
A Systematic Review of Endothelial Dysfunction in Chronic Venous Disease—Inflammation, Oxidative Stress, and Shear Stress
 , ,
, , 
Abstract
1. Introduction
2. Methods
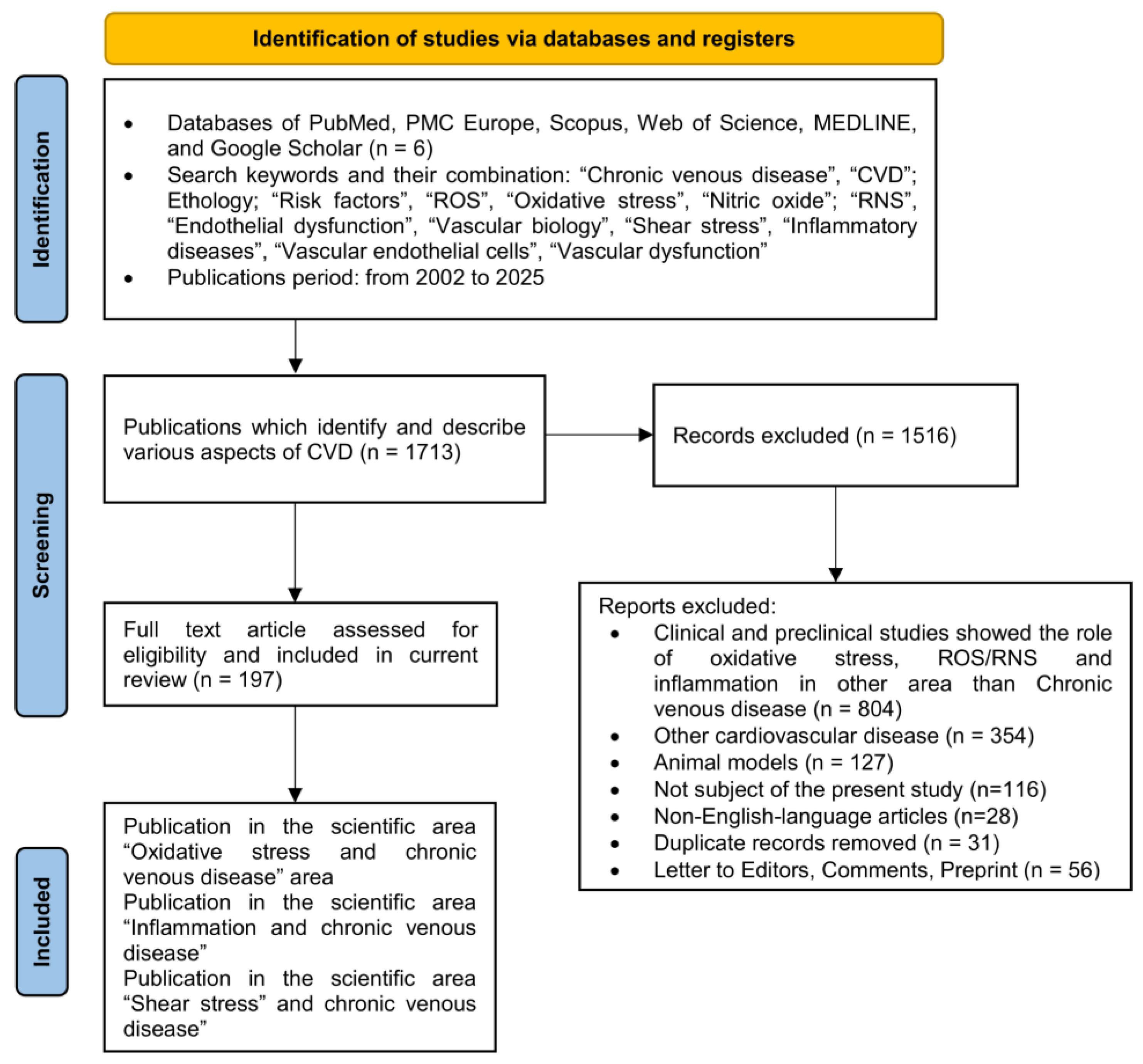
2.1. Literature Search and Data Collection Process
2.1.1. Inclusion Criteria
2.1.2. Exclusion Criteria
3. Results
3.1. CVD Etiology and CEAP Classification
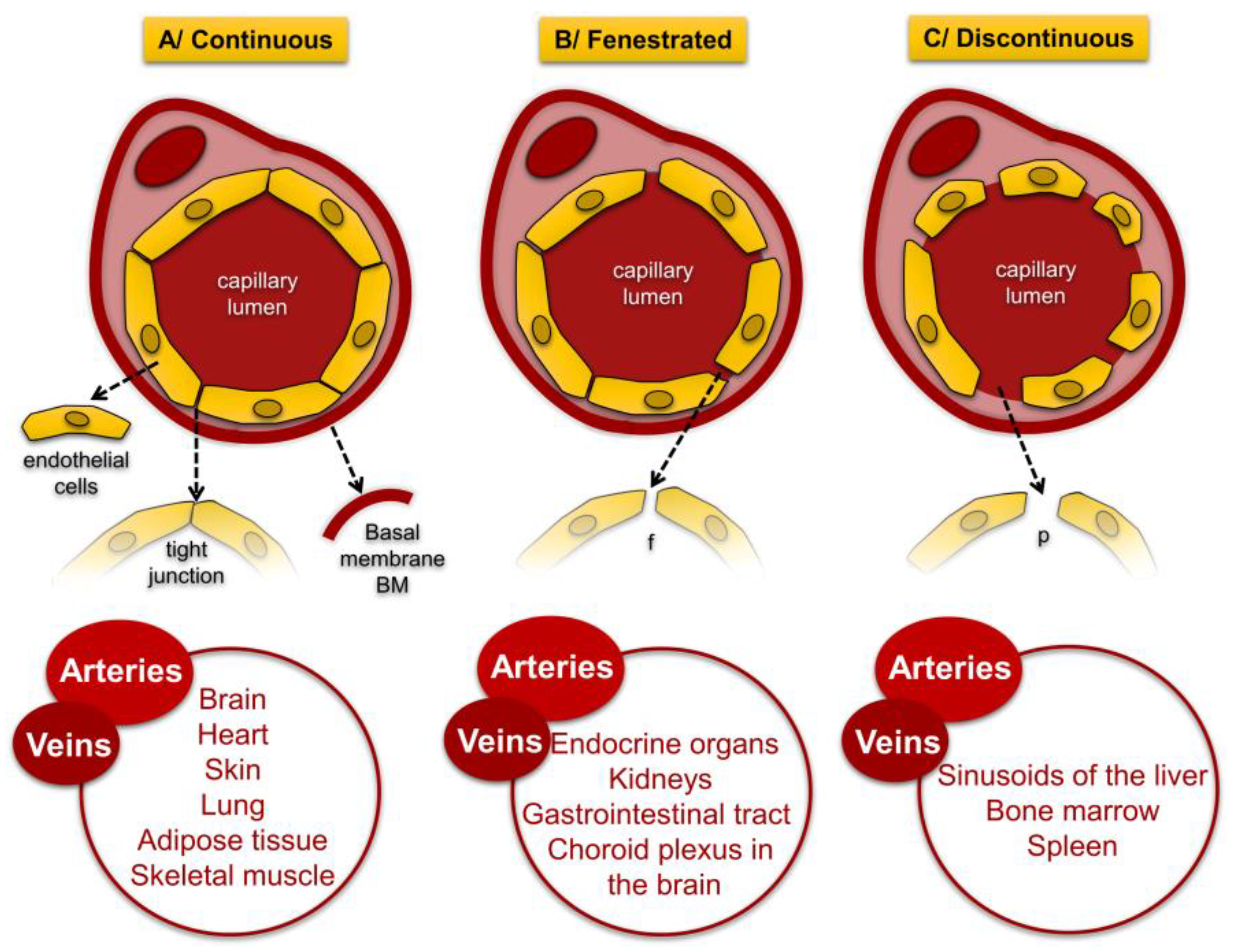
3.2. Structure of Venous Vessels
4. Morphology and Function of the Vascular Endothelium
4.1. Endothelial Cell Morphology
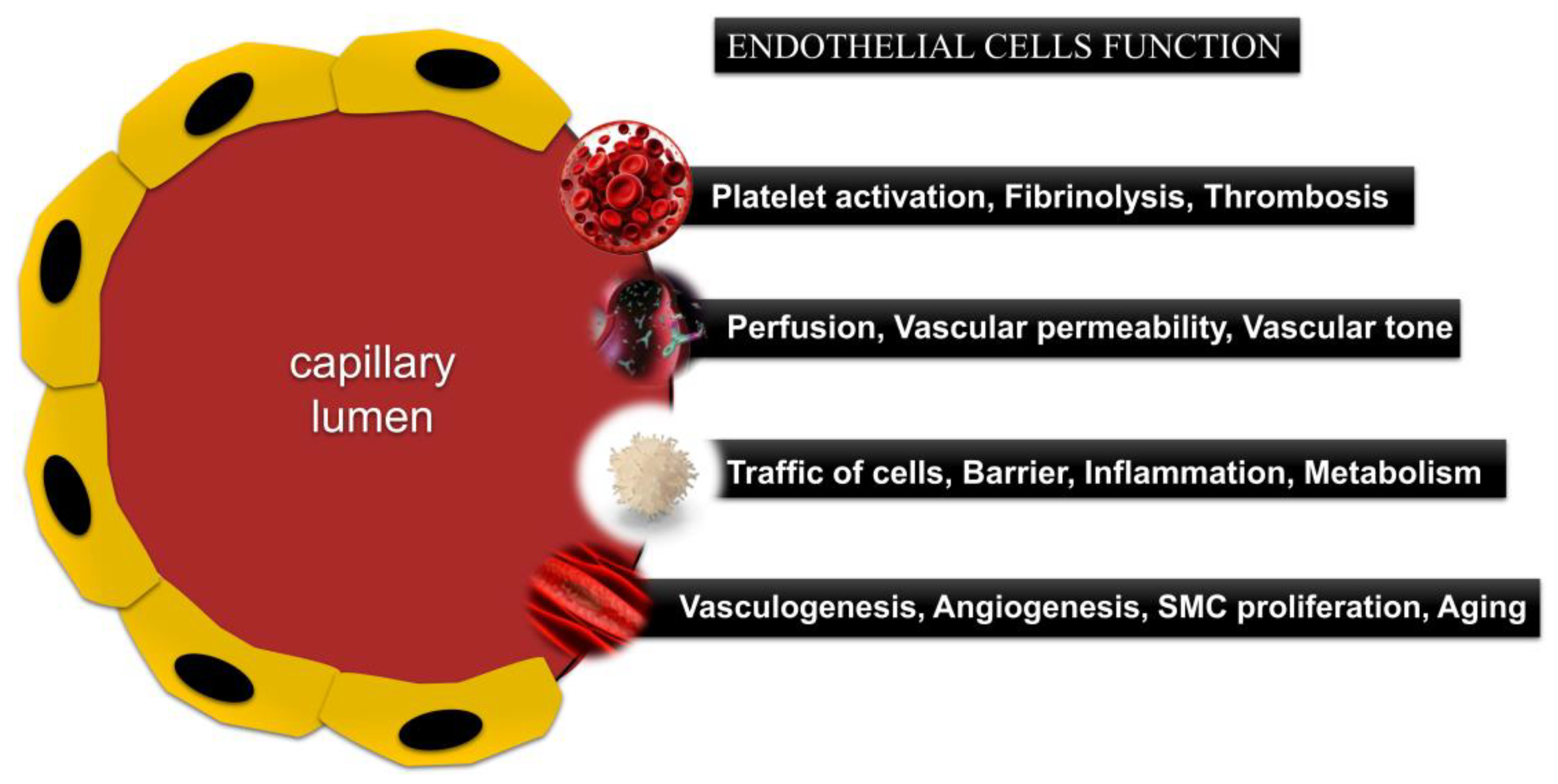
4.2. Functions of the Endothelium
4.3. Endothelial Dysfunction in CVD
4.3.1. Role of Inflammation in Pathogenesis of CVD
4.3.2. Role of ROS
4.3.3. Role of NO
4.4. Basement Membrane—Structure and Function
4.5. Shear Stress
5. Discussion
6. Therapeutic Aim and Strategy Outside of Mechanical Intervention
6.1. Venoactive Drugs with Inhibitory and Antioxidant Effects for Targeted Therapy
6.2. Personalized Medicine, Stratification and Assessment of the Risk of Complications
6.3. Prevention, Diet, and Physical Activity
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CVD | Chronic venous disease |
| ED | Endothelial dysfunction |
| OS | Oxidative stress |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| SS | Shear stress |
| VSMCs | Vascular smooth muscle cells |
| ECs | Endothelial cells |
| CEAP | Clinical-Etiology-Anatomy-Pathophysiology classification |
| DVT | Deep vein thrombosis |
| PTS | Post-thrombotic syndrome |
| BM | Basement membrane |
| NOS | Nitric oxide synthases |
References
- Netala, V.R.; Teertam, S.K.; Li, H.; Zhang, Z. A Comprehensive Review of Cardiovascular Disease Management: Cardiac Biomarkers, Imaging Modalities, Pharmacotherapy, Surgical Interventions, and Herbal Remedies. Cells 2024, 13, 1471. [Google Scholar] [CrossRef] [PubMed]
- Frąk, W.; Wojtasińska, A.; Lisińska, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines 2022, 10, 1938. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Hahad, O.; Lelieveld, J.; Aschner, M.; Nieuwenhuijsen, M.J.; Landrigan, P.J.; Daiber, A. Soil and water pollution and cardiovascular disease. Nat. Rev. Cardiol. 2025, 22, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Eklof, B.; Perrin, M.; Delis, K.T.; Rutherford, R.B.; Gloviczki, P. American Venous Forum; European Venous Forum; International Union of Phlebology; American College of Phlebology; International Union of Angiology. Updated terminology of chronic venous disorders: The VEIN-TERM transatlantic interdisciplinary consensus document. J. Vasc. Surg. 2009, 49, 498–501. [Google Scholar] [CrossRef]
- Criqui, M.H.; Jamosmos, M.; Fronek, A.; Denenberg, J.O.; Langer, R.D.; Bergan, J.; Golomb, B.A. Chronic venous disease in an ethnically diverse population: The San Diego Population Study. Am. J. Epidemiol. 2003, 158, 448–456. [Google Scholar] [CrossRef]
- Rabe, E.; Guex, J.J.; Puskas, A.; Scuderi, A. Epidemiology of chronic venous disorders in geographically diverse populations: Results from the Vein Consult Program. Int. Angiol. A J. Int. Union Angiol. 2012, 31, 105–115. [Google Scholar]
- Salim, S.; Machin, M.; Patterson, B.O.; Onida, S.; Davies, A.H. Global epidemiology of chronic venous disease: A systematic review with pooled prevalence analysis. Ann. Surg. 2021, 274, 971–976. [Google Scholar] [CrossRef]
- Preisner, K.; Hetjens, S. Risk Factors and Preventive Measures for Cardiovascular Diseases. J. Clin. Med. 2024, 13, 3308. [Google Scholar] [CrossRef]
- Bays, H.E. Ten things to know about ten cardiovascular disease risk factors (“ASPC Top Ten—2020”). Am. J. Prev. Cardiol. 2020, 1, 100003. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, W.; Liu, J.; Xie, M.; Liu, Q.; Li, S. Vascular complications of diabetes: A narrative review. Medicine 2023, 102, e35285. [Google Scholar] [CrossRef]
- Arrebola-Moreno, A.L.; Laclaustra, M.; Kaski, J.C. Noninvasive assessment of endothelial function in clinical practice. Rev. Esp. Cardiol. 2012, 65, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Fiore, A.; Masiglat, J.; Cavuoti, T.; Romandini, M.; Nappi, P.; Avtaar Singh, S.S.; Couetil, J.-P. Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review. Biomedicines 2022, 10, 2884. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef]
