
Claudin-17 Deficiency Drives Vascular Permeability and Inflammation Causing Lung Injury

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
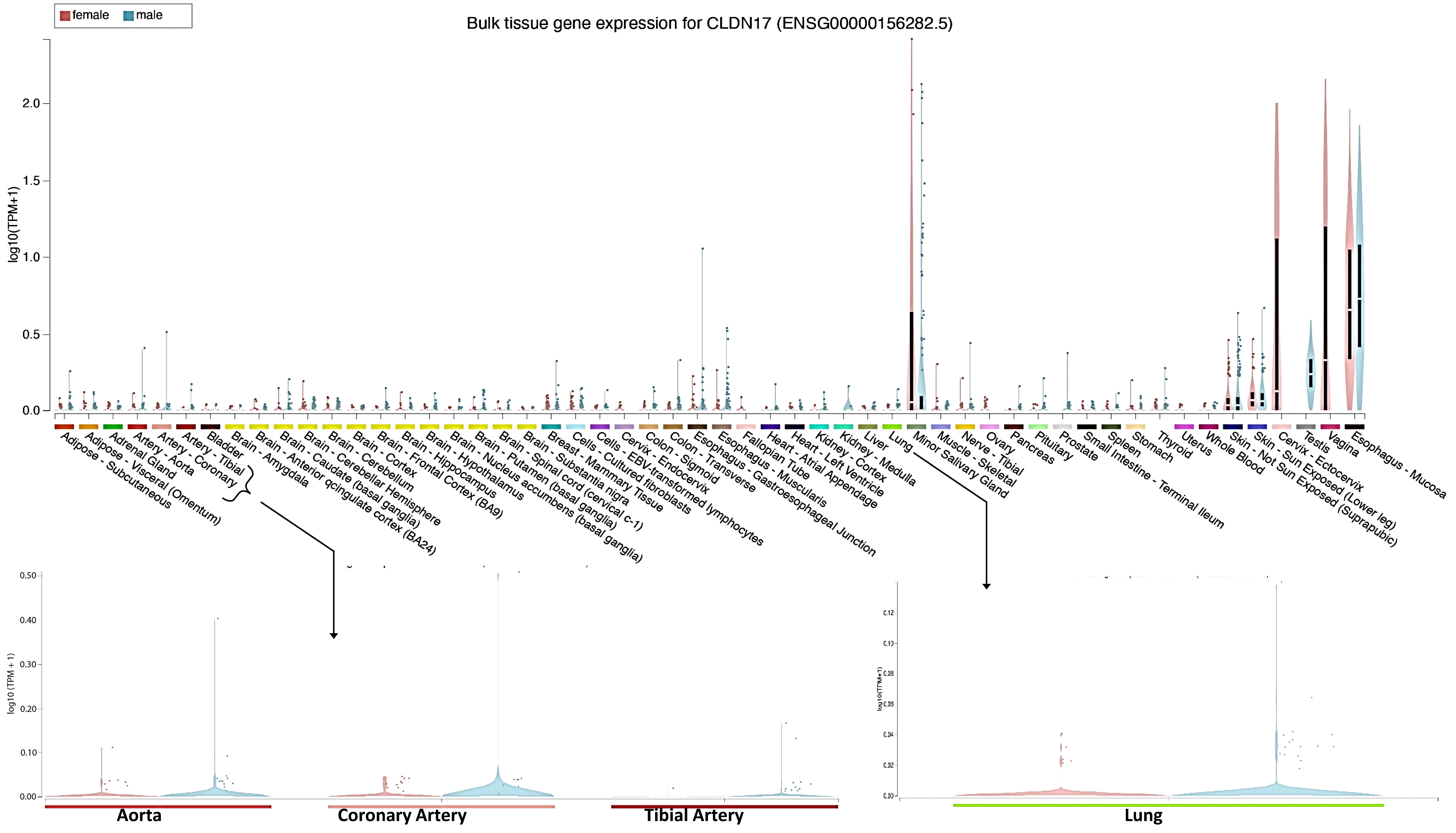
2.1. Cldn17 Is Expressed in Various Tissues, Albeit at Low Levels
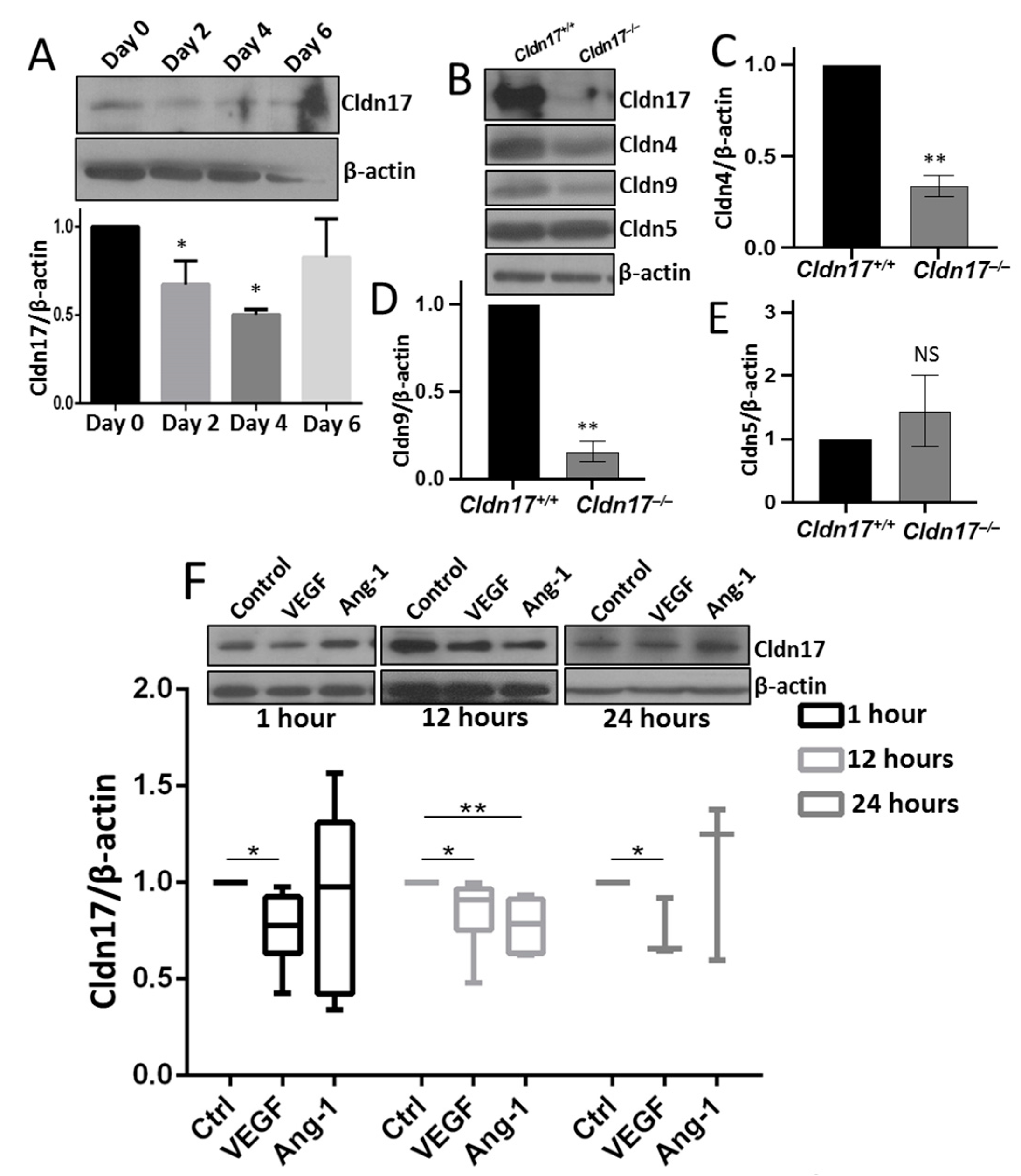
2.2. Reduced Cldn17 Expression in Lipopolysaccharide-Administered Mouse Lungs
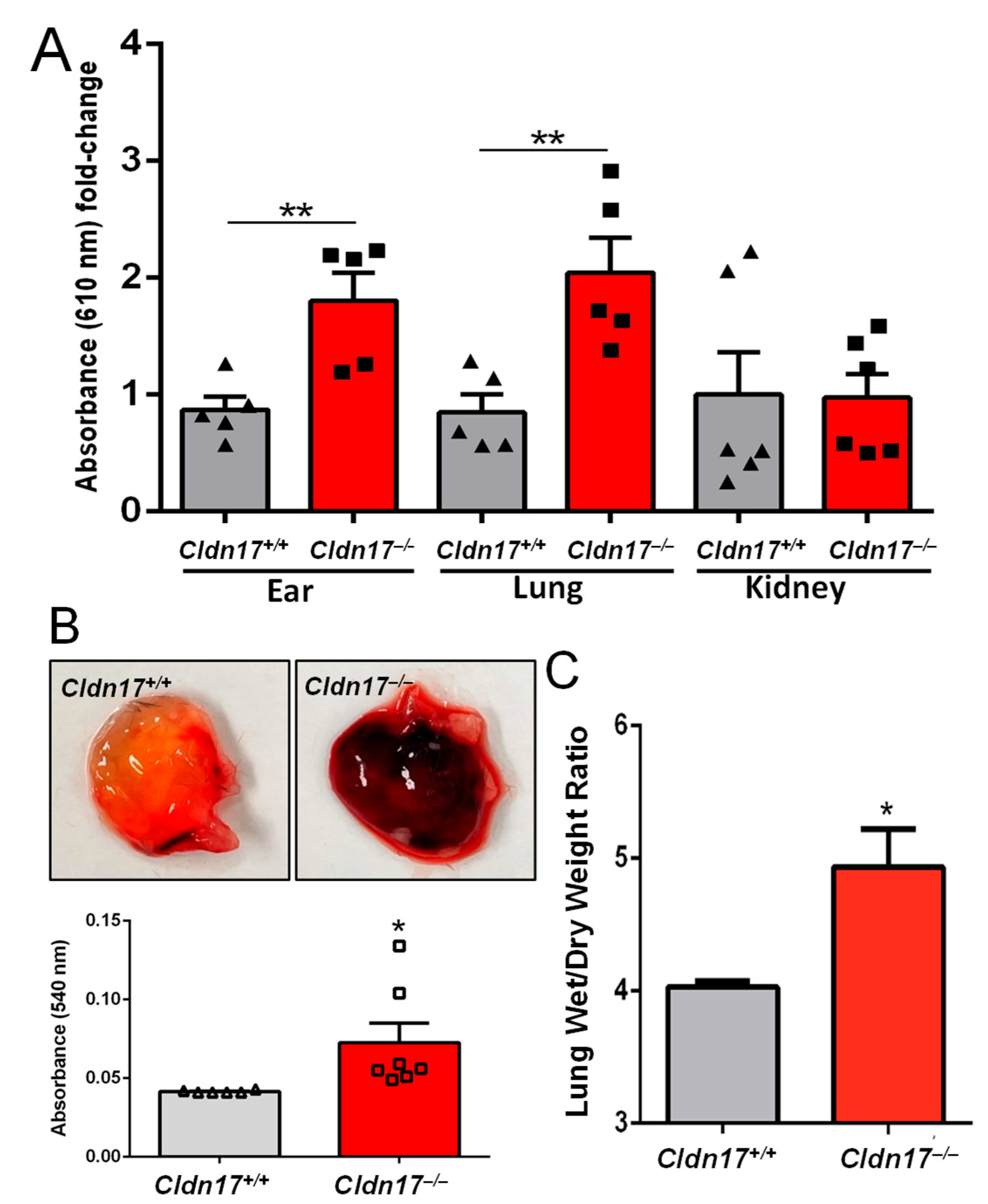
2.3. Cldn17 Deficiency Results in Vascular Permeability and Pulmonary Edema
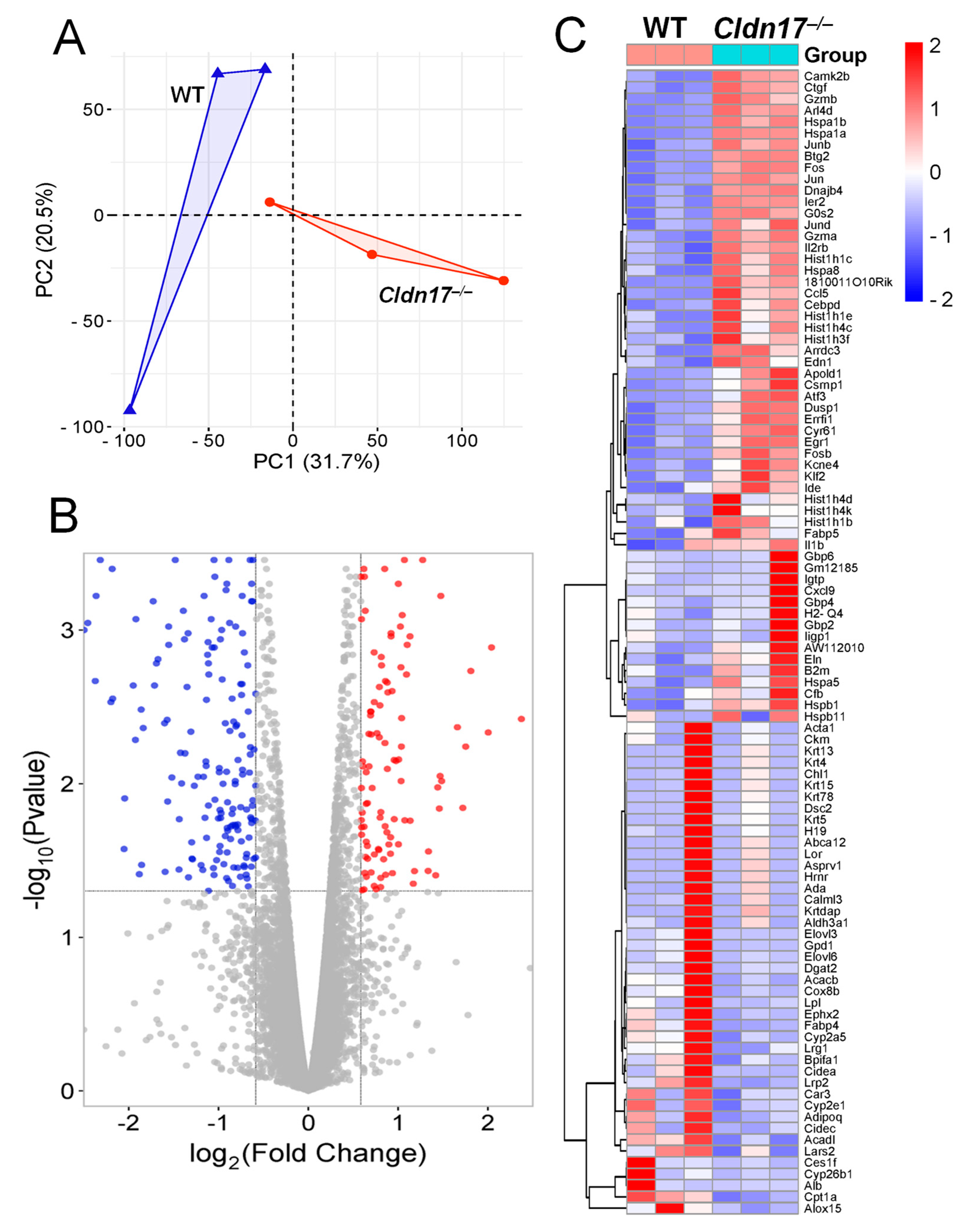
2.4. Principal Component Analysis and Differential Gene Expression Identify Transcriptional Shifts and Dysregulated Genes in Cldn17−/− Lungs
2.5. Gene Ontology and Pathway Analysis Reveal Significant Enrichment in Vascular, Immune, and Metabolic Pathways in Cldn17−/− Lungs
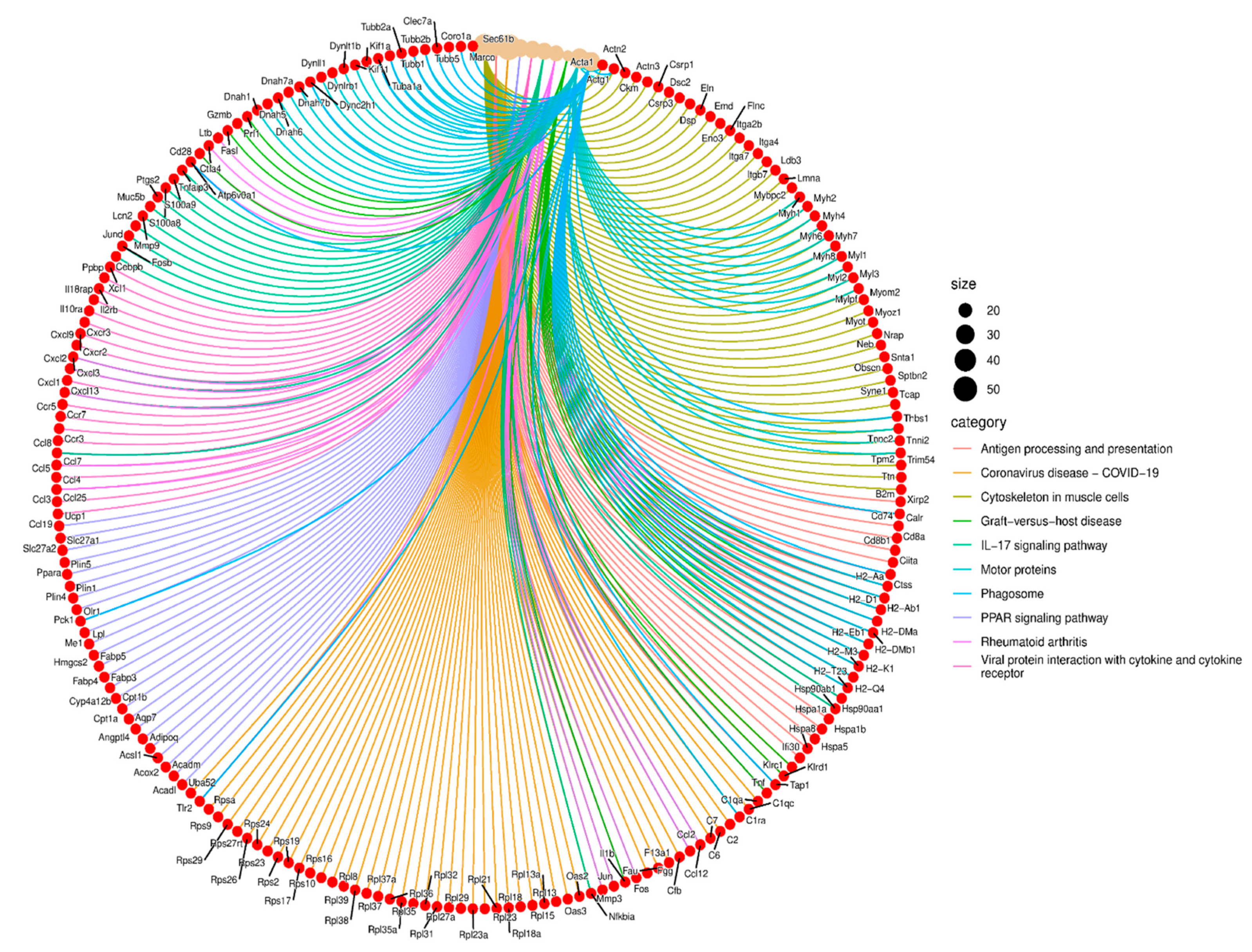
2.6. Functional Enrichment Analysis Reveals Key Pathways Affected by Cldn17 Deletion
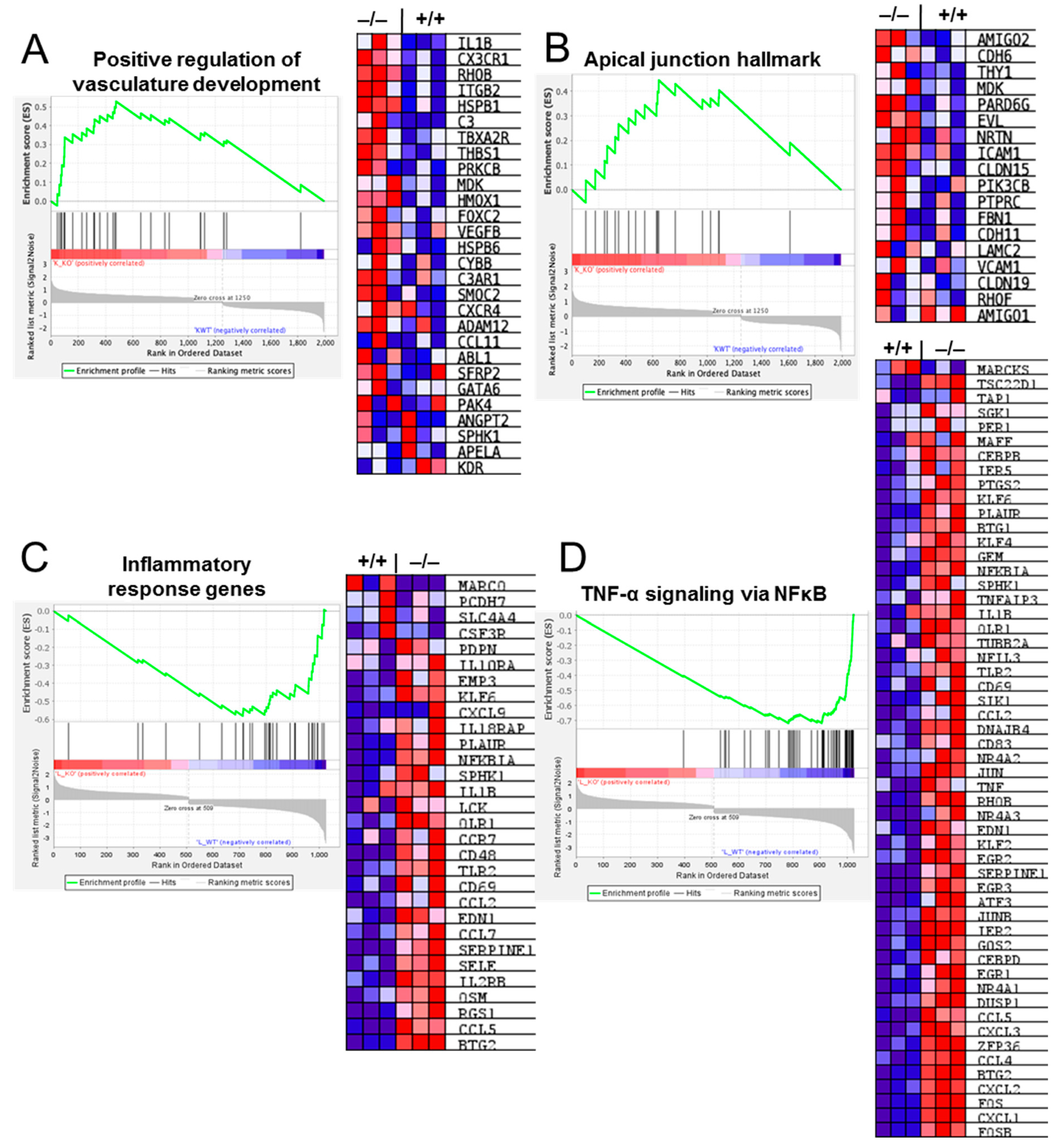
2.7. Gene Set Enrichment Analysis (GSEA) Reveals Deregulation of Pathways Involved in Vascular and Inflammatory Modulation in Cldn17−/− Compared with WT Mouse Lungs
2.8. Cldn17 Deficiency Did Not Affect the Expression of Other Cell–Cell Junction Proteins in Mouse Lungs
2.9. Cldn17 Deficiency Exacerbates LPS-Induced Lung Injury
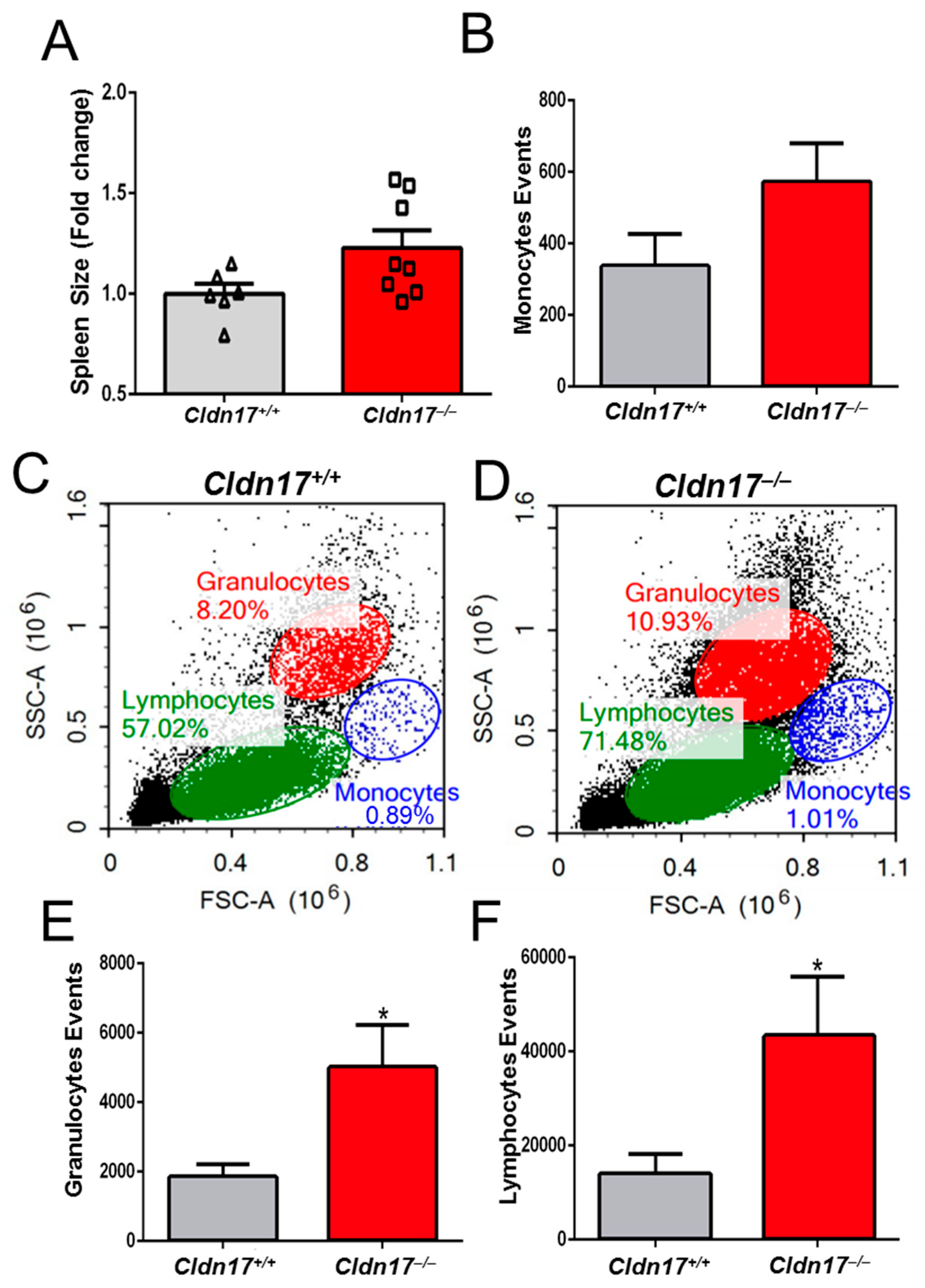
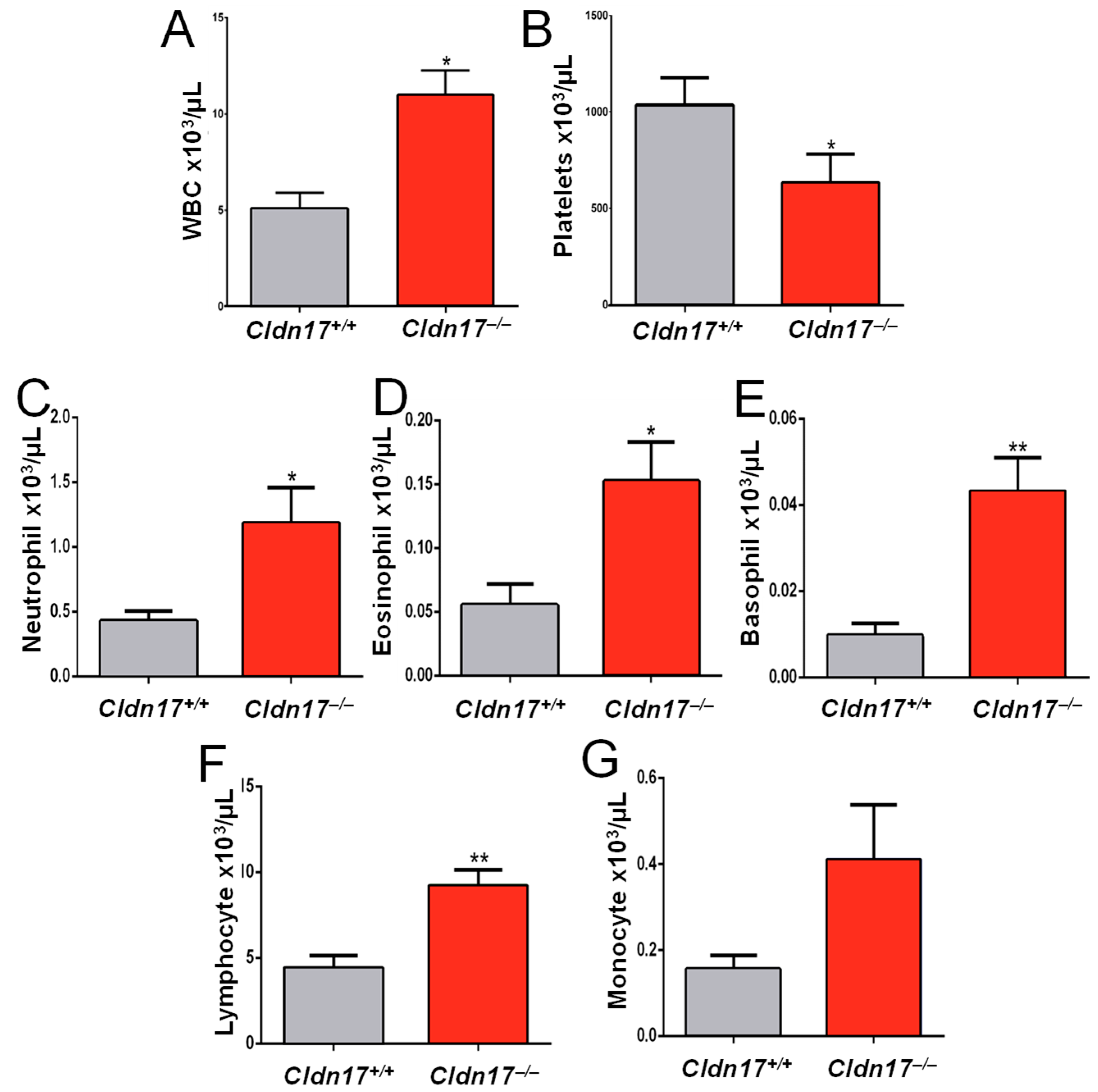
2.10. Cldn17 Deficiency Leads to Inflammation and Leukocytosis in Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture and Reagents
4.3. Western Blot Analysis
4.4. Miles Assay
4.5. Matrigel Assay
4.6. Analysis of Lung Injury and Pulmonary Edema
4.7. Flow Cytometry Analysis
