
Nlrc4 Inflammasome Expression After Acute Myocardial Infarction in Rats

 , ,
, ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Experimental Groups
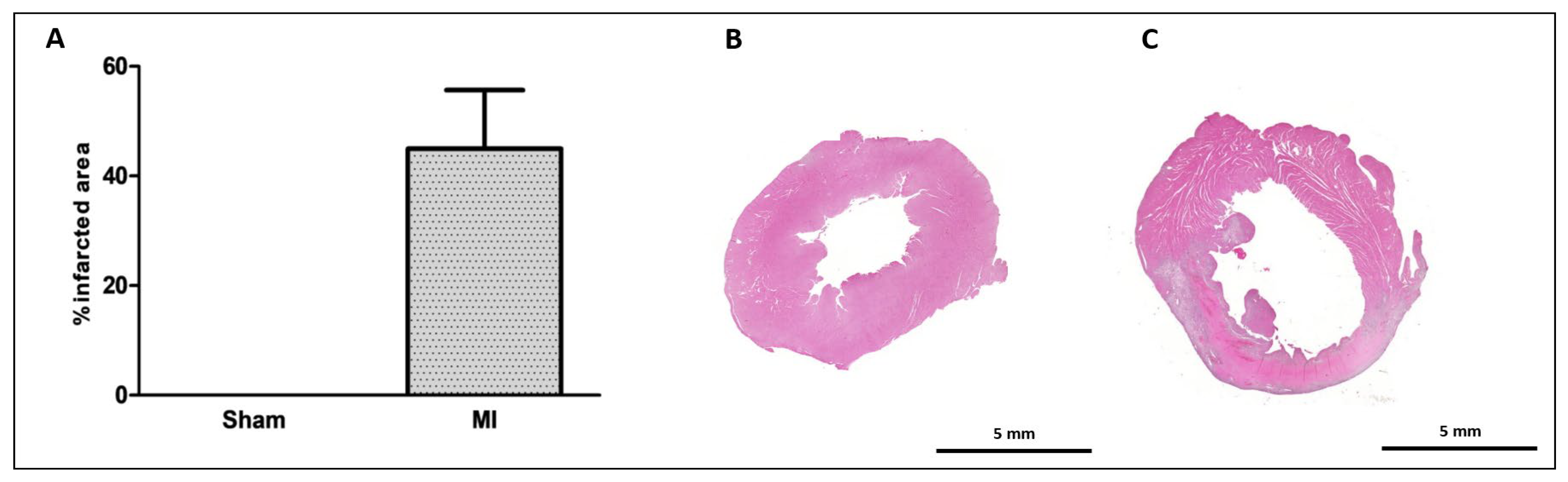
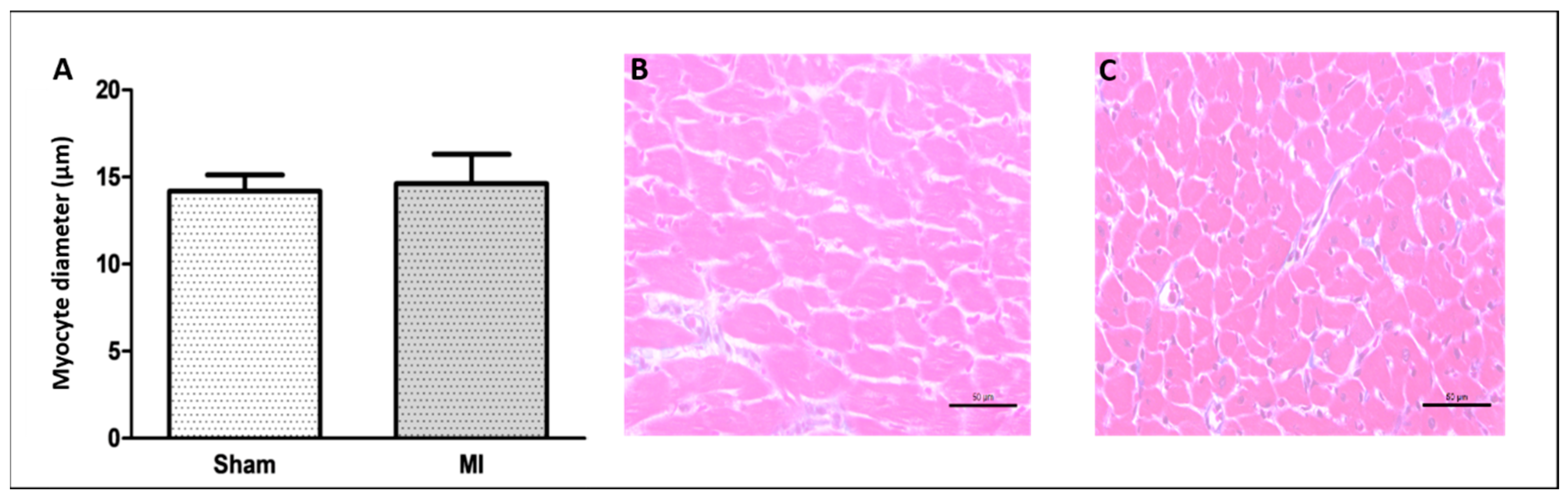
2.2. Histological Study
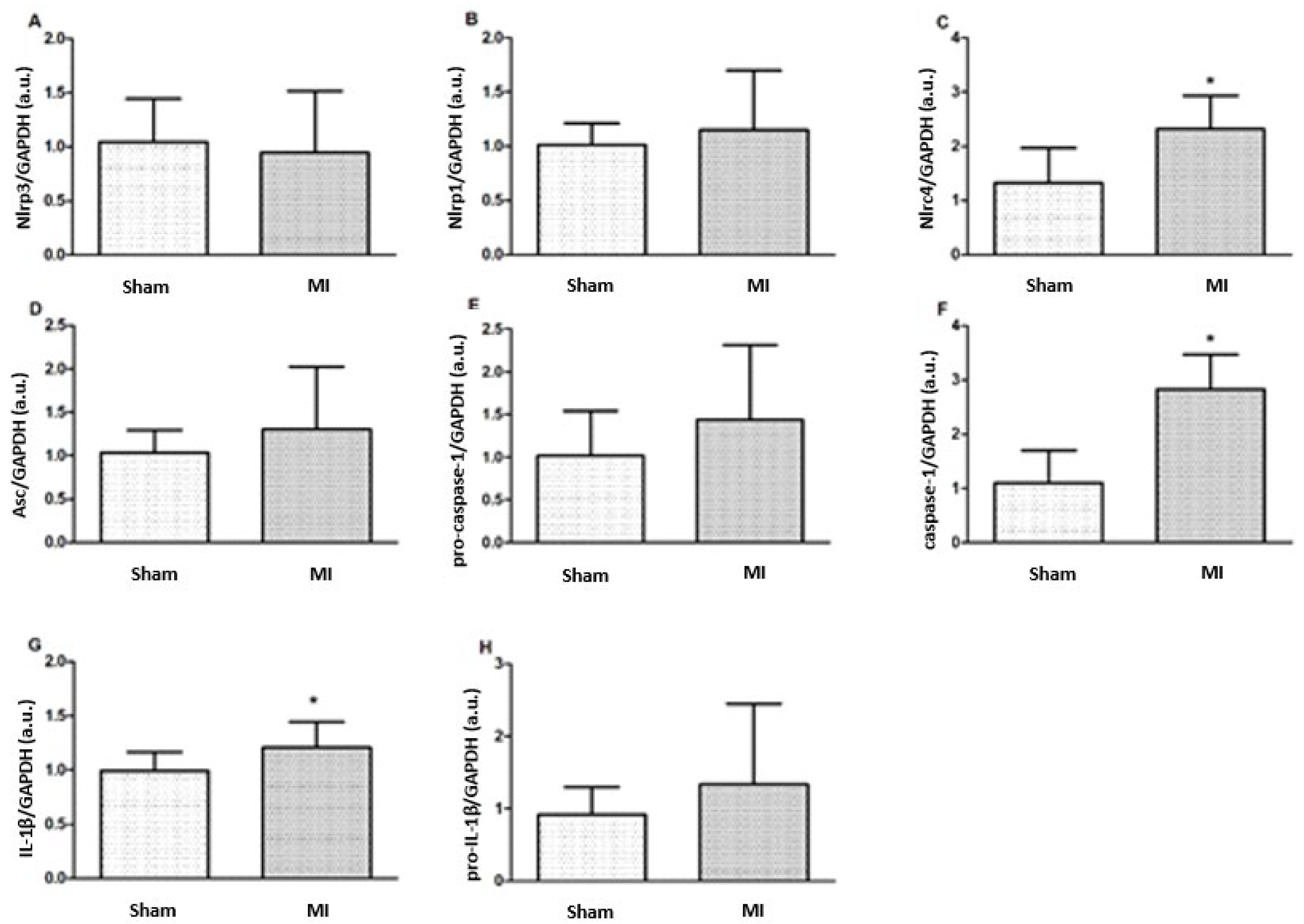
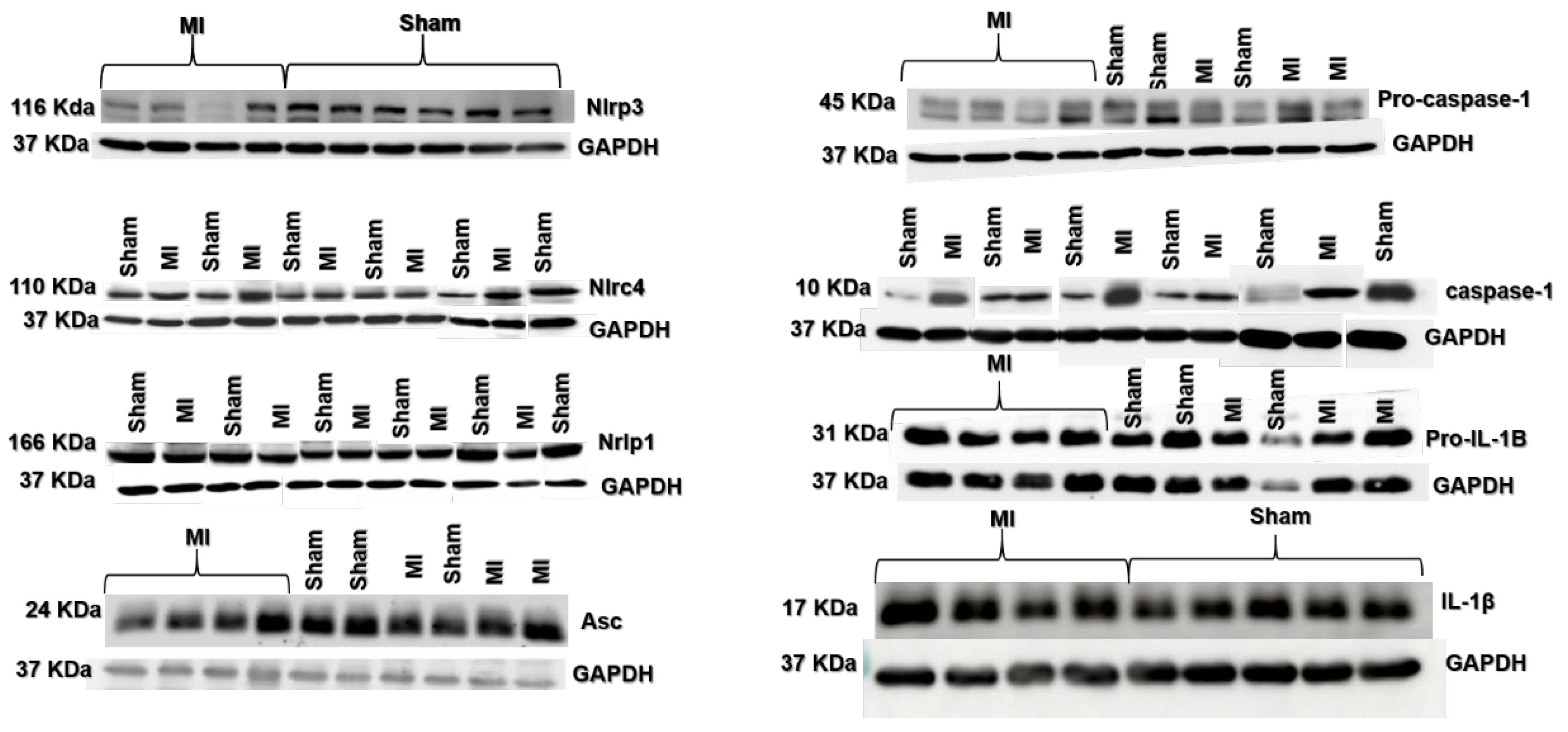
2.3. Protein Expression
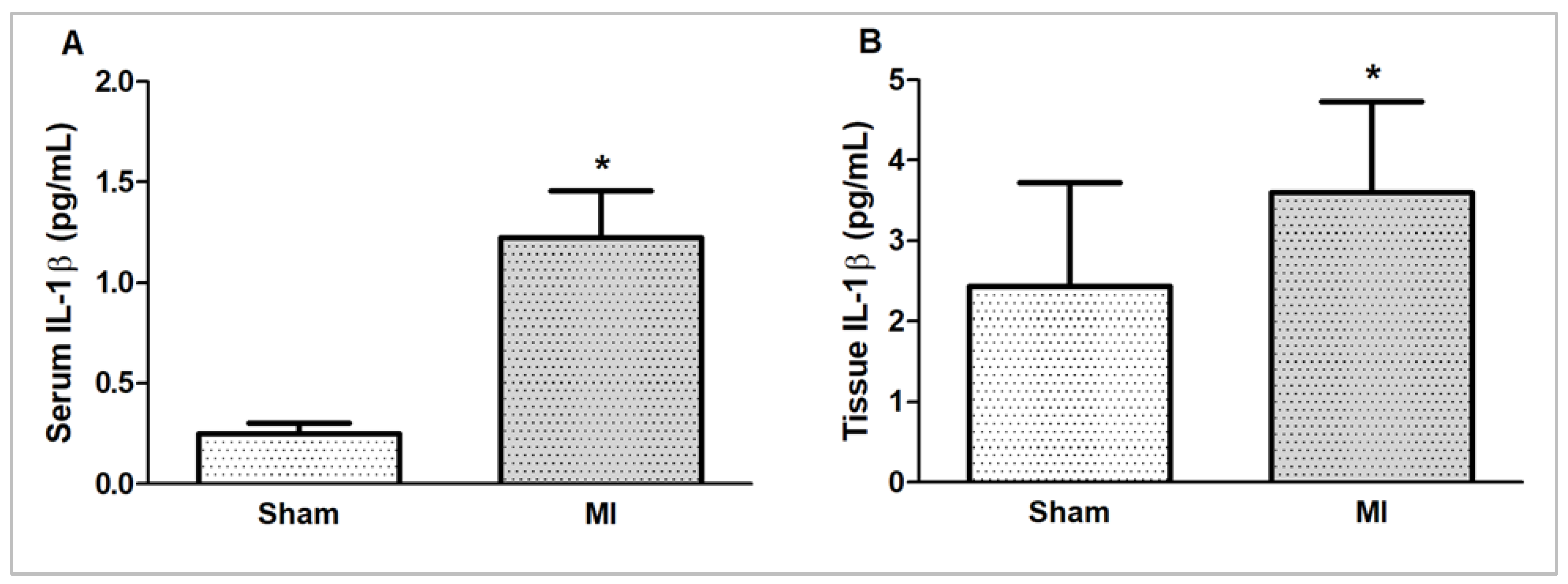
2.4. IL-1β Concentration
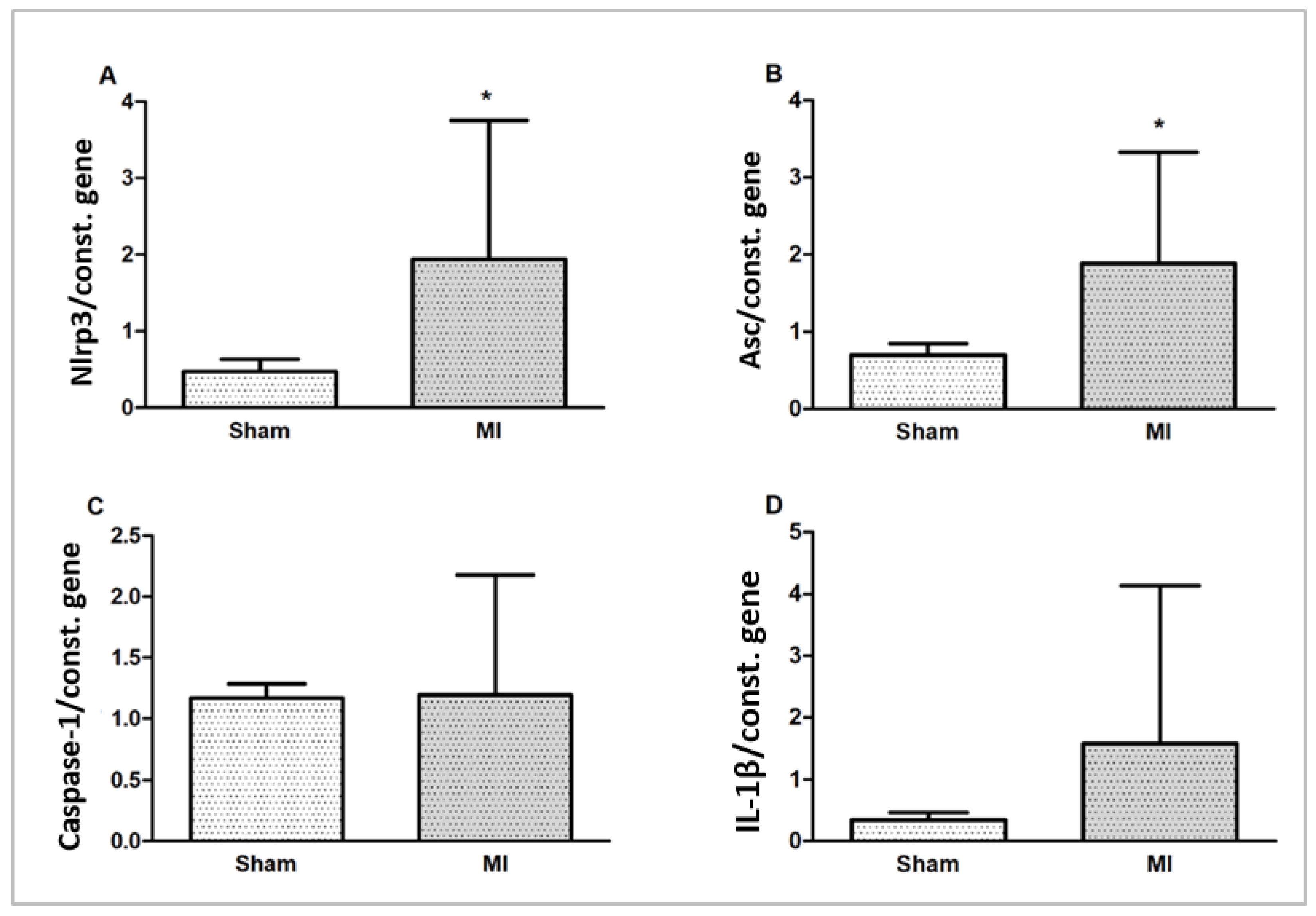
2.5. Gene Expression
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Study Protocol
4.2. Tissue Collection
4.3. Infarction Size
4.4. Histological Analysis
4.5. Western Blot
4.6. Cytokine Concentration
4.7. Gene Expression by qRT-PCR
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, S.S.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Gibbs, B.B.; Beaton, A.Z.; Boehme, A.K.; et al. 2024 Heart disease and stroke statistics: A report of US and Global Data from the American Heart Association. Circulation 2024, 149, e347–e913. [Google Scholar] [PubMed]
- Frangogiannis, N.G. Inflammation in cardiac injury, repair and regeneration. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yang, K.; Zhao, M.; Liu, W.; Zhang, X.; Chi, J.; Shi, Z.; Zhang, X.; Fu, Y.; Liu, Y.; et al. Calcium-sensing receptor on neutrophil promotes myocardial apoptosis and fibrosis after acute myocardial infarction via NLRP3 inflammasome activation. Can. J. Cardiol. 2020, 36, 893–905. [Google Scholar] [CrossRef]
- Gatto, M.; Mota, G.A.F.; Okoshi, M.P. Influência do sistema imunológico nas doenças cardiovasculares. Arq. Bras. Cardiol. 2023, 120, e20230398. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 inflammasome inhibitors in cardiovascular diseases. Molecules 2021, 26, 976. [Google Scholar] [CrossRef]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, K.; Zhu, L. Emerging roles of inflammasomes in cardiovascular diseases. Front. Immunol. 