- Paolini, L.; Guida, F.; Calvaruso, A.; Andreozzi, L.; Pierantoni, L.; Lanari, M.; Fabi, M. Endothelial Dysfunction: Molecular Mechanisms and Therapeutic Strategies in Kawasaki Disease. Int. J. Mol. Sci. 2024, 25, 13322. [Google Scholar] [CrossRef]
- Immanuel, J.; Yun, S. Vascular Inflammatory Diseases and Endothelial Phenotypes. Cells 2023, 12, 1640. [Google Scholar] [CrossRef]
- Ambrosino, P.; Calcaterra, I.; Molino, A.; Moretta, P.; Lupoli, R.; Spedicato, G.A.; Papa, A.; Motta, A.; Maniscalco, M.; Di Minno, M.N.D. Persistent Endothelial Dysfunction in Post-Acute COVID-19 Syndrome: A Case-Control Study. Biomedicines 2021, 9, 957. [Google Scholar] [CrossRef]
- Castro-Ferreira, R.; Cardoso, R.; Leite-Moreira, A.; Mansilha, A. The Role of Endothelial Dysfunction and Inflammation in Chronic Venous Disease. Ann. Vasc. Surg. 2018, 46, 380–393. [Google Scholar] [CrossRef]
- Batinac, T.; Batičić, L.; Kršek, A.; Knežević, D.; Marcucci, E.; Sotošek, V.; Ćurko-Cofek, B. Endothelial Dysfunction and Cardiovascular Disease: Hyperbaric Oxygen Therapy as an Emerging Therapeutic Modality? J. Cardiovasc. Dev. Dis. 2024, 11, 408. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Aldosari, S.; Awad, M.; Harrington, E.O.; Sellke, F.W.; Abid, M.R. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7, 14. [Google Scholar] [CrossRef]
- Schulz, E.; Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2004, 287, R1014–R1030. [Google Scholar] [CrossRef] [PubMed]
- Yuyun, M.F.; Ng, L.L.; Ng, G.A. Endothelial dysfunction, endothelial nitric oxide bioavailability, tetrahydrobiopterin, and 5-methyltetrahydrofolate in cardiovascular disease. Where are we with therapy? Microvasc. Res. 2018, 119, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y. Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease. Antioxidants 2022, 11, 1958. [Google Scholar] [CrossRef]
- Panda, P.; Verma, H.K.; Lakkakula, S.; Merchant, N.; Kadir, F.; Rahman, S.; Jeffree, M.S.; Lakkakula, B.V.K.S.; Rao, P.V. Biomarkers of Oxidative Stress Tethered to Cardiovascular Diseases. Oxid. Med. Cell Longev. 2022, 2022, 9154295. [Google Scholar] [CrossRef]
- Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Nitration of Proteins, Lipids and DNA by Peroxynitrite Derivatives-Chemistry Involved and Biological Relevance. Stresses 2022, 2, 53–64. [Google Scholar] [CrossRef]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free. Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef]
- Tain, Y.-L.; Hsu, C.-N. Oxidative Stress-Induced Hypertension of Developmental Origins: Preventive Aspects of Antioxidant Therapy. Antioxidants 2022, 11, 511. [Google Scholar] [CrossRef]
- Vattemi, G.; Mechref, Y.; Marini, M.; Tonin, P.; Minuz, P.; Grigoli, L.; Guglielmi, V.; Klouckova, I.; Chiamulera, C.; Meneguzzi, A.; et al. Increased protein nitration in mitochondrial diseases: Evidence for vessel wall involvement. Mol. Cell. Proteom. 2011, 10, M110.002964. [Google Scholar] [CrossRef] [PubMed]
- Fels, B.; Kusche-Vihrog, K. It takes more than two to tango: Mechanosignaling of the endothelial surface. Pflug. Arch.-Eur. J. Physiol. 2020, 472, 419–433. [Google Scholar] [CrossRef]
- Ando, J.; Yamamoto, K. Hemodynamic forces, endothelial mechanotransduction, and vascular diseases. Magn. Reson. Med. Sci. 2022, 21, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA statement. Ann. Intern. Med. 2009, 151, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Lurie, F.; Passman, M.; Meisner, M.; Dalsing, M.; Masuda, E.; Welch, H.; Bush, R.L.; Blebea, J.; Carpentier, P.H.; De Maeseneer, M.; et al. The 2020 update of the CEAP classification system and reporting standards. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Saleem, T. An Overview of Specific Considerations in Chronic Venous Disease and Iliofemoral Venous Stenting. J. Pers. Med. 2023, 13, 331. [Google Scholar] [CrossRef]
- Kistner, R.L.; Eklöf, B.O. Classification and etiology of chronic venous disease. In Handbook of Venous and Lymphatic Disorders; CRC Press: Boca Raton, FL, USA, 2017; pp. 39–49. [Google Scholar]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases Writing Group. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J. Menopausal Hormone Replacement Therapy and Reduction of All-Cause Mortality and Cardiovascular Disease: It Is About Time and Timing. Cancer J. 2022, 28, 208–223. [Google Scholar] [CrossRef]