4.8. RNA-Seq and Bioinformatic Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park-Windhol, C.; D’Amore, P.A. Disorders of Vascular Permeability. Annu. Rev. Pathol. 2016, 11, 251–281. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Felgueiras, J.; Fardilha, M. Signaling pathways in anchoring junctions of epithelial cells: Cell-to-cell and cell-to-extracellular matrix interactions. J. Recept. Signal Transduct. Res. 2015, 35, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Aird, W.C. Endothelial cell gene regulation. Trends Cardiovasc. Med. 2005, 15, 174–184. [Google Scholar] [CrossRef]
- Sahni, S.K. Endothelial cell infection and hemostasis. Thromb. Res. 2007, 119, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.; Yap, A.S. Tensile Forces and Mechanotransduction at Cell-Cell Junctions. Curr. Biol. 2018, 28, R445–R457. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Bradfield, P.F.; Imhof, B.A. Tight junction dynamics: The role of junctional adhesion molecules (JAMs). Cell Tissue Res. 2014, 355, 701–715. [Google Scholar] [CrossRef]
- Sawada, N. Tight junction-related human diseases. Pathol. Int. 2013, 63, 1–12. [Google Scholar] [CrossRef]
- Runkle, E.A.; Mu, D. Tight junction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef]
- Adil, M.S.; Narayanan, S.P.; Somanath, P.R. Cell-cell junctions: Structure and regulation in physiology and pathology. Tissue Barriers 2021, 9, 1848212. [Google Scholar] [CrossRef]
- Gao, F.; Artham, S.; Sabbineni, H.; Al-Azayzih, A.; Peng, X.D.; Hay, N.; Adams, R.H.; Byzova, T.V.; Somanath, P.R. Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell Mol. Life Sci. 2016, 73, 3917–3933. [Google Scholar] [CrossRef]
