

In Silico Analysis Identified Putative Pathogenic Missense Single Nucleotide Polymorphisms (SNPs) in the Human HNF1A Gene


Abstract
1. Introduction
2. Results
2.1. Variant Recruitment and Selection
2.2. Predicting Pathogenicity of Variants
2.3. Determining Variant Frequency
2.4. Predicting Protein Secondary Structure
2.5. Predicting Protein Stability
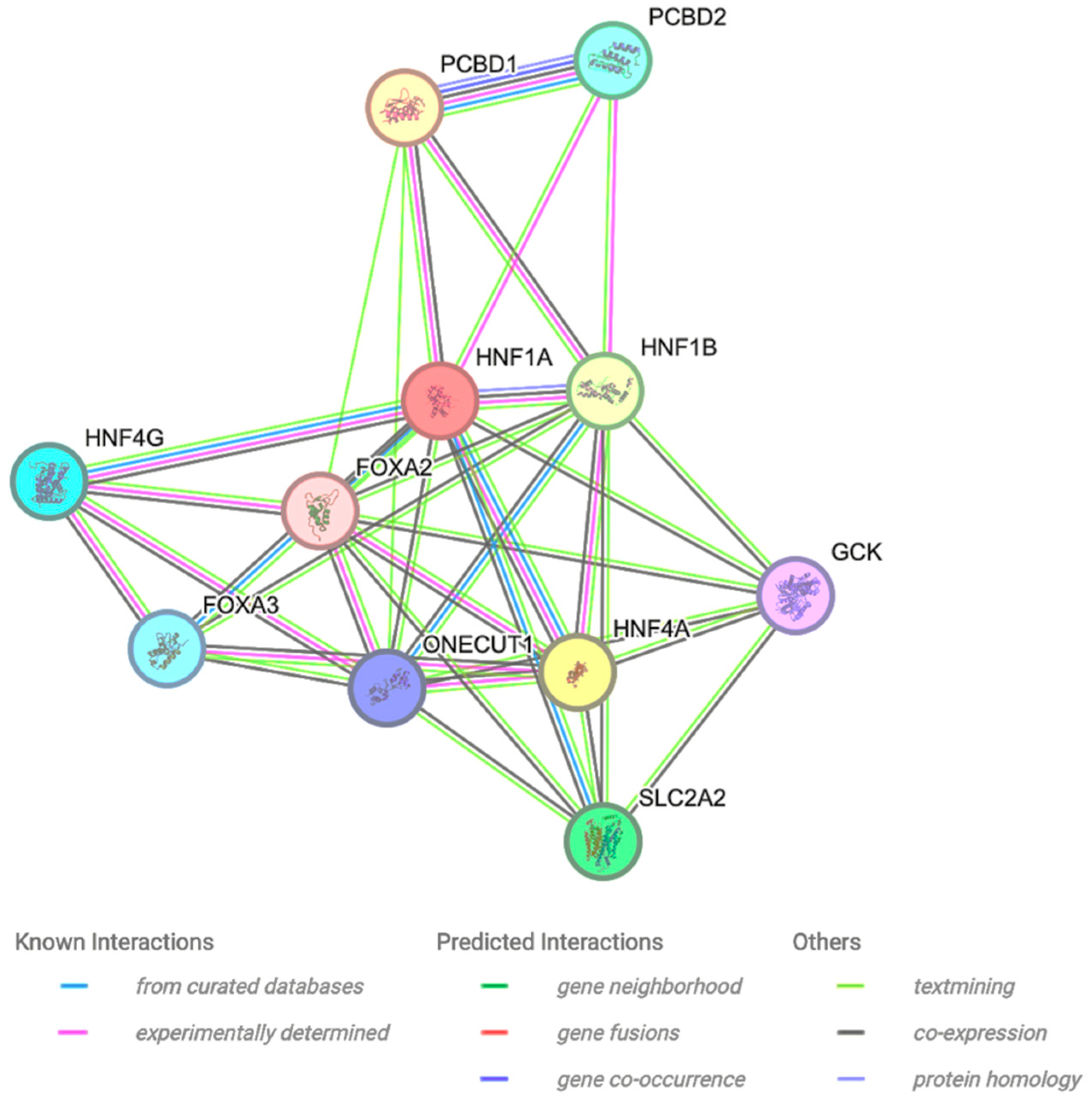
2.6. Protein–Protein Interaction
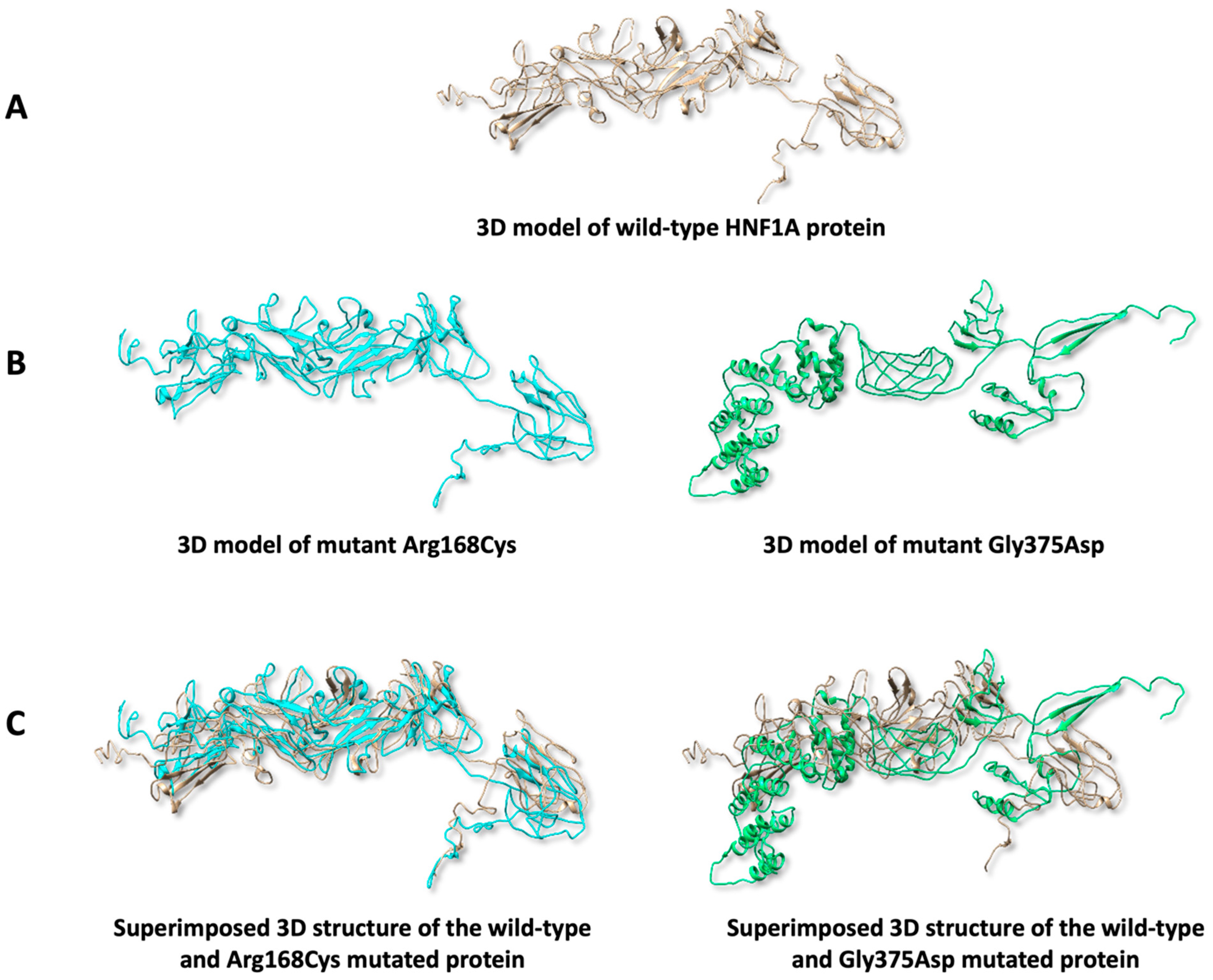
2.7. Predicting Protein 3D Structure
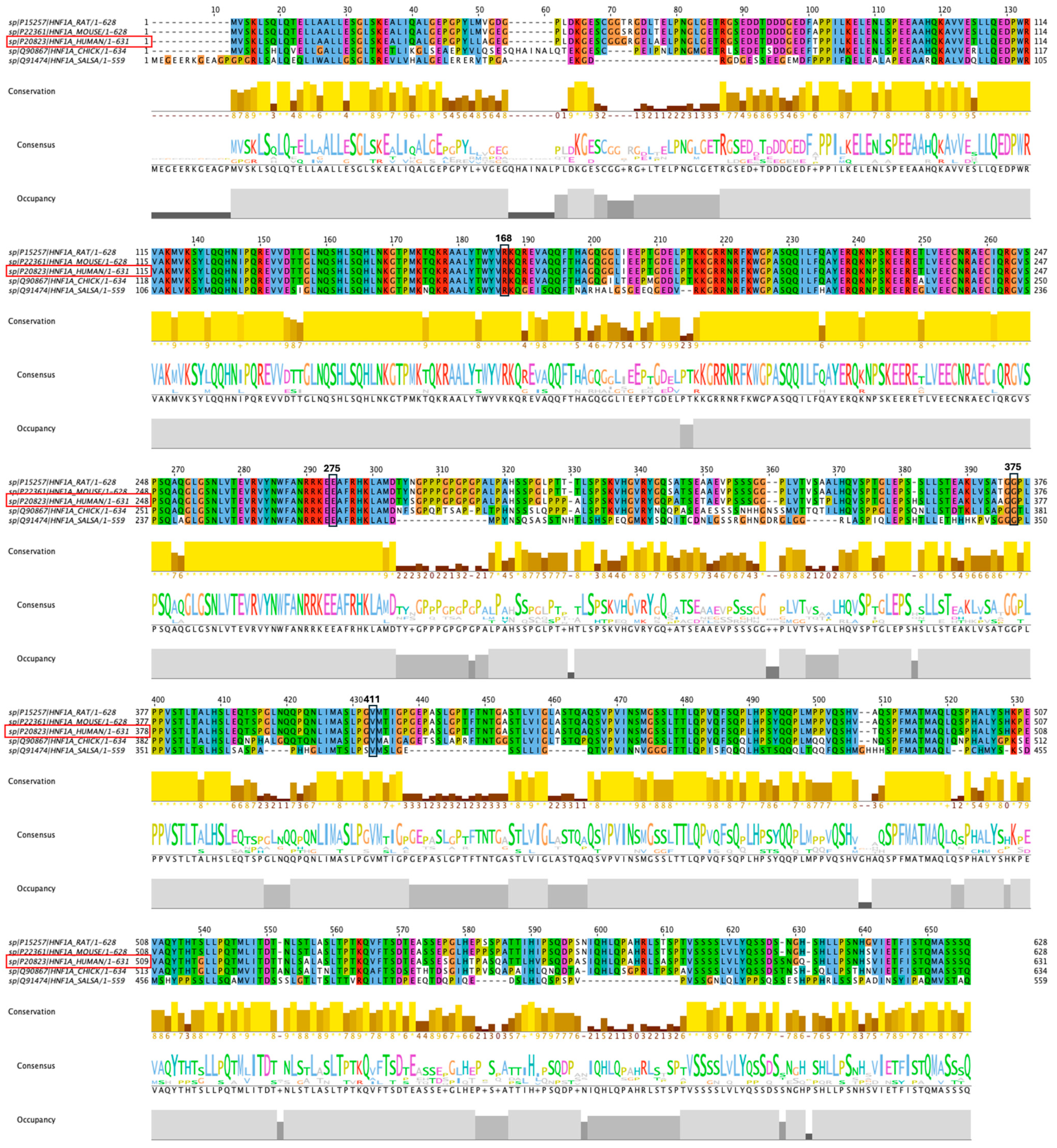
2.8. Conservation of the HNF1A Amino Acids
3. Materials and Methods
3.1. Variants Recruitment and Selection
3.2. Predicting Pathogenicity of Variants
3.3. Determining Variant Frequency
3.4. Predicting Protein Secondary Structure
3.5. Predicting Protein Stability
3.6. Protein–Protein Interaction
3.7. Predicting Protein 3D Structure
3.8. Conservation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fajans, S.S.; Bell, G.I. MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011, 34, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.C.; Neves, J.S.; Pérez, A.; Carvalho, D. Maturity-onset diabetes of the young: From a molecular basis perspective toward the clinical phenotype and proper management. Endocrinol. Diabetes Nutr. 2020, 67, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Pihoker, C.; Gilliam, L.K.; Ellard, S.; Dabelea, D.; Davis, C.; Dolan, L.M.; Greenbaum, C.J.; Imperatore, G.; Lawrence, J.M.; Marcovina, S.M.; et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: Results from the SEARCH for diabetes in Youth. J. Clin. Endocrinol. Metab. 