- Machuca, J.N.; Rosales-Alvarez, C.P. Cardiovascular Disease in Women and the Role of Hormone Replacement Therapy. Cureus 2024, 16, e69752. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; Álvarez-Mon, M.A.; Chaowen, C.; Ruiz-Grande, F.; Pekarek, L.; Monserrat, J.; Asúnsolo, A.; García-Honduvilla, N.; et al. Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management. J. Clin. Med. 2021, 10, 3239. [Google Scholar] [CrossRef]
- Mangwani, J.; Roberts, V.; Shannak, O.; Divall, P.; Srinivasan, A.; Dias, J. Epidemiology and Diagnosis of Post-Thrombotic Syndrome: Qualitative Synthesis with a Systematic Review. J. Clin. Med. 2023, 12, 5896. [Google Scholar] [CrossRef]
- Meissner, M.H.; Eklof, B.; Smith, P.C.; Dalsing, M.C.; DePalma, R.G.; Gloviczki, P.; Moneta, G.; Neglén, P.; O’ Donnell, T.; Partsch, H.; et al. Secondary chronic venous disorders. J. Vasc. Surg. 2007, 46, S68–S83. [Google Scholar] [CrossRef] [PubMed]
- Ozcinar, E.; Dikmen, N.; Kayan, A.; Kandemir, M.; Saricaoglu, M.C. Pharmacomechanical Thrombectomy and Catheter-Directed Thrombolysis, with or without Iliac Vein Stenting, in the Treatment of Acute Iliofemoral Deep Vein Thrombosis. J. Cardiovasc. Dev. Dis. 2024, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Galanaud, J.P.; Bertoletti, L.; Amitrano, M.; Fernández-Capitán, C.; Pedrajas, J.M.; Rosa, V.; Barrón, M.; Lorenzo, A.; Madridano, O.; Quéré, I.; et al. Predictors of Post-Thrombotic Ulcer after Acute DVT: The RIETE Registry. Thromb. Haemost. 2018, 118, 320–328. [Google Scholar] [CrossRef]
- Kahn, S.R.; Galanaud, J.P.; Vedantham, S.; Ginsberg, J.S. Guidance for the prevention and treatment of the post-thrombotic syndrome. J. Thromb. Thrombolysis 2016, 41, 144–153. [Google Scholar] [CrossRef]
- Santler, B.; Goerge, T. Chronic venous insufficiency—A review of pathophysiology, diagnosis, and treatment. J. Dtsch. Dermatol. Ges. 2017, 15, 538–556. [Google Scholar] [CrossRef]
- Mangiafico, M.; Costanzo, L. Superficial Venous Thrombosis: A Comprehensive Review. Healthcare 2024, 12, 500. [Google Scholar] [CrossRef]
- Anwar, M.A.; Georgiadis, K.A.; Shalhoub, J.; Lim, C.S.; Gohel, M.S.; Davies, A.H. A review of familial, genetic, and congenital aspects of primary varicose vein disease. Circ. Cardiovasc. Genet. 2012, 5, 460–466. [Google Scholar] [CrossRef]
- Banzic, I.; Brankovic, M.; Maksimović, Ž.; Davidović, L.; Marković, M.; Rančić, Z. Parkes Weber syndrome-Diagnostic and management paradigms: A systematic review. Phlebology 2017, 32, 371–383. [Google Scholar] [CrossRef]
- Mansour, S.; Brice, G.W.; Jeffery, S.; Mortimer, P. Lymphedema-Distichiasis Syndrome; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Mansour, S., Brice, G.W., Jeffery, S., Mortimer, P., Eds.; University of Washington: Seattle, WA, USA, 2005. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1457/ (accessed on 29 March 2005).
- Wilkinson, N.R.; Cervi, E.; Wagner, B.; Morris-Rosendahl, D.; Baker, D.; Flora, H.; von Klemperer, K.; Andrew, T.; Ghali, N.; van Dijk, F.S. Vascular Ehlers-Danlos syndrome in children: Evaluating the importance of diagnosis and follow-up during childhood. Eur. J. Hum. Genet. 2024, 33, 368–376. [Google Scholar] [CrossRef]
- Henderson, A.R.; Choi, H.; Lee, E. Blood and Lymphatic Vasculatures On-Chip Platforms and Their Applications for Organ-Specific In Vitro Modeling. Micromachines 2020, 11, 147. [Google Scholar] [CrossRef]
- Hennigs, J.K.; Matuszcak, C.; Trepel, M.; Körbelin, J. Vascular Endothelial Cells: Heterogeneity and Targeting Approaches. Cells 2021, 10, 2712. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y. Architecture of the Blood Vessels. In Biology of Vascular Smooth Muscle; Springer: Singapore, 2022; pp. 3–17. [Google Scholar] [CrossRef]
- Kazemzadeh, G.; Jirofti, N.; Kazemi Mehrjerdi, H.; Rajabioun, M.; Alamdaran, S.A.; Mohebbi-Kalhori, D.; Mirbagheri, M.S.; Taheri, R. A review on developments of in-vitro and in-vivo evaluation of hybrid PCL-based natural polymers nanofibers scaffolds for vascular tissue engineering. J. Ind. Text. 2022, 52, 15280837221128314. [Google Scholar] [CrossRef]