- Krug, S.M.; Gunzel, D.; Conrad, M.P.; Rosenthal, R.; Fromm, A.; Amasheh, S.; Schulzke, J.D.; Fromm, M. Claudin-17 forms tight junction channels with distinct anion selectivity. Cell Mol. Life Sci. 2012, 69, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Adil, M.S.; Parvathagiri, V.; Verma, A.; Liu, F.; Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Claudin-17 Deficiency in Mice Results in Kidney Injury Due to Electrolyte Imbalance and Oxidative Stress. Cells 2022, 11, 1782. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef]
- Frank, P.G.; Lisanti, M.P. ICAM-1: Role in inflammation and in the regulation of vascular permeability. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H926–H927. [Google Scholar] [CrossRef]
- Yang, S.; Yu, M.; Sun, L.; Xiao, W.; Yang, X.; Sun, L.; Zhang, C.; Ma, Y.; Yang, H.; Liu, Y.; et al. Interferon-gamma-induced intestinal epithelial barrier dysfunction by NF-kappaB/HIF-1alpha pathway. J. Interferon Cytokine Res. 2014, 34, 195–203. [Google Scholar] [CrossRef]
- Shan, S.; Alanazi, A.H.; Han, Y.; Zhang, D.; Liu, Y.; Narayanan, S.P.; Somanath, P.R. Pro-Inflammatory Characteristics of Extracellular Vesicles in the Vitreous of Type 2 Diabetic Patients. Biomedicines 2024, 12, 2053. [Google Scholar] [CrossRef]
- Kumari, L.; Yadav, R.; Kumar, Y.; Bhatia, A. Role of tight junction proteins in shaping the immune milieu of malignancies. Expert. Rev. Clin. Immunol. 2024, 20, 1305–1321. [Google Scholar] [CrossRef]