2022, 13, 834289. [Google Scholar] [CrossRef]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia–reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Bracey, N.A.; Beck, P.L.; Muruve, D.A.; Hirota, S.A.; Guo, J.; Jabagi, H.; Wright, J.R., Jr.; Macdonald, J.A.; Lees-Miller, J.P.; Roach, D.; et al. The NLRP3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 2013, 98, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Li, X.; Pang, P.; Hu, X.L.; Yu, S.T.; Liu, Y.N.; Li, X.; Wang, N.; Wang, J.H.; Xiao, W.; et al. Kanglexin, a novel anthraquinone compound, protects against myocardial ischemic injury in mice by suppressing NLRP3 and pyroptosis. Acta Pharmacol. Sin. 2020, 41, 319–326. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Pontillo, A.; Girardelli, M.; Kamada, A.J.; Pancotto, J.A.T.; Donadi, E.A.; Crovella, S.; Sandrin-Garcia, P. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity 2012, 45, 271–278. [Google Scholar] [CrossRef]
- Pontillo, A.; Catamo, E.; Arosio, B.; Mari, D.; Crovella, S. NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 277–281. [Google Scholar] [CrossRef]
- Johansson, Å.; Eriksson, N.; Becker, R.C.; Storey, R.F.; Himmelmann, A.; Hagström, E.; Varenhorst, C.; Axelsson, T.; Barratt, B.J.; James, S.K.; et al. NLRC4 inflammasome is an important regulator of interleukin-18 levels in patients with acute coronary syndromes: Genome-Wide Association Study in the PLATelet inhibition and patient Outcomes Trial (PLATO). Circ. Cardiovasc. Genet. 2015, 8, 498–506. [Google Scholar] [CrossRef]
- Onódi, Z.; Ruppert, M.; Kucsera, D.; Sayour, A.A.; Tóth, V.E.; Koncsos, G.; Novák, J.; Brenner, G.B.; Makkos, A.; Baranyai, T.; et al. AIM2-driven inflammasome activation in heart failure. Cardiovasc. Res. 2021, 117, 2639–2651. [Google Scholar] [CrossRef]