2013, 98, 4055–4062. [Google Scholar] [CrossRef]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin. Diabetes Endocrinol. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Behl, R.; Malhotra, N.; Joshi, V.; Poojary, S.; Middha, S.; Gupta, S.; Olaonipekun, A.B.; Okoye, I.; Wagh, B.; Biswas, D.; et al. Meta-analysis of HNF1A-MODY3 variants among human population. J. Diabetes Metab. Disord. 2022, 21, 1037–1046. [Google Scholar] [CrossRef]
- Valkovicova, T.; Skopkova, M.; Stanik, J.; Gasperikova, D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr. Regul. 2019, 53, 110–134. [Google Scholar] [CrossRef]
- Firdous, P.; Nissar, K.; Ali, S.; Ganai, B.A.; Shabir, U.; Hassan, T.; Masoodi, S.R. Genetic testing of maturity-onset diabetes of the young current status and future perspectives. Front. Endocrinol. 2018, 9, 253. [Google Scholar] [CrossRef]
- Iafusco, F.; Maione, G.; Mazzaccara, C.; Di Candia, F.; Mozzillo, E.; Franzese, A.; Tinto, N. NGS analysis revealed digenic heterozygous GCK and HNF1A variants in a child with mild hyperglycemia: A case report. Diagnostics 2021, 11, 1164. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulyte, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.G.; Sharp, G.W.G. Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes. Metab. Res. Rev. 2002, 18, 451–463. [Google Scholar] [CrossRef]
- Bae, J.S.; Kim, T.H.; Kim, M.Y.; Park, J.M.; Ahn, Y.H. Transcriptional Regulation of Glucose Sensors in Pancreatic β-Cells and Liver: An Update. Sensors 2010, 10, 5031–5053. [Google Scholar] [CrossRef] [PubMed]