- Gao, F.; Alwhaibi, A.; Artham, S.; Verma, A.; Somanath, P.R. Endothelial Akt1 loss promotes prostate cancer metastasis via beta-catenin-regulated tight-junction protein turnover. Br. J. Cancer 2018, 118, 1464–1475. [Google Scholar] [CrossRef]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Regulation of blood-retinal barrier cell-junctions in diabetic retinopathy. Pharmacol. Res. 2020, 161, 105115. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Liu, F.; Ford, E.; Caldwell, R.B.; Narayanan, S.P.; Somanath, P.R. Triciribine attenuates pathological neovascularization and vascular permeability in a mouse model of proliferative retinopathy. Biomed. Pharmacother. 2023, 162, 114714. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sabbineni, H.; Artham, S.; Somanath, P.R. Modulation of long-term endothelial-barrier integrity is conditional to the cross-talk between Akt and Src signaling. J. Cell Physiol. 2017, 232, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Alanazi, A.H.; Chastain, D.B.; Rudraraju, M.; Parvathagiri, V.; Shan, S.; Lin, X.; Henao-Martinez, A.F.; Franco-Paredes, C.; Narayanan, S.P.; Somanath, P.R. A multi-arm, parallel, preclinical study investigating the potential benefits of acetazolamide, candesartan, and triciribine in combination with fluconazole for the treatment of cryptococcal meningoencephalitis. Eur. J. Pharmacol. 2023, 960, 176177. [Google Scholar] [CrossRef]
- Conrad, M.P.; Piontek, J.; Gunzel, D.; Fromm, M.; Krug, S.M. Molecular basis of claudin-17 anion selectivity. Cell Mol. Life Sci. 2016, 73, 185–200. [Google Scholar] [CrossRef]
- Klar, J.; Piontek, J.; Milatz, S.; Tariq, M.; Jameel, M.; Breiderhoff, T.; Schuster, J.; Fatima, A.; Asif, M.; Sher, M.; et al. Altered paracellular cation permeability due to a rare CLDN10B variant causes anhidrosis and kidney damage. PLoS Genet. 2017, 13, e1006897. [Google Scholar] [CrossRef]