- Durga Devi, T.; Babu, M.; Mäkinen, P.; Kaikkonen, M.U.; Heinaniemi, M.; Laakso, H.; Ylä-Herttuala, E.; Rieppo, L.; Liimatainen, T.; Naumenko, N.; et al. Aggravated postinfarct heart failure in Type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. Am. J. Pathol. 2017, 187, 2659–2673. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.M.; Gomes, M.J.; Brandao, B.B.; Pagan, L.U.; Gatto, M.; Damatto, F.C.; Rodrigues, E.A.; Pontes, T.H.D.; Borim, P.A.; Fernandes, A.A.H.; et al. Effects of resistance exercise on slow-twitch soleus muscle of infarcted rats. Antioxidants 2023, 12, 291. [Google Scholar] [CrossRef] [PubMed]
- Minicucci, M.F.; Azevedo, P.S.; Martinez, P.F.; Lima, A.R.R.; Bonomo, C.; Guizoni, D.M.; Polegato, B.F.; Okoshi, M.P.; Okoshi, K.; Matsubara, B.B.; et al. Critical infarct size to induce ventricular remodeling, cardiac dysfunction and heart failure in rats. Int. J. Cardiol. 2011, 151, 242–243. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- Broz, P.; Von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femminò, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid. Med. Cell Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-mediated cardiomyocyte pyroptosis promotes myocardial I/R injury. Circ. Res. 2021, 129, 383–396. [Google Scholar] [CrossRef]
- Frantz, S. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J. Mol. Cell Cardiol. 2003, 35, 685–694. [Google Scholar] [CrossRef]
- Pomerantz, B.J.; Reznikov, L.L.; Harken, A.H.; Dinarello, C.A. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1β. Proc. Natl. Acad. Sci. USA 2001, 98, 2871–2876. [Google Scholar] [CrossRef]
- Yue, R.; Zheng, Z.; Luo, Y.; Wang, X.; Lv, M.; Qin, D.; Tan, Q.; Zhang, Y.; Wang, T.; Hu, H. NLRP3-mediated pyroptosis aggravates pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction in mice: Cardioprotective role of irisin. Cell Death Discov. 