- Munjal, A.; Bordoni, B. Histology, Vascular. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554407/ (accessed on 9 April 2025).
- Hernández-Cabré, L.; Ulldemolins-Rams, M.; Vilanova-Corsellas, J.; Torras, C. Effects of Channelling a Peripherally Inserted Central Venous Catheter on Blood Flow. Fluids 2024, 9, 245. [Google Scholar] [CrossRef]
- Camasão, D.B.; Mantovani, D.J.M.T.B. The mechanical characterization of blood vessels and their substitutes in the continuous quest for physiological-relevant performances. A critical review. Mater. Today Bio 2021, 10, 100106. [Google Scholar] [CrossRef]
- Rodrigues, S.F.; Granger, D.N. Blood cells and endothelial barrier function. Tissue Barriers 2015, 3, e978720. [Google Scholar] [CrossRef]
- Larionov, A.; Hammer, C.M.; Fiedler, K.; Filgueira, L. Dynamics of Endothelial Cell Diversity and Plasticity in Health and Disease. Cells 2024, 13, 1276. [Google Scholar] [CrossRef]
- Aquino, J.B.; Sierra, R.; Montaldo, L.A. Diverse cellular origins of adult blood vascular endothelial cells. Dev. Biol. 2021, 477, 117–132. [Google Scholar] [CrossRef]
- Inglebert, M.; Locatelli, L.; Tsvirkun, D.; Sinha, P.; Maier, J.A.; Misbah, C.; Bureau, L. The effect of shear stress reduction on endothelial cells: A microfluidic study of the actin cytoskeleton. Biomicrofluidics 2020, 14, 024115. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Savkovic, V.; Li, H.; Obradovic, D.; Masieri, F.F.; Bartella, A.K.; Zimmerer, R.; Simon, J.-C.; Etz, C.; Lethaus, B. The Angiogenic Potential of Mesenchymal Stem Cells from the Hair Follicle Outer Root Sheath. J. Clin. Med. 2021, 10, 911. [Google Scholar] [CrossRef]
- Félétou, M. The Endothelium, Part I: Multiple Functions of the Endothelial Cells--Focus on Endothelium-Derived Vasoactive Mediators; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011; pp. 1–306. [Google Scholar]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Kiouptsi, K.; Reinhardt, C. Physiological roles of the von Willebrand factor-factor VIII interaction. In Vertebrate and Invertebrate Respiratory Proteins, Lipoproteins and other Body Fluid Proteins; Springer: Berlin/Heidelberg, Germany; pp. 437–464. [CrossRef]
- Filippini, A.; Tamagnone, L.; D’Alessio, A. Endothelial Cell Metabolism in Vascular Functions. Cancers 2022, 14, 1929. [Google Scholar] [CrossRef] [PubMed]
- Mojzisch, A.; Brehm, M.A. The Manifold Cellular Functions of von Willebrand Factor. Cells 2021, 10, 2351. [Google Scholar] [CrossRef] [PubMed]
- Beds, I.R.V. Phenotypic heterogeneity of the endothelium. System 2007, 100, 174–190. [Google Scholar] [CrossRef]
- Wolf, K.; Hu, H.; Isaji, T.; Dardik, A. Molecular identity of arteries, veins, and lymphatics. J. Vasc. Surg. 2019, 69, 253–262. [Google Scholar] [CrossRef]
- Foo, S.S.; Turner, C.J.; Adams, S.; Compagni, A.; Aubyn, D.; Kogata, N.; Lindblom, P.; Shani, M.; Zicha, D.; Adams, R.H. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 2006, 124, 161–173. [Google Scholar] [CrossRef]
- Citrin, K.M.; Chaube, B.; Fernández-Hernando, C.; Suárez, Y. Intracellular endothelial cell metabolism in vascular function and dysfunction. Trends Endocrinol. Metab. 2024. [Google Scholar] [CrossRef]
- Nguyen, J.; Lin, Y.Y.; Gerecht, S. The next generation of endothelial differentiation: Tissue-specific ECs. Cell Stem Cell 2021, 28, 1188–1204. [Google Scholar] [CrossRef]
- Mou, X.; Leeman, S.M.; Roye, Y.; Miller, C.; Musah, S. Fenestrated Endothelial Cells across Organs: Insights into Kidney Function and Disease. Int. J. Mol. Sci. 2024, 25, 9107. [Google Scholar] [CrossRef]
- Sluiter, T.J.; van Buul, J.D.; Huveneers, S.; Quax, P.H.A.; de Vries, M.R. Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis. Biomedicines 2021, 9, 328. [Google Scholar] [CrossRef]
- Bkaily, G.; Jacques, D. Morphological and Functional Remodeling of Vascular Endothelium in Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 1998. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M. Chapter 2, Multiple Functions of the Endothelial Cells. In The Endothelium, Part I: Multiple Functions of the Endothelial Cells--Focus on Endothelium-Derived Vasoactive Mediators; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011. Available online: https://www.ncbi.nlm.nih.gov/books/NBK57148/ (accessed on 1 January 2011).