- Kind, L.; Molnes, J.; Tjora, E.; Raasakka, A.; Myllykoski, M.; Colclough, K.; Saint-Martin, C.; Adelfalk, C.; Dusatkova, P.; Pruhova, S.; et al. Molecular mechanism of HNF-1A–mediated HNF4A gene regulation and promoter-driven HNF4A-MODY diabetes. JCI Insight 2024, 9, e175278. [Google Scholar] [CrossRef]
- Chi, Y.I.; Frantz, J.D.; Oh, B.C.; Hansen, L.; Dhe-Paganon, S.; Shoelson, S.E. Diabetes mutations delineate an atypical POU domain in HNF-1alpha. Mol. Cell 2002, 10, 1129–1137. [Google Scholar] [CrossRef]
- Bach, I.; Yaniv, M. More potent transcriptional activators or a transdominant inhibitor of the HNF1 homeoprotein family are generated by alternative RNA processing. EMBO J. 1993, 12, 4229–4242. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Bjørkhaug, L.; Bratland, A.; Njølstad, P.R.; Molven, A. Functional dissection of the HNF-1alpha transcription factor: A study on nuclear localization and transcriptional activation. DNA Cell Biol. 2005, 24, 661–669. [Google Scholar] [CrossRef]
- Worth, C.L.; Gong, S.; Blundell, T.L. Structural and functional constraints in the evolution of protein families. Nat. Rev. Mol. Cell Biol. 2009, 10, 709–720. [Google Scholar] [CrossRef]
- Simon, A.L.; Stone, E.A.; Sidow, A. Inference of functional regions in proteins by quantification of evolutionary constraints. Proc. Natl. Acad. Sci. USA 2002, 99, 2912–2917. [Google Scholar] [CrossRef]
- Zhang, C.; Xie, F.; Li, L.; Zhang, C.; Zhang, Y.; Ying, W.; Liu, L.; Yan, X.; Yin, F.; Zhang, L. Hepatocyte nuclear factor 1 alpha (HNF1A) regulates transcription of O-GlcNAc transferase in a negative feedback mechanism. FEBS Lett. 2019, 593, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Bellanné-Chantelot, C.; Carette, C.; Riveline, J.P.; Valéro, R.; Gautier, J.F.; Larger, E.; Reznik, Y.; Ducluzeau, P.H.; Sola, A.; Hartemann-Heurtier, A.; et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes 