
Therapeutic Potential of Tricyclic Pyridazinone-Based Molecules: An Overview

 , , ,
, , ,
Abstract
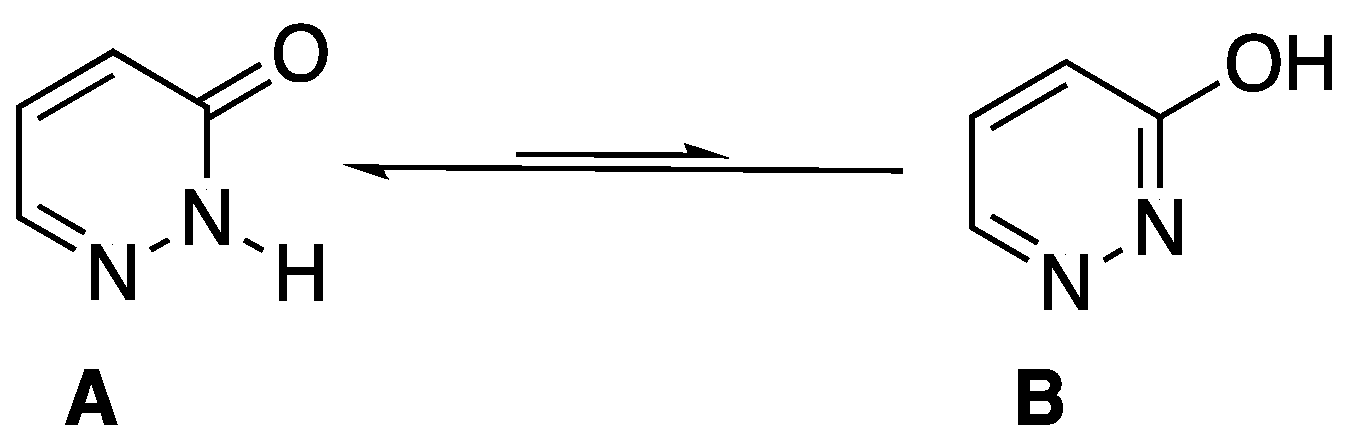
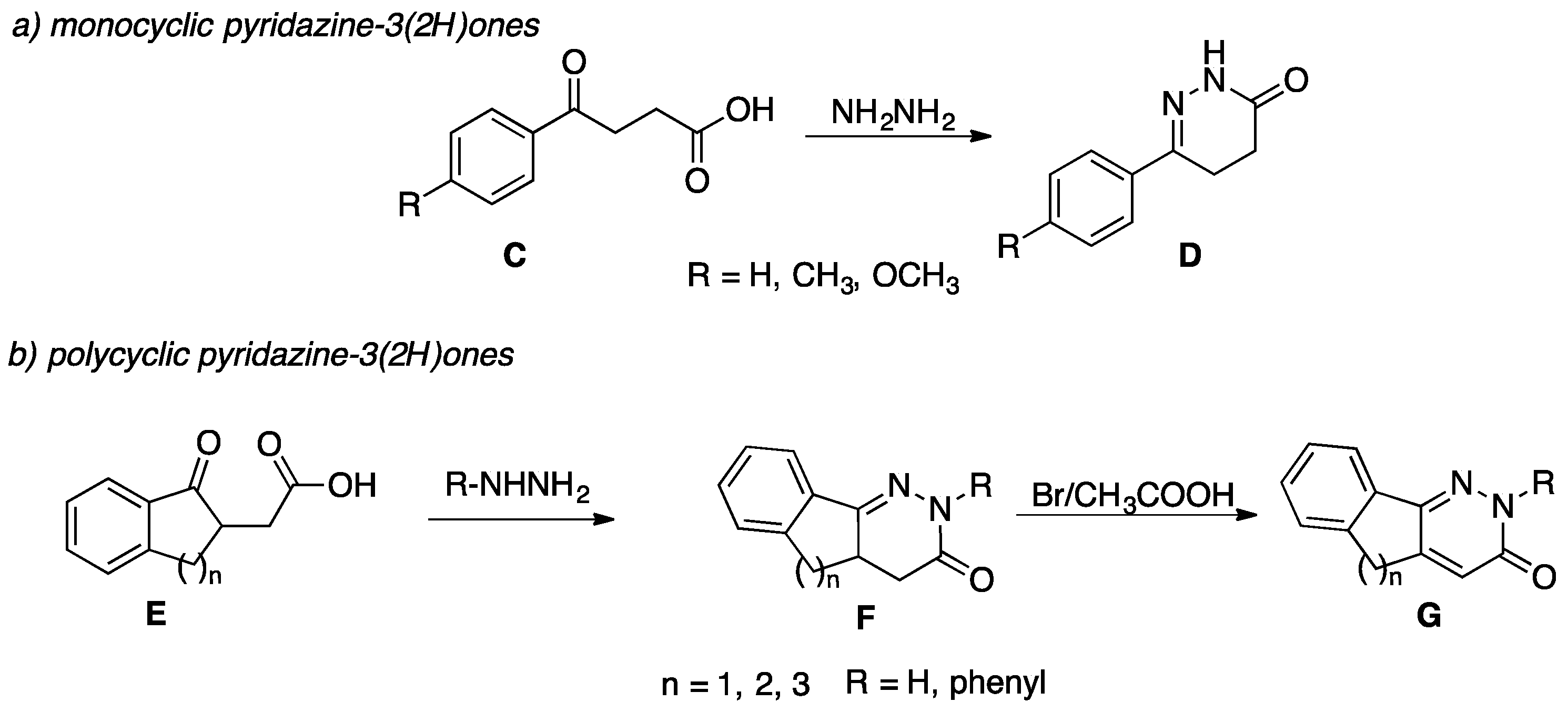