- Ray, A.; Maharana, K.C.; Meenakshi, S.; Singh, S. Endothelial dysfunction and its relation in different disorders: Recent update. Health Sci. Rev. 2023, 7, 100084. [Google Scholar] [CrossRef]
- Poredos, P.; Jezovnik, M.K. Endothelial dysfunction and venous thrombosis. Angiology 2018, 69, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef]
- Barrera-Vázquez, O.S.; Escobar-Ramírez, J.L.; Santiago-Mejía, J.; Carrasco-Ortega, O.F.; Magos-Guerrero, G.A. Discovering Potential Compounds for Venous Disease Treatment through Virtual Screening and Network Pharmacology Approach. Molecules 2023, 28, 7937. [Google Scholar] [CrossRef]
- Gwozdzinski, L.; Pieniazek, A.; Bernasinska-Slomczewska, J.; Hikisz, P.; Gwozdzinski, K. Alterations in the Plasma and Red Blood Cell Properties in Patients with Varicose Vein: A Pilot Study. Cardiol. Res. Pract. 2021, 2021, 5569961. [Google Scholar] [CrossRef]
- Ghaderian, S.M.H.; Khodaii, Z. Tissue remodeling investigation in varicose veins. Int. J. Mol. Cell. Med. 2012, 1, 50. [Google Scholar]
- Münzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Siekierzycka, A.; Płoska, A.; Dobrucki, I.T.; Kalinowski, L. Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules 2021, 11, 982. [Google Scholar] [CrossRef]
- Balta, S. Endothelial dysfunction and inflammatory markers of vascular disease. Curr. Vasc. Pharmacol. 2021, 19, 243–249. [Google Scholar] [CrossRef]
- Cheng, H.; Zhong, W.; Wang, L.; Zhang, Q.; Ma, X.; Wang, Y.; Wang, S.; He, C.; Wei, Q.; Fu, C. Effects of shear stress on vascular endothelial functions in atherosclerosis and potential therapeutic approaches. Biomed. Pharmacother. 2023, 158, 114198. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, A.; Shishkova, D.; Markova, V.; Markova, Y.; Frolov, A.; Lazebnaya, A.; Oshchepkova, K.; Perepletchikova, D.; Smirnova, D.; Basovich, L.; et al. Proteomic Profiling of Endothelial Cell Secretomes After Exposure to Calciprotein Particles Reveals Downregulation of Basement Membrane Assembly and Increased Release of Soluble CD59. Int. J. Mol. Sci. 2024, 25, 11382. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease–a 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef]
- Peng, Z.; Shu, B.; Zhang, Y.; Wang, M. Endothelial response to pathophysiological stress. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e233–e243. [Google Scholar] [CrossRef]
- Dri, E.; Lampas, E.; Lazaros, G.; Lazarou, E.; Theofilis, P.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Endothelial Dysfunction. Life 2023, 13, 1420. [Google Scholar] [CrossRef]
- Zhao, K.; Zeng, Z.; He, Y.; Zhao, R.; Niu, J.; Sun, H.; Li, S.; Dong, J.; Jing, Z.; Zhou, J. Recent advances in targeted therapy for inflammatory vascular diseases. J. Control. Release 2024, 372, 730–750. [Google Scholar] [CrossRef]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.F. An evolving new paradigm: Endothelial cells—Conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61. [Google Scholar] [CrossRef]
- de Oliveira, M.G.; Nadruz Jr, W.; Monica, F.Z. Endothelial and vascular smooth muscle dysfunction in hypertension. Biochem. Pharmacol. 2022, 205, 115263. [Google Scholar] [CrossRef]
- Birnhuber, A.; Fliesser, E.; Gorkiewicz, G.; Zacharias, M.; Seeliger, B.; David, S.; Welte, T.; Schmidt, J.; Olschewski, H.; Wygrecka, M.; et al. Between inflammation and thrombosis: Endothelial cells in COVID-19. Eur. Respir. J. 2021, 58, 2100377. [Google Scholar] [CrossRef]
- Carter, K.; Shah, E.; Waite, J.; Rana, D.; Zhao, Z.-Q. Pathophysiology of Angiotensin II-Mediated Hypertension, Cardiac Hypertrophy, and Failure: A Perspective from Macrophages. Cells 2024, 13, 2001. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Khalil, R.A. Mechanisms of Lower Extremity Vein Dysfunction in Chronic Venous Disease and Implications in Management of Varicose Veins. Vessel. Plus. 2021, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- del Rio Solá, L.; Aceves, M.; Dueñas, A.I.; González-Fajardo, J.A.; Vaquero, C.; Crespo, M.S.; García-Rodríguez, C. Varicose veins show enhanced chemokine expression. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.R.; Yerly, A.; van der Vorst, E.P.; Baumgartner, I.; Bernhard, S.M.; Schindewolf, M.; Döring, Y. Inflammatory mediators in atherosclerotic vascular remodeling. Front. Cardiovasc. Med. 2022, 9, 868934. [Google Scholar] [CrossRef]
- Fraile-Martinez, O.; García-Montero, C.; Gomez-Lahoz, A.M.; Sainz, F.; Bujan, J.; Barrena-Blázquez, S.; López-González, L.; Díaz-Pedrero, R.; Álvarez-Mon, M.; García-Honduvilla, N.; et al. Evidence of Inflammatory Network Disruption in Chronic Venous Disease: An Analysis of Circulating Cytokines and Chemokines. Biomedicines 2025, 13, 150. [Google Scholar] [CrossRef]
- Alias, S.; Lang, I.M. Coagulation and the vessel wall in pulmonary embolism. Pulm. Circ. 2013, 3, 728–738. [Google Scholar] [CrossRef]
- Chang, J.C. Novel Classification of Thrombotic Disorders Based on Molecular Hemostasis and Thrombogenesis Producing Primary and Secondary Phenotypes of Thrombosis. Biomedicines 2022, 10, 2706. [Google Scholar] [CrossRef]
- Niculae, C.-M.; Hristea, A.; Moroti, R. Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review. Biomedicines 2023, 11, 929. [Google Scholar] [CrossRef]
- Chang, J.C.; Hawley, H.B. Vaccine-Associated Thrombocytopenia and Thrombosis: Venous Endotheliopathy Leading to Venous Combined Micro-Macrothrombosis. Medicina 2021, 57, 1163. [Google Scholar] [CrossRef]