2021, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, P.; Wen, W.; Bai, X.; Zhang, Y.; Liu, M.; Wang, L.; Wu, Y.; Yuan, Z.; Zhou, J. IL-17A contributes to myocardial ischemic injury by activating NLRP3 inflammasome in macrophages through AMPKα/p38MAPK/ERK1/2 signal pathway in mice. Mol. Immunol. 2019, 105, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Xu, D.; Zhong, L.; Wang, Y.; Wang, W.; Xu, H.; Han, X.; Min, J.; Wu, G.; Huang, W.; et al. Ubiquitin-specific protease 25 improves myocardial ischemia-reperfusion injury by deubiquitinating NLRP3 and negatively regulating NLRP3 inflammasome activity in cardiomyocytes. Clin. Transl. Med. 2025, 15, e70243. [Google Scholar] [CrossRef]
- Li, J.-P.; Qiu, S.; Tai, G.-J.; Liu, Y.-M.; Wei, W.; Fu, M.-M.; Fang, P.Q.; Otieno, J.N.; Battulga, T.; Li, X.X.; et al. NLRP3 inflammasome-modulated angiogenic function of EPC via PI3K/Akt/mTOR pathway in diabetic myocardial infarction. Cardiovasc. Diabetol. 2025, 24, 6. [Google Scholar] [CrossRef]
- Cui, L.G.; Zhai, M.-M.; Yin, J.-J.; Wang, Z.-M.; Wang, S.-H.; Zhou, Y.-J.; Li, P.P.; Wang, Y.; Xia, L.; Wang, P.; et al. Targeting the ALKBH5-NLRP3 positive feedback loop alleviates cardiomyocyte pyroptosis after myocardial infarction. Eur. J. Pharmacol. 2025, 989, 177247. [Google Scholar] [CrossRef]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 inflammasome in acute myocardial infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Okoshi, M.P.; Matsubara, L.S.; Franco, M.; Cicogna, A.C.; Matsubara, B.B. Myocyte necrosis is the basis for fibrosis in renovascular hypertensive rats. Braz. J. Med. Biol. Res. 1997, 30, 1135–1144. [Google Scholar] [CrossRef]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. Methylation of apoptosis-associated speck-like protein with a caspase recruitment domain and outcomes in heart failure. J. Card. Fail. 2016, 22, 340–346. [Google Scholar] [CrossRef]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P.Y. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yan, W.; Zhu, H.; Zhai, J.; Xue, M.; Zheng, C. NLRC4 promotes the cGAS-STING signaling pathway by facilitating CBL-mediated K63-linked polyubiquitination of TBK1. J. Med. Virol. 2023, 95, e29013. [Google Scholar] [CrossRef] [PubMed]
- Guey, B.; Bodnar, M.; Manié, S.N.; Tardivel, A.; Petrilli, V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc. Natl. Acad. Sci. USA 2014, 111, 17254–17259. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia–reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment–elevation myocardial infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef]
- Garvin, A.M.; Jackson, M.A.; Korzick, D.H. Inhibition of programmed necrosis limits infarct size through altered mitochondrial and immune responses in the aged female rat heart. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H1434–H1442. [Google Scholar] [CrossRef]
- Guizoni, D.M.; Oliveira-Junior, S.A.; Noor, S.L.R.; Pagan, L.U.; Martinez, P.F.; Lima, A.R.R.; Gomes, M.J.; Damatto, R.L.; Cezar, M.D.; Bonomo, C.; et al. Effects of late exercise on cardiac remodeling and myocardial calcium handling proteins in rats with moderate and large size myocardial infarction. Int. J. Cardiol. 2016, 221, 406–412. [Google Scholar] [CrossRef]
- Okoshi, K.; Cezar, M.D.M.; Polin, M.A.M.; Paladino, J.R.; Martinez, P.F.; Oliveira, S.A., Jr.; Lima, A.R.R.; Damatto, R.L.; Paiva, S.A.R.; Zornoff, L.A.M.; et al. Influence of intermittent fasting on myocardial infarction-induced cardiac remodeling. BMC Cardiovasc. Disord. 2019, 19, 126. [Google Scholar] [CrossRef]
- Gomes, M.J.; Pagan, L.U.; Lima, A.R.R.; Reyes, D.R.A.; Martinez, P.F.; Damatto, F.C.; Pontes, T.H.D.; Rodrigues, E.A.; Souza, L.M.; Tosta, I.F.; et al. Effects of aerobic and resistance exercise on cardiac remodelling and skeletal muscle oxidative stress of infarcted rats. J. Cell Mol. Med. 2020, 24, 5352–5362. [Google Scholar] [CrossRef]