2008, 57, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Harries, L.W.; Ellard, S.; Stride, A.; Morgan, N.G.; Hattersley, A.T.; Vaxillaire, M.; Tuomi, T.; Barbetti, E.; Njolstad, P.R.; Hansen, T.; et al. Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum. Mol. Genet. 2006, 15, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Sneha, P.; Thirumal, K.D.; George Priya Doss, C.; Siva, R.; Zayed, H. Determining the role of missense mutations in the POU domain of HNF1A that reduce the DNA-binding affinity: A computational approach. PLoS ONE 2017, 12, e0174953. [Google Scholar]
- Zoldoš, V.; Horvat, T.; Novokmet, M.; Cuenin, C.; Mužinić, A.; Pučić, M.; Hufman, J.E.; Gornik, O.; Polašek, O.; Campbell, H.; et al. Epigenetic silencing of HNF1A associates with changes in the composition of the human plasma N-glycome. Epigenetics 2012, 7, 164–172. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Polyphen2 | SNPs&Go | MetaSNP | SIFT | Panther | PHD SNP | SNAP2 | AlphaMissense | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S.No | Chr:bp | Alleles | AA | AA Coord | Pred | Prob | Pred | Prob | Pred | Score | Pred | Score | Pred | Preservation Time | Pred | Score | Pred | Score | Pre | Score |
| I | 120989008 | C/T | Cys/Arg | 168 | ProD | 1 | D | 0.994 | D | 0.56 | D | 0.03 | D | 0.693 | D | 0.678 | D | 0.625 | P | 0.97 |
| II | 120994274 | A/C | Glu/Ala | 275 | ProD | 1 | D | 0.989 | D | 0.502 | D | 0.01 | D | 0.729 | D | 0.562 | D | 0.695 | P | 0.85 |
| III | 120996557 | G/T | Gly/Asp | 375 | ProD | 0.998 | D | 0.992 | D | 0.728 | D | 0 | D | 0.698 | D | 0.68 | D | 0.745 | P | 0.92 |