- Berselli, A.; Alberini, G.; Benfenati, F.; Maragliano, L. Computational study of ion permeation through claudin-4 paracellular channels. Ann. N. Y. Acad. Sci. 2022, 1516, 162–174. [Google Scholar] [CrossRef]
- Xu, Y.N.; Deng, M.S.; Liu, Y.F.; Yao, J.; Xiao, Z.Y. Tight junction protein CLDN17 serves as a tumor suppressor to reduce the invasion and migration of oral cancer cells by inhibiting epithelial-mesenchymal transition. Arch. Oral. Biol. 2022, 133, 105301. [Google Scholar] [CrossRef]
- Li, Z.H.; Xia, T.H.; Kang, Z.J.; Deng, X.; Wang, Y. Expression and significance of tight junction proteins in the kidney in a mouse model of renal ischemia-reperfusion injury. Zhongguo Dang Dai Er Ke Za Zhi 2018, 20, 1055–1062. [Google Scholar] [CrossRef]
- Sun, L.; Feng, L.; Cui, J. Increased expression of claudin-17 promotes a malignant phenotype in hepatocyte via Tyk2/Stat3 signaling and is associated with poor prognosis in patients with hepatocellular carcinoma. Diagn. Pathol. 2018, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.; Piontek, J.; Rosenthal, R.; Gunzel, D.; Krug, S.M. Tight junctions of the proximal tubule and their channel proteins. Pflug. Arch. 2017, 469, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Li, W.; Wang, H.; Wang, G. The distinct expression patterns of claudin-10, -14, -17 and E-cadherin between adjacent non-neoplastic tissues and gastric cancer tissues. Diagn. Pathol. 2013, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Domenech, P.; Perez, T.; Saldarini, A.; Uad, P.; Musso, C.G. Kidney-lung pathophysiological crosstalk: Its characteristics and importance. Int. Urol. Nephrol. 2017, 49, 1211–1215. [Google Scholar] [CrossRef]