- Souza, L.M.; Okoshi, M.P.; Gomes, M.J.; Gatto, M.; Rodrigues, E.A.; Pontes, T.H.D.; Damatto, F.C.; Oliveira, L.R.S.; Borim, P.A.; Lima, A.R.R.; et al. Efeitos do exercício aeróbico tardio na remodelação cardíaca de ratos com infarto do miocárdio pequeno. Arq. Bras. Cardiol. 2021, 116, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Pagan, L.U.; Damatto, R.L.; Gomes, M.J.; Lima, A.R.R.; Cezar, M.D.M.; Damatto, F.C.; Reyes, D.R.A.; Caldonazo, T.M.M.; Polegato, B.F.; Okoshi, M.P.; et al. Low-intensity aerobic exercise improves cardiac remodelling of adult spontaneously hypertensive rats. J. Cell Mol. Med. 2019, 23, 6504–6507. [Google Scholar] [CrossRef] [PubMed]
- Reyes, D.R.A.; Gomes, M.J.; Rosa, C.M.; Pagan, L.U.; Damatto, F.C.; Damatto, R.L.; Depra, I.; Campos, D.H.S.; Fernandez, A.A.H.; Martinez, P.F.; et al. N-acetylcysteine influence on oxidative stress and cardiac remodeling in rats during transition from compensated left ventricular hypertrophy to heart failure. Cell Physiol. Biochem. 2017, 44, 2310–2321. [Google Scholar] [CrossRef] [PubMed]
- Damatto, R.L.; Lima, A.R.R.; Martinez, P.F.; Cezar, M.D.M.; Okoshi, K.; Okoshi, M.P. Myocardial myostatin in spontaneously hypertensive rats with heart failure. Int. J. Cardiol. 2016, 215, 384–387. [Google Scholar] [CrossRef]
- Rosa, C.M.; Campos, D.H.S.; Reyes, D.R.A.; Damatto, F.C.; Kurosaki, L.Y.; Pagan, L.U.; Gomes, M.J.; Corrêa, C.R.; Fernandes, A.A.H.; Okoshi, M.P.; et al. Effects of the SGLT2 inhibition on cardiac remodeling in streptozotocin-induced diabetic rats, a model of Type 1 diabetes mellitus. Antioxidants 2022, 11, 982. [Google Scholar] [CrossRef]
- Pagan, L.U.; Gomes, M.J.; Damatto, R.L.; Lima, A.R.R.; Cezar, M.D.M.; Damatto, F.C.; Reyes, D.R.A.; Campos, D.H.S.; Caldonazo, T.M.M.; Polegato, B.F.; et al. Aerobic exercise during advance stage of uncontrolled arterial hypertension. Front. Physiol. 2021, 12, 675778. [Google Scholar] [CrossRef]
