M.J. Notch ligands Delta 1 and Jagged1 transmit distinct signals to T-cell precursors. Blood 2005, 105, 1440–1447. [Google Scholar] [CrossRef]
- Balciunaite, G.; Ceredig, R.; Rolink, A.G. The earliest subpopulation of mouse thymocytes contains potent T, significant macrophage, and natural killer cell but no B-lymphocyte potential. Blood 2005, 105, 1930–1936. [Google Scholar] [PubMed]
- Glatman Zaretsky, A.; Taylor, J.J.; King, I.L.; Marshall, F.A.; Mohrs, M.; Pearce, E.J. T follicular helper cells differentiate from th2 cells in response to helminth antigens. J. Exp. Med. 2009, 206, 991–999. [Google Scholar]
- Singh, K.; Gatzka, M.; Peters, T.; Borkner, L.; Hainzl, A.; Wang, H.; Sindrilaru, A.; Scharffetter-Kochanek, K. Reduced cd18 levels drive regulatory t cell conversion into th17 cells in the cd18hypo pl/j mouse model of psoriasis. J. Immunol. 2013, 190, 2544–2553. [Google Scholar] [PubMed]
- García-Ramírez, I.; Bhatia, S.; Rodríguez-Hernández, G.; González-Herrero, I.; Walter, C.; González de Tena-Dávila, S.; Parvin, S.; Haas, O.; Woessmann, W.; Stanulla, M.; et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018, 37, e98783. [Google Scholar] [PubMed]
- Martín-Lorenzo, A.; Auer, F.; Chan, L.N.; García-Ramírez, I.; González-Herrero, I.; Rodríguez-Hernández, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of Pax5 Exploits Sca1-BCR-ABLp190 Susceptibility to Confer the Metabolic Shift Essential for pB-ALL. Cancer Res. 2018, 78, 2669–2679. [Google Scholar]
- Perez-Caro, M.; Cobaleda, C.; Gonzalez-Herrero, I.; Vicente-Duenas, C.; Bermejo-Rodriguez, C.; Sanchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sánchez-Martín, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar]
- Brown, G.; Sanchez, L.; Sanchez-Garcia, I. Lineage decision-making within normal hematopoietic and leukaemic stem cells. Int. J. Mol. Sci. 2020, 21, 2247. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G. Cell Lineage Affiliation During Hematopoiesis. Int. J. Mol. Sci. 2025, 26, 3346. https://doi.org/10.3390/ijms26073346
Brown G. Cell Lineage Affiliation During Hematopoiesis. International Journal of Molecular Sciences. 2025; 26(7):3346. https://doi.org/10.3390/ijms26073346
Chicago/Turabian StyleBrown, Geoffrey. 2025. "Cell Lineage Affiliation During Hematopoiesis" International Journal of Molecular Sciences 26, no. 7: 3346. https://doi.org/10.3390/ijms26073346
APA StyleBrown, G. (2025). Cell Lineage Affiliation During Hematopoiesis. International Journal of Molecular Sciences, 26(7), 3346. https://doi.org/10.3390/ijms26073346