- Malek, M.; Hassanshahi, J.; Fartootzadeh, R.; Azizi, F.; Shahidani, S. Nephrogenic acute respiratory distress syndrome: A narrative review on pathophysiology and treatment. Chin. J. Traumatol. 2018, 21, 4–10. [Google Scholar] [CrossRef]
- Singbartl, K. Renal-pulmonary crosstalk. Contrib. Nephrol. 2011, 174, 65–70. [Google Scholar] [CrossRef]
- Matuschak, G.M.; Lechner, A.J. Acute lung injury and the acute respiratory distress syndrome: Pathophysiology and treatment. Mo. Med. 2010, 107, 252–258. [Google Scholar]
- Andres-Hernando, A.; Dursun, B.; Altmann, C.; Ahuja, N.; He, Z.; Bhargava, R.; Edelstein, C.E.; Jani, A.; Hoke, T.S.; Klein, C.; et al. Cytokine production increases and cytokine clearance decreases in mice with bilateral nephrectomy. Nephrol. Dial. Transpl. 2012, 27, 4339–4347. [Google Scholar] [CrossRef]
- Ramirez, G.A.; Coletto, L.A.; Sciorati, C.; Bozzolo, E.P.; Manunta, P.; Rovere-Querini, P.; Manfredi, A.A. Ion Channels and Transporters in Inflammation: Special Focus on TRP Channels and TRPC6. Cells 2018, 7, 70. [Google Scholar] [CrossRef]
- Rom, S.; Heldt, N.A.; Gajghate, S.; Seliga, A.; Reichenbach, N.L.; Persidsky, Y. Author Correction: Hyperglycemia and advanced glycation end products disrupt BBB and promote occludin and claudin-5 protein secretion on extracellular microvesicles. Sci. Rep. 2020, 10, 18828. [Google Scholar] [CrossRef]