- Tonon, C.R.; Monte, M.G.; Balin, P.S.; Fujimori, A.S.S.; Ribeiro, A.P.D.; Ferreira, N.F.; Vieira, N.M.; Cabral, R.P.; Okoshi, M.P.; Okoshi, K.; et al. Liraglutide pretreatment does not improve acute doxorubicin-induced cardiotoxicity in rats. Int. J. Mol. Sci. 2024, 25, 5833. [Google Scholar] [CrossRef]
- Carvalho, M.R.; Mendonça, M.L.M.; Oliveira, J.M.L.; Romanenghi, R.B.; Morais, C.S.; Ota, G.E.; Lima, A.R.R.; Oliveira, R.J.; Filiú, W.F.O.; Okoshi, K.; et al. Influence of high-intensity interval training and intermittent fasting on myocardium apoptosis pathway and cardiac morphology of healthy rats. Life Sci. 2021, 264, 118697. [Google Scholar] [CrossRef]
- Cezar, M.D.M.; Damatto, R.L.; Martinez, P.F.; Lima, A.R.R.; Campos, D.H.S.; Rosa, C.M.; Guizoni, D.M.; Bonomo, C.; Cicogna, A.C.; Gimenes, R.; et al. Aldosterone blockade reduces mortality without changing cardiac remodeling in spontaneously hypertensive rats. Cell Physiol. Biochem. 2013, 32, 1275–1287. [Google Scholar] [CrossRef]
- Carvalho, R.F.; Dariolli, R.; Justulin Junior, L.A.; Sugizaki, M.M.; Politi Okoshi, M.; Cicogna, A.C.; Felisbino, S.L.; Dal Pai-Silva, M. Heart failure alters matrix metalloproteinase gene expression and activity in rat skeletal muscle: Heart failure alters MMP gene expression and activity in rat skeletal muscle. Int. J. Exp. Pathol. 2006, 87, 437–443. [Google Scholar] [CrossRef]
- Reyes, D.R.A.; Gomes, M.J.; Rosa, C.M.; Pagan, L.U.; Zanati, S.G.; Damatto, R.L.; Rodrigues, E.A.; Carvalho, R.F.; Fernandes, A.A.H.; Martinez, P.F.; et al. Exercise during transition from compensated left ventricular hypertrophy to heart failure in aortic stenosis rats. J. Cell Mol. Med. 2019, 23, 1235–1245. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham (n = 15) | MI (n = 16) | |