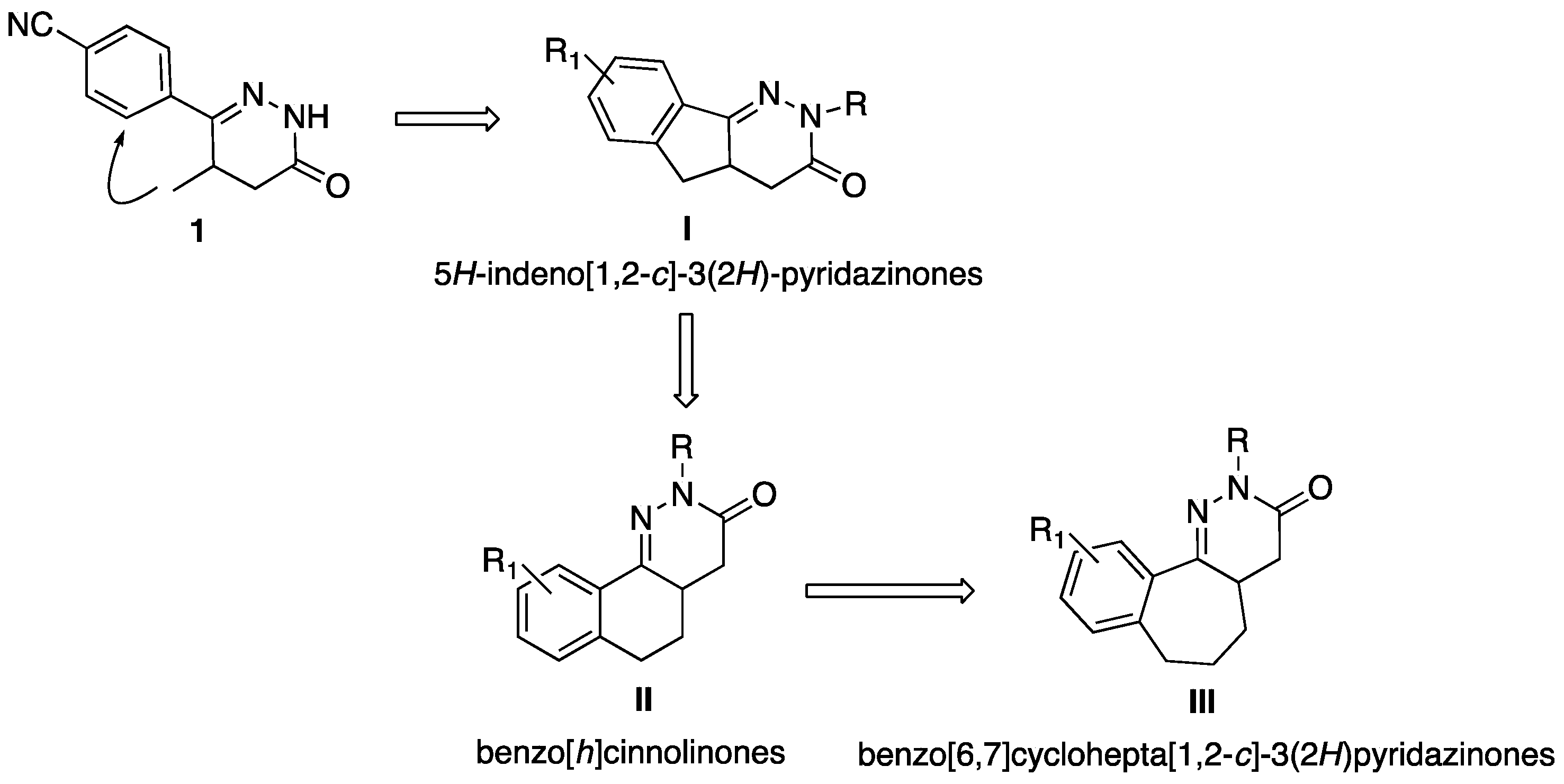
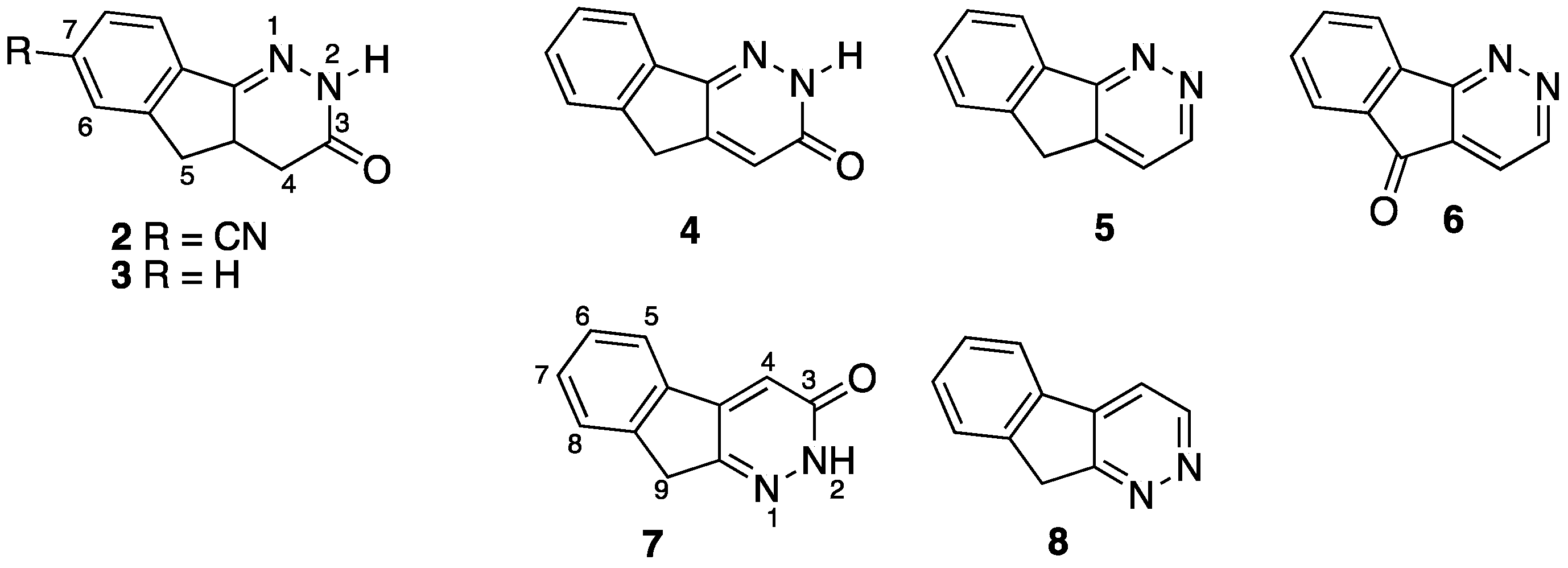
:1. Introduction