- Kozlov, A.V.; Javadov, S.; Sommer, N. Cellular ROS and Antioxidants: Physiological and Pathological Role. Antioxidants 2024, 13, 602. [Google Scholar] [CrossRef]
- Khan, M.; Ali, S.; Al Azzawi, T.N.I.; Saqib, S.; Ullah, F.; Ayaz, A.; Zaman, W. The Key Roles of ROS and RNS as a Signaling Molecule in Plant–Microbe Interactions. Antioxidants 2023, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Abdelazim, A.M.; Abomughaid, M.M. Oxidative stress: An overview of past research and future insights. All Life 2024, 17, 2316092. [Google Scholar] [CrossRef]
- Panayotova, M. Oxidative stress and inflammatory bowel disease in pediatrics. Trakia J. Sci 2023, 21, 375. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Soto, M.E.; Castrejón-Tellez, V.; Rubio-Ruiz, M.E.; Manzano Pech, L.; Guarner-Lans, V. Oxidative, reductive, and nitrosative stress effects on epigenetics and on posttranslational modification of enzymes in cardiometabolic diseases. Oxidative Med. Cell. Longev. 2020, 1, 8819719. [Google Scholar] [CrossRef]
- Smetanina, M.A.; Oscorbin, I.P.; Shadrina, A.S.; Sevost’ianova, K.S.; Korolenya, V.A.; Gavrilov, K.A.; Shevela, A.I.; Shirshova, A.N.; Oskina, N.A.; Zolotukhin, I.A.; et al. Quantitative and structural characteristics of mitochondrial DNA in varicose veins. Vasc. Pharmacol. 2022, 145, 107021. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437–478. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef]
- Cipriano, A.; Viviano, M.; Feoli, A.; Milite, C.; Sarno, G.; Castellano, S.; Sbardella, G. NADPH oxidases: From molecular mechanisms to current inhibitors. J. Med. Chem. 2023, 66, 11632–11655. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.; Briones, A. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular mechanisms of ischemia–reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Meephat, S.; Prasatthong, P.; Potue, P.; Bunbupha, S.; Pakdeechote, P.; Maneesai, P. Diosmetin Ameliorates Vascular Dysfunction and Remodeling by Modulation of Nrf2/HO-1 and p-JNK/p-NF-κB Expression in Hypertensive Rats. Antioxidants 2021, 10, 1487. [Google Scholar] [CrossRef]
- Minjares, M.; Wu, W.; Wang, J.-M. Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders. Cells 2023, 12, 1341. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, J. Deciphering Oxidative Stress in Cardiovascular Disease Progression: A Blueprint for Mechanistic Understanding and Therapeutic Innovation. Antioxidants 2025, 14, 38. [Google Scholar] [CrossRef]
- He, M.; Wang, D.; Xu, Y.; Jiang, F.; Zheng, J.; Feng, Y.; Cao, J.; Zhou, X. Nitric Oxide-Releasing Platforms for Treating Cardiovascular Disease. Pharmaceutics 2022, 14, 1345. [Google Scholar] [CrossRef]
- Carlström, M.; Weitzberg, E.; Lundberg, J.O. Nitric Oxide Signaling and Regulation in the Cardiovascular System: Recent Advances. Pharmacol. Rev. 2024, 76, 1038–1062. [Google Scholar] [CrossRef]
- Chandimali, N.; Bak, S.G.; Park, E.H.; Lim, H.J.; Won, Y.S.; Kim, E.K.; Park, S.I.; Lee, S.J. Free radicals and their impact on health and antioxidant defenses: A review. Cell Death Discov. 2025, 11, 19. [Google Scholar] [CrossRef]
- Zhu, H.-Y.; Hong, F.-F.; Yang, S.-L. The Roles of Nitric Oxide Synthase/Nitric Oxide Pathway in the Pathology of Vascular Dementia and Related Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 4540. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; De Leon-Oliva, D.; Gimeno-Longas, M.J.; Boaru, D.L.; Fraile-Martinez, O.; García-Montero, C.; de Castro, A.V.; Barrena-Blázquez, S.; López-González, L.; Amor, S.; et al. Vascular Calcification: Molecular Networking, Pathological Implications and Translational Opportunities. Biomolecules 2024, 14, 275. [Google Scholar] [CrossRef]
- Triggle, C.R.; Samuel, S.M.; Ravishankar, S.; Marei, I.; Arunachalam, G.; Ding, H. The endothelium: Influencing vascular smooth muscle in many ways. Can. J. Physiol. Pharmacol. 2012, 90, 713–738. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular oxidative stress: Impact and therapeutic approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, H.; Tang, B.; Luo, Y.; Yang, Y.; Zhong, X.; Chen, S.; Xu, X.; Huang, S.; Liu, C. Macrophages in cardiovascular diseases: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 130. [Google Scholar] [CrossRef]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C., IV; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular endothelial cells and innate immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Valaitienė, J.; Laučytė-Cibulskienė, A. Oxidative Stress and Its Biomarkers in Cardiovascular Diseases. Artery Res. 2024, 30, 18. [Google Scholar] [CrossRef]
- Penna, C.; Pagliaro, P. Endothelial Dysfunction: Redox Imbalance, NLRP3 Inflammasome, and Inflammatory Responses in Cardiovascular Diseases. Antioxidants 2025, 14, 256. [Google Scholar] [CrossRef]
- Kulovic-Sissawo, A.; Tocantins, C.; Diniz, M.S.; Weiss, E.; Steiner, A.; Tokic, S.; Madreiter-Sokolowski, C.T.; Pereira, S.P.; Hiden, U. Mitochondrial Dysfunction in Endothelial Progenitor Cells: Unraveling Insights from Vascular Endothelial Cells. Biology 2024, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef]
- Urbanczyk, M.; Zbinden, A.; Schenke-Layland, K. Organ-specific endothelial cell heterogenicity and its impact on regenerative medicine and biomedical engineering applications. Adv. Drug Deliv. Rev. 2022, 186, 114323. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflügers Arch.-Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef]