- Paul, D.; Baena, V.; Ge, S.; Jiang, X.; Jellison, E.R.; Kiprono, T.; Agalliu, D.; Pachter, J.S. Appearance of claudin-5(+) leukocytes in the central nervous system during neuroinflammation: A novel role for endothelial-derived extracellular vesicles. J. Neuroinflamm. 2016, 13, 292. [Google Scholar] [CrossRef] [PubMed]
- Baluna, R.; Vitetta, E.S. Vascular leak syndrome: A side effect of immunotherapy. Immunopharmacology 1997, 37, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Boghossian, N.S.; Sicko, R.J.; Kay, D.M.; Rigler, S.L.; Caggana, M.; Tsai, M.Y.; Yeung, E.H.; Pankratz, N.; Cole, B.R.; Druschel, C.M.; et al. Rare copy number variants implicated in posterior urethral valves. Am. J. Med. Genet. A 2016, 170, 622–633. [Google Scholar] [CrossRef]
- Sabbineni, H.; Verma, A.; Artham, S.; Anderson, D.; Amaka, O.; Liu, F.; Narayanan, S.P.; Somanath, P.R. Pharmacological inhibition of beta-catenin prevents EndMT in vitro and vascular remodeling in vivo resulting from endothelial Akt1 suppression. Biochem. Pharmacol. 2019, 164, 205–215. [Google Scholar] [CrossRef]
- Artham, S.; Verma, A.; Newsome, A.S.; Somanath, P.R. Patients with acute respiratory distress syndrome exhibit increased stromelysin1 activity in the blood samples. Cytokine 2020, 131, 155086. [Google Scholar] [CrossRef] [PubMed]
- Adil, M.S.; Somanath, P.R. Vascular Permeability Assays In Vivo. Methods Mol. Biol. 2020, 2367, 165–175. [Google Scholar] [CrossRef]
- Chen, J.; Somanath, P.R.; Razorenova, O.; Chen, W.S.; Hay, N.; Bornstein, P.; Byzova, T.V. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005, 11, 1188–1196. [Google Scholar] [CrossRef]
- Artham, S.; Gao, F.; Verma, A.; Alwhaibi, A.; Sabbineni, H.; Hafez, S.; Ergul, A.; Somanath, P.R. Endothelial stromelysin1 regulation by the forkhead box-O transcription factors is crucial in the exudative phase of acute lung injury. Pharmacol. Res. 2019, 141, 249–263. [Google Scholar] [CrossRef]