2. Anti-Inflammatory/Analgesic/Antipyretic Activities
3. Cardiovascular Activity
4. Antiulcer Activity
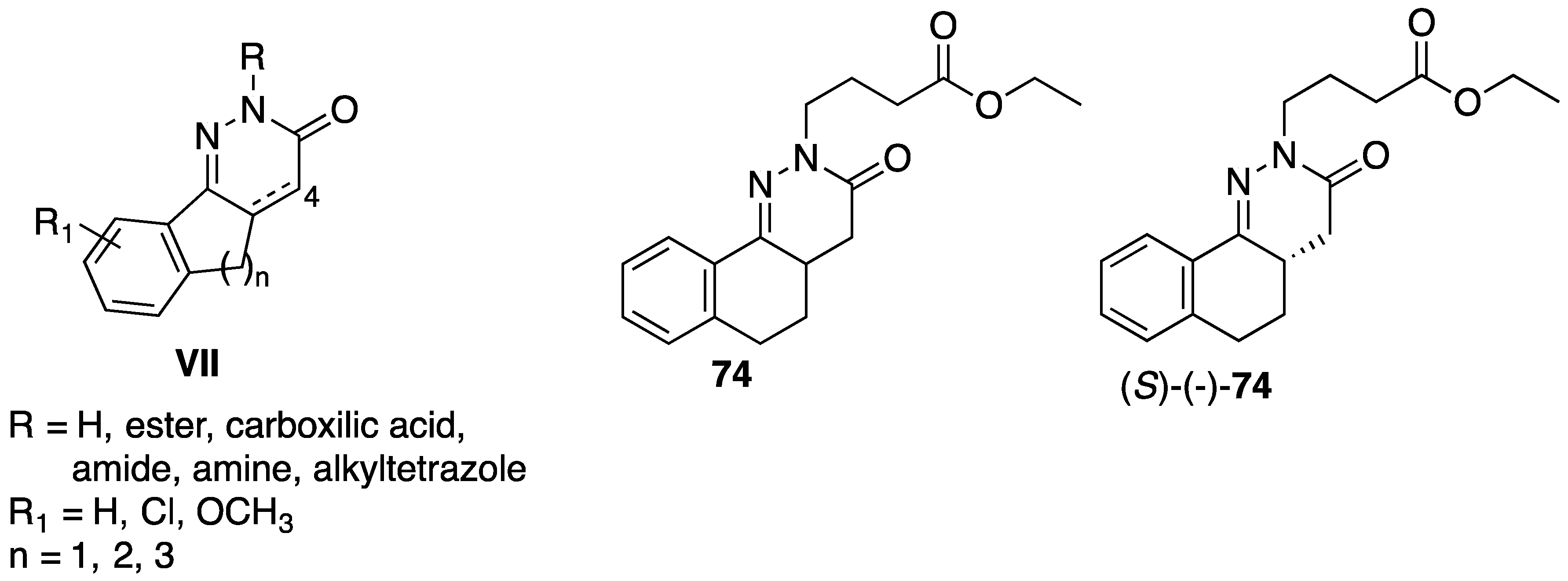
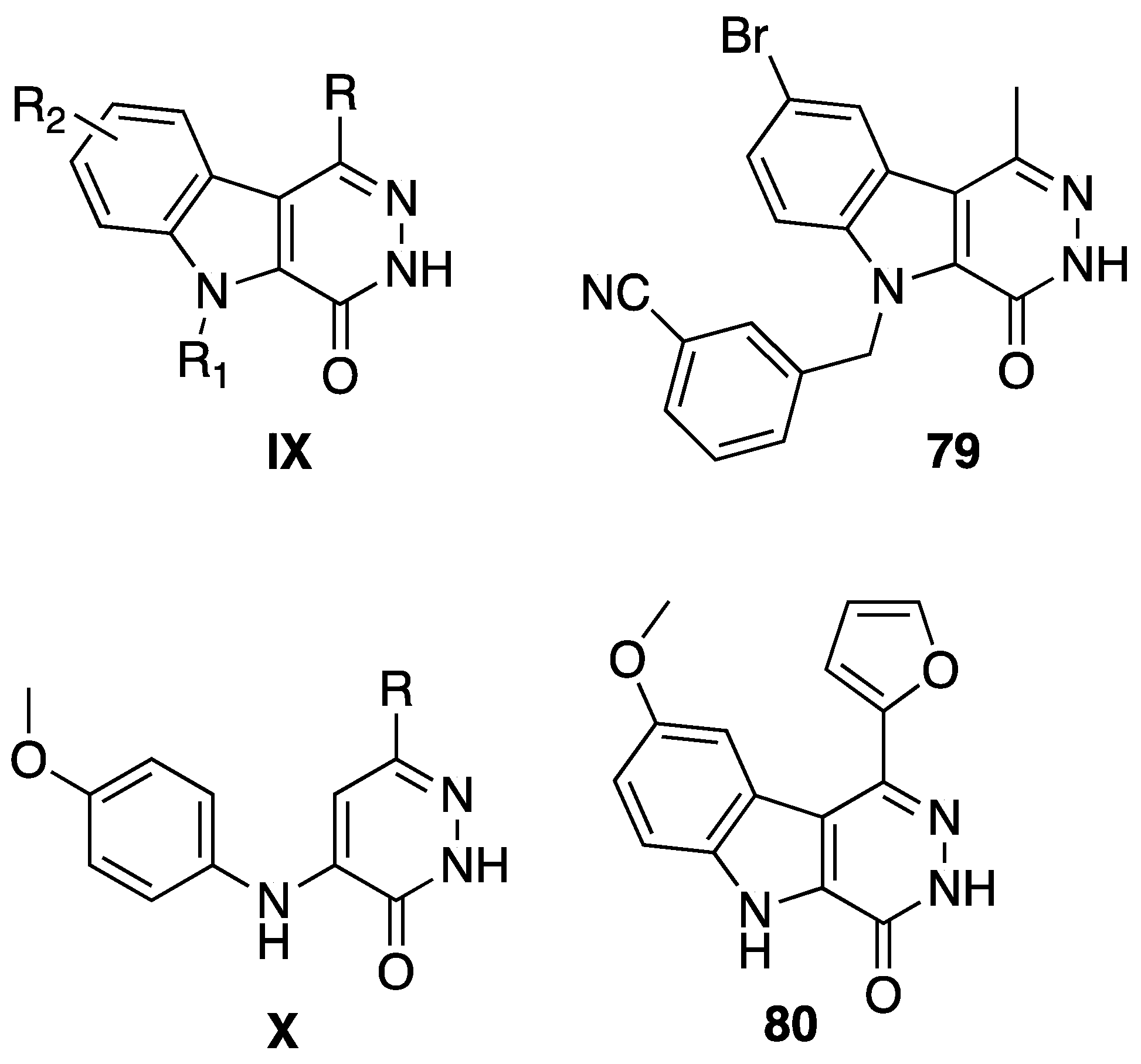