- Wilkie, I.C. Basement Membranes, Brittlestar Tendons, and Their Mechanical Adaptability. Biology 2024, 13, 375. [Google Scholar] [CrossRef]
- LeBleu, V.S.; MacDonald, B.; Kalluri, R. Structure and function of basement membranes. Exp. Biol. Med. 2007, 232, 1121–1129. [Google Scholar] [CrossRef]
- Jayadev, R.; Sherwood, D.R. Basement membranes. Curr. Biol. 2017, 27, R207–R211. [Google Scholar] [CrossRef]
- Sekiguchi, R.; Yamada, K.M. Basement membranes in development and disease. Curr Top Dev Biol. 2018, 130, 143–191. [Google Scholar] [CrossRef]
- Ishihara, J.; Ishihara, A.; Fukunaga, K.; Sasaki, K.; White, M.J.; Briquez, P.S.; Hubbell, J.A. Laminin heparin-binding peptides bind to several growth factors and enhance diabetic wound healing. Nat. Commun. 2018, 9, 2163. [Google Scholar] [CrossRef] [PubMed]
- Urschel, K.; Tauchi, M.; Achenbach, S.; Dietel, B. Investigation of Wall Shear Stress in Cardiovascular Research and in Clinical Practice—From Bench to Bedside. Int. J. Mol. Sci. 2021, 22, 5635. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, F.; Mai, P.; Peng, Y.; Shu, X.; Nie, R.; Zhang, H. Mechanism Analysis of Vascular Calcification Based on Fluid Dynamics. Diagnostics 2023, 13, 2632. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Investig. 2005, 85, 9–23. [Google Scholar] [CrossRef]
- Marziano, C.; Genet, G.; Hirschi, K.K. Vascular endothelial cell specification in health and disease. Angiogenesis 2021, 24, 213–236. [Google Scholar] [CrossRef]
- Davis, M.J.; Earley, S.; Li, Y.S.; Chien, S. Vascular mechanotransduction. Physiol. Rev. 2023, 103, 1247–1421. [Google Scholar] [CrossRef]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hellenic J. Cardiol. 2005, 46, 9–15. [Google Scholar]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef]
- Garoffolo, G.; Pesce, M. Mechanotransduction in the Cardiovascular System: From Developmental Origins to Homeostasis and Pathology. Cells 2019, 8, 1607. [Google Scholar] [CrossRef]
- Hamrangsekachaee, M.; Wen, K.; Yazdani, N.; Willits, R.K.; Bencherif, S.A.; Ebong, E.E. Endothelial glycocalyx sensitivity to chemical and mechanical sub-endothelial substrate properties. Front. Bioeng. Biotechnol. 2023, 11, 1250348. [Google Scholar] [CrossRef]
- Bryan, M.T.; Duckles, H.; Feng, S.; Hsiao, S.T.; Kim, H.R.; Serbanovic-Canic, J.; Evans, P.C. Mechanoresponsive networks controlling vascular inflammation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2199–2205. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zheng, X.; Lin, S.; Zhang, Y.; Wu, J.; Li, Y. Mechanotransduction regulates the interplays between alveolar epithelial and vascular endothelial cells in lung. Front. Physiol. 2022, 13, 818394. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.A.; Uryash, A.; Lopez, J.R. Non-Invasive Pulsatile Shear Stress Modifies Endothelial Activation; A Narrative Review. Biomedicines 2022, 10, 3050. [Google Scholar] [CrossRef]
- Wang, X.; He, B. Endothelial dysfunction: Molecular mechanisms and clinical implications. MedComm 2024, 5, e651. [Google Scholar] [CrossRef]
- Milošević, N.; Rütter, M.; David, A. Endothelial cell adhesion molecules-(un) Attainable targets for nanomedicines. Front. Med. Technol. 2022, 4, 846065. [Google Scholar] [CrossRef]
- Li, Y.; Chen, M.; Chang, W. Roles of the nucleus in leukocyte migration. J. Leukoc. Biol. 2022, 112, 771–783. [Google Scholar] [CrossRef]
- Medrano-Bosch, M.; Simón-Codina, B.; Jiménez, W.; Edelman, E.R.; Melgar-Lesmes, P. Monocyte-endothelial cell interactions in vascular and tissue remodeling. Front. Immunol. 2023, 14, 1196033. [Google Scholar] [CrossRef]
- Virdis, A.; Dell’Agnello, U.; Taddei, S. Impact of inflammation on vascular disease in hypertension. Maturitas 2014, 78, 179–183. [Google Scholar] [CrossRef]
- Stephan, D.; Roger, A.; Aghzadi, J.; Carmona, S.; Picard, C.; Dales, J.-P.; Desplat-Jégo, S. TWEAK and TNFα, Both TNF Ligand Family Members and Multiple Sclerosis-Related Cytokines, Induce Distinct Gene Response in Human Brain Microvascular Endothelial Cells. Genes 2022, 13, 1714. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Ojdana, D.; Safiejko, K.; Lipska, A.; Sacha, P.; Wieczorek, P.; Radziwon, P.; Dadan, J.; Tryniszewska, E. The inflammatory reaction during chronic venous disease of lower limbs. Folia Histochem. Et Cytobiol. 2009, 47, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dhalla, N.S. The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 1082. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, F.; Zhu, L.; Yang, H.; Pan, D.; Liu, Y.; Qu, X.; Gu, Y.; Li, X.; Chen, S. Low shear stress induces endothelial cell apoptosis and monocyte adhesion by upregulating PECAM 1 expression. Mol. Med. Rep. 2020, 21, 2580–2588. [Google Scholar] [CrossRef] [PubMed]
- Smetanina, M.A.; Korolenya, V.A.; Kel, A.E.; Sevostyanova, K.S.; Gavrilov, K.A.; Shevela, A.I.; Filipenko, M.L. Epigenome-Wide Changes in the Cell Layers of the Vein Wall When Exposing the Venous Endothelium to Oscillatory Shear Stress. Epigenomes 2023, 7, 8. [Google Scholar] [CrossRef]
- Pickett, J.R.; Wu, Y.; Zacchi, L.F.; Ta, H.T. Targeting endothelial vascular cell adhesion molecule-1 in atherosclerosis: Drug discovery and development of vascular cell adhesion molecule-1–directed novel therapeutics. Cardiovasc. Res. 2023, 119, 2278–2293. [Google Scholar] [CrossRef]
- Singh, A.; Zahra, F. Chronic Venous Insufficiency. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK587341/ (accessed on 27 April 2023).