- Artham, S.; Verma, A.; Alwhaibi, A.; Adil, M.S.; Manicassamy, S.; Munn, D.H.; Somanath, P.R. Delayed Akt suppression in the lipopolysaccharide-induced acute lung injury promotes resolution that is associated with enhanced effector regulatory T cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L750–L761. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adil, M.S.; Parvathagiri, V.; Alanazi, A.H.; Khulood, D.; Narayanan, S.P.; Somanath, P.R. Claudin-17 Deficiency Drives Vascular Permeability and Inflammation Causing Lung Injury. Int. J. Mol. Sci. 2025, 26, 3612. https://doi.org/10.3390/ijms26083612
Adil MS, Parvathagiri V, Alanazi AH, Khulood D, Narayanan SP, Somanath PR. Claudin-17 Deficiency Drives Vascular Permeability and Inflammation Causing Lung Injury. International Journal of Molecular Sciences. 2025; 26(8):3612. https://doi.org/10.3390/ijms26083612
Chicago/Turabian StyleAdil, Mir S., Varun Parvathagiri, Abdulaziz H. Alanazi, Daulat Khulood, S. Priya Narayanan, and Payaningal R. Somanath. 2025. "Claudin-17 Deficiency Drives Vascular Permeability and Inflammation Causing Lung Injury" International Journal of Molecular Sciences 26, no. 8: 3612. https://doi.org/10.3390/ijms26083612
APA StyleAdil, M. S., Parvathagiri, V., Alanazi, A. H., Khulood, D., Narayanan, S. P., & Somanath, P. R. (2025). Claudin-17 Deficiency Drives Vascular Permeability and Inflammation Causing Lung Injury. International Journal of Molecular Sciences, 26(8), 3612. https://doi.org/10.3390/ijms26083612