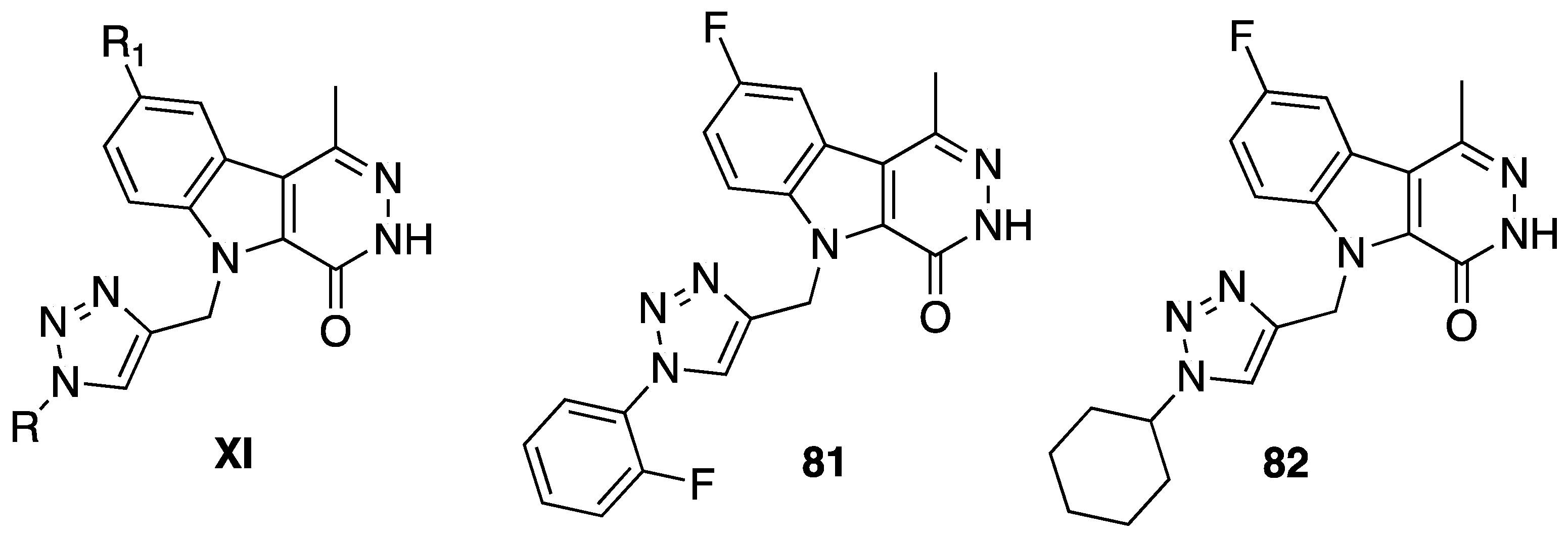
5. Anticancer Activity
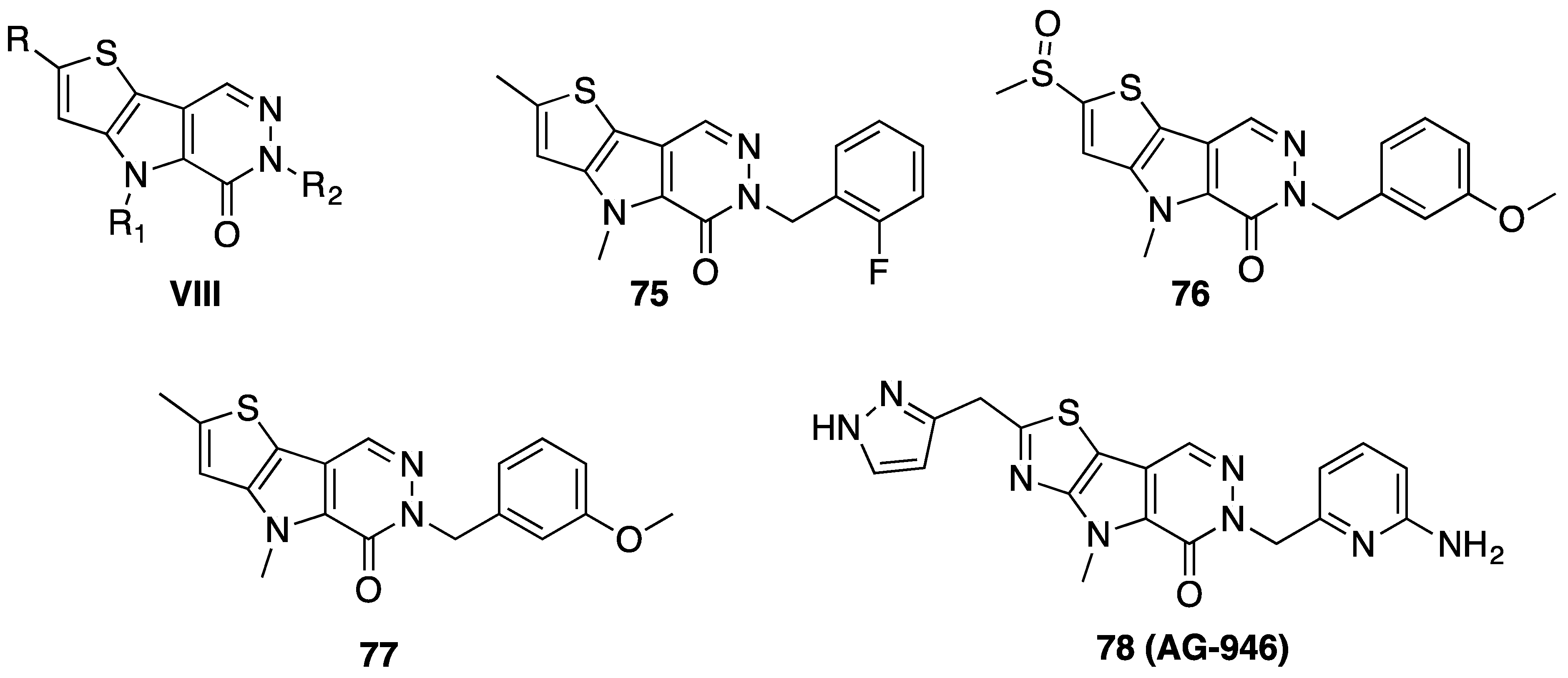