| IV | 120996664 | G/T | Val/Phe | 411 | ProD | 1 | D | 0.993 | D | 0.669 | D | 0.02 | D | 0.602 | D | 0.609 | D | 0.69 | LP | 0.78 |
| I-Mutant | MuPro | ||||||
|---|---|---|---|---|---|---|---|
| Variant No. | rs ID | AA | AA Coord | Stability | RI | Stability | Score |
| I | rs764434453 | Cys/Arg | 168 | decrease | 3 | decrease | −0.57 |
| II | rs199890776 | Glu/Ala | 275 | decrease | 2 | decrease | −0.37 |
| III | rs1315462017 | Gly/Asp | 375 | decrease | 5 | decrease | −0.24 |
| IV | rs767284188 | Val/Phe | 411 | decrease | 9 | decrease | −1.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldharee, H.; Hamdan, H.Z. In Silico Analysis Identified Putative Pathogenic Missense Single Nucleotide Polymorphisms (SNPs) in the Human HNF1A Gene. Int. J. Mol. Sci. 2025, 26, 3768. https://doi.org/10.3390/ijms26083768
Aldharee H, Hamdan HZ. In Silico Analysis Identified Putative Pathogenic Missense Single Nucleotide Polymorphisms (SNPs) in the Human HNF1A Gene. International Journal of Molecular Sciences. 2025; 26(8):3768. https://doi.org/10.3390/ijms26083768
Chicago/Turabian StyleAldharee, Hitham, and Hamdan Z. Hamdan. 2025. "In Silico Analysis Identified Putative Pathogenic Missense Single Nucleotide Polymorphisms (SNPs) in the Human HNF1A Gene" International Journal of Molecular Sciences 26, no. 8: 3768. https://doi.org/10.3390/ijms26083768
APA StyleAldharee, H., & Hamdan, H. Z. (2025). In Silico Analysis Identified Putative Pathogenic Missense Single Nucleotide Polymorphisms (SNPs) in the Human HNF1A Gene. International Journal of Molecular Sciences, 26(8), 3768. https://doi.org/10.3390/ijms26083768