|---|---|---|
| BW (g) | 263 ± 31 | 249 ± 16 |
| LV (g) | 0.55 (0.49–0.65) | 0.59 (0.57–0.64) |
| LV/BW (mg/g) | 2.17 ± 0.15 | 2.44 ± 0.17 * |
| RV (g) | 0.19 ± 0.03 | 0.20 ± 0.03 |
| RV/BW (mg/g) | 0.74 ± 0.09 | 0.80 ± 0.11 |
| Atria (g) | 0.07 ± 0.01 | 0.11 ± 0.03 * |
| Atria/BW (mg/g) | 0.27 ± 0.05 | 0.45 ± 0.12 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borim, P.A.; Gatto, M.; Mota, G.A.F.; Meirelles, A.L.B.; dos Santos, A.C.C.; Pagan, L.U.; Ojopi, E.P.B.; Rodrigues, E.A.; Souza, L.M.; Damatto, F.C.; et al. Nlrc4 Inflammasome Expression After Acute Myocardial Infarction in Rats. Int. J. Mol. Sci. 2025, 26, 3697. https://doi.org/10.3390/ijms26083697
Borim PA, Gatto M, Mota GAF, Meirelles ALB, dos Santos ACC, Pagan LU, Ojopi EPB, Rodrigues EA, Souza LM, Damatto FC, et al. Nlrc4 Inflammasome Expression After Acute Myocardial Infarction in Rats. International Journal of Molecular Sciences. 2025; 26(8):3697. https://doi.org/10.3390/ijms26083697
Chicago/Turabian StyleBorim, Patricia Aparecida, Mariana Gatto, Gustavo Augusto Ferreira Mota, Ana Luiza Barioni Meirelles, Anna Clara Consorti dos Santos, Luana Urbano Pagan, Elida Paula Benquique Ojopi, Eder Anderson Rodrigues, Lidiane Moreira Souza, Felipe Cesar Damatto, and et al. 2025. "Nlrc4 Inflammasome Expression After Acute Myocardial Infarction in Rats" International Journal of Molecular Sciences 26, no. 8: 3697. https://doi.org/10.3390/ijms26083697
APA StyleBorim, P. A., Gatto, M., Mota, G. A. F., Meirelles, A. L. B., dos Santos, A. C. C., Pagan, L. U., Ojopi, E. P. B., Rodrigues, E. A., Souza, L. M., Damatto, F. C., Oliveira, L. R. d. S., Zornoff, L. A. M., Okoshi, K., & Okoshi, M. P. (2025). Nlrc4 Inflammasome Expression After Acute Myocardial Infarction in Rats. International Journal of Molecular Sciences, 26(8), 3697. https://doi.org/10.3390/ijms26083697