- Anuforo, A.; Evbayekha, E.; Agwuegbo, C.; Okafor, T.L.; Antia, A.; Adabale, O.; Ugoala, O.S.; Okorare, O.; Phagoora, J.; Alagbo, H.O.; et al. Superficial Venous Disease—An Updated Review. Ann. Vasc. Surg. 2024, 105, 106–124. [Google Scholar] [CrossRef]
- Wang, S.-H.; Shyu, V.B.-H.; Chiu, W.-K.; Huang, R.-W.; Lai, B.-R.; Tsai, C.-H. An Overview of Clinical Examinations in the Evaluation and Assessment of Arterial and Venous Insufficiency Wounds. Diagnostics 2023, 13, 2494. [Google Scholar] [CrossRef]
- Avcı Işık, S.; Budak Ertürk, E.; Akay, H.T.; Karahan, A.; Akpınar, D.; Karslıoğlu, A.O. Analysis of Venous Insufficiency Risk Factors and Demographic Characteristics among Nurses: An Analytical Cross-Sectional Study. Medicina 2024, 60, 1498. [Google Scholar] [CrossRef]
- Jaworucka-Kaczorowska, A.; Simka, M. Anatomical, Pathophysiological, and Clinical Aspects of Extra-Pelvic Varicose Veins of Pelvic Origin. Diagnostics 2025, 15, 245. [Google Scholar] [CrossRef]
- Costa, D.; Ielapi, N.; Minici, R.; Peluso, A.; Bracale, U.M.; Andreucci, M.; Serra, R. Risk Factors for Bleeding Varicose Veins in Patients with Chronic Venous Disease. Medicina 2023, 59, 1034. [Google Scholar] [CrossRef]
- Chee, Y.J.; Dalan, R.; Cheung, C. The Interplay Between Immunity, Inflammation and Endothelial Dysfunction. Int. J. Mol. Sci. 2025, 26, 1708. [Google Scholar] [CrossRef] [PubMed]
- Rieger, J.; Kaessmeyer, S.; Al Masri, S.; Hünigen, H.; Plendl, J. Endothelial cells and angiogenesis in the horse in health and disease—A review. Anat. Histol. Embryol. 2020, 49, 656–678. [Google Scholar] [CrossRef] [PubMed]
- Alsaigh, T.; Fukaya, E. Varicose veins and chronic venous disease. Cardiol. Clin. 2021, 39, 567–581. [Google Scholar] [CrossRef]
- Saberianpour, S.; Modaghegh, M.H.S.; Rahimi, H.; Kamyar, M.M. Role of mechanosignaling on pathology of varicose vein. Biophys. Rev. 2021, 13, 139–145. [Google Scholar] [CrossRef]
- Gonzalez-Avila, G.; Sommer, B.; Flores-Soto, E.; Aquino-Galvez, A. Hypoxic Effects on Matrix Metalloproteinases’ Expression in the Tumor Microenvironment and Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 16887. [Google Scholar] [CrossRef]
- Matei, S.C.; Matei, M.; Anghel, F.M.; Derban, M.D.; Olariu, A.; Olariu, S. Impact of statin treatment on patients diagnosed with chronic venous disease. Morphological analysis of the venous wall and clinical implications. Phlebology 2022, 37, 188–195. [Google Scholar] [CrossRef]
- Gianesini, S.; Rimondi, E.; Raffetto, J.D.; Melloni, E.; Pellati, A.; Menegatti, E.; Avruscio, G.P.; Bassetto, F.; Costa, A.L.; Rockson, S. Human collecting lymphatic glycocalyx identification by electron microscopy and immunohistochemistry. Sci. Rep. 2023, 13, 3022. [Google Scholar] [CrossRef]
- Moore, K.H.; Murphy, H.A.; George, E.M. The glycocalyx: A central regulator of vascular function. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2021, 320, R508–R518. [Google Scholar] [CrossRef]
- Diaz, J.A.; Gianesini, S.; Khalil, R.A. Glycocalyx disruption, endothelial dysfunction and vascular remodeling as underlying mechanisms and treatment targets of chronic venous disease. Int. Angiol. A J. Int. Union Angiol. 2024, 43, 563–590. [Google Scholar] [CrossRef]
- Rosendorff, C. Effects of LDL cholesterol on vascular function. J. Hum. Hypertens 2002, 16, S26–S28. [Google Scholar] [CrossRef]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Balzan, S.; Papa, A. LOX-1 variants modulate the severity of cardiovascular disease: State of the art and future directions. Mol. Cell. Biochem. 2024, 479, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Munno, M.; Mallia, A.; Greco, A.; Modafferi, G.; Banfi, C.; Eligini, S. Radical Oxygen Species, Oxidized Low-Density Lipoproteins, and Lectin-like Oxidized Low-Density Lipoprotein Receptor 1: A Vicious Circle in Atherosclerotic Process. Antioxidants 2024, 13, 583. [Google Scholar] [CrossRef] [PubMed]
- Vlajinac, H.D.; Marinkovic, J.M.; Maksimovic, M.Z.; Matic, P.A.; Radak, D.J. Body mass index and primary chronic venous disease–a cross-sectional study. European J. Vasc. Endovasc. Surg. 2013, 45, 293–298. [Google Scholar] [CrossRef]
- Davies, A.H. The seriousness of chronic venous disease: A review of real-world evidence. Adv. Ther. 2019, 36, 5–12. [Google Scholar] [CrossRef]
- Deol, Z.K.; Lakhanpal, S.; Franzon, G.; Pappas, P.J. Effect of obesity on chronic venous insufficiency treatment outcomes. Journal of Vascular Surgery: Venous Lymphat. Disord. 2020, 8, 617–628. [Google Scholar] [CrossRef]
- Zottola, Z.R.; Geiger, J.T.; Choo, G.E.; Kedwai, B.J.; Balceniuk, M.D.; Ellis, J.L.; Doyle, A.J.; Newhall, K.A. Obese patients with CEAP (clinical, etiology, anatomy, pathophysiology) C2 and C3 disease show enhanced symptom improvement after endovenous thermal ablation. J. Vasc. Surg. Venous Lymphat. Disord. 2024, 12, 101873. [Google Scholar] [CrossRef]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, adipose tissue and vascular dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef]
- Petrascu, F.-M.; Matei, S.-C.; Margan, M.-M.; Ungureanu, A.-M.; Olteanu, G.-E.; Murariu, M.-S.; Olariu, S.; Marian, C. The Impact of Inflammatory Markers and Obesity in Chronic Venous Disease. Biomedicines 2024, 12, 2524. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Exclusion Criteria | |
|---|---|---|
| Topic | Papers correspond to research question | Not correspond |
| Specify databases | PubMed, PMC Europe, Scopus, WoS, MEDLINE, Google Scholar, grey literatures | Others |
| Search interval | from 2002 to 2025 (with priority last 10 years) | Before 2000 |
| Population/Target group | Studies in humans/patients, cell lines, biological samples | Animal studies |
| Study design (papers) | Scoping, Narrative, Systematic Review, Meta-Analysis, Case Report, Full articles | Others |
| Study design (language) | Original studies in English | Others |
| Accessed review protocol | PRISMA, PICOS, Cochrane Handbook, etc. | Others |
| Prospective Register | PROSPERO protocols | Others |
| Outcomes | Outcomes papers that provide 1/an unbiased and exhaustive overview and 2/summarize the current scientific data on the role of vascular endothelial dysfunction in chronic venous disease, associated with inflammation, oxidative stress, and shear stress | Others |
| Declare competing interests | The authors declare no competing interests | Others |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrashev, H.; Abrasheva, D.; Nikolov, N.; Ananiev, J.; Georgieva, E. A Systematic Review of Endothelial Dysfunction in Chronic Venous Disease—Inflammation, Oxidative Stress, and Shear Stress. Int. J. Mol. Sci. 2025, 26, 3660. https://doi.org/10.3390/ijms26083660
Abrashev H, Abrasheva D, Nikolov N, Ananiev J, Georgieva E. A Systematic Review of Endothelial Dysfunction in Chronic Venous Disease—Inflammation, Oxidative Stress, and Shear Stress. International Journal of Molecular Sciences. 2025; 26(8):3660. https://doi.org/10.3390/ijms26083660
Chicago/Turabian StyleAbrashev, Hristo, Despina Abrasheva, Nadelin Nikolov, Julian Ananiev, and Ekaterina Georgieva. 2025. "A Systematic Review of Endothelial Dysfunction in Chronic Venous Disease—Inflammation, Oxidative Stress, and Shear Stress" International Journal of Molecular Sciences 26, no. 8: 3660. https://doi.org/10.3390/ijms26083660
APA StyleAbrashev, H., Abrasheva, D., Nikolov, N., Ananiev, J., & Georgieva, E. (2025). A Systematic Review of Endothelial Dysfunction in Chronic Venous Disease—Inflammation, Oxidative Stress, and Shear Stress. International Journal of Molecular Sciences, 26(8), 3660. https://doi.org/10.3390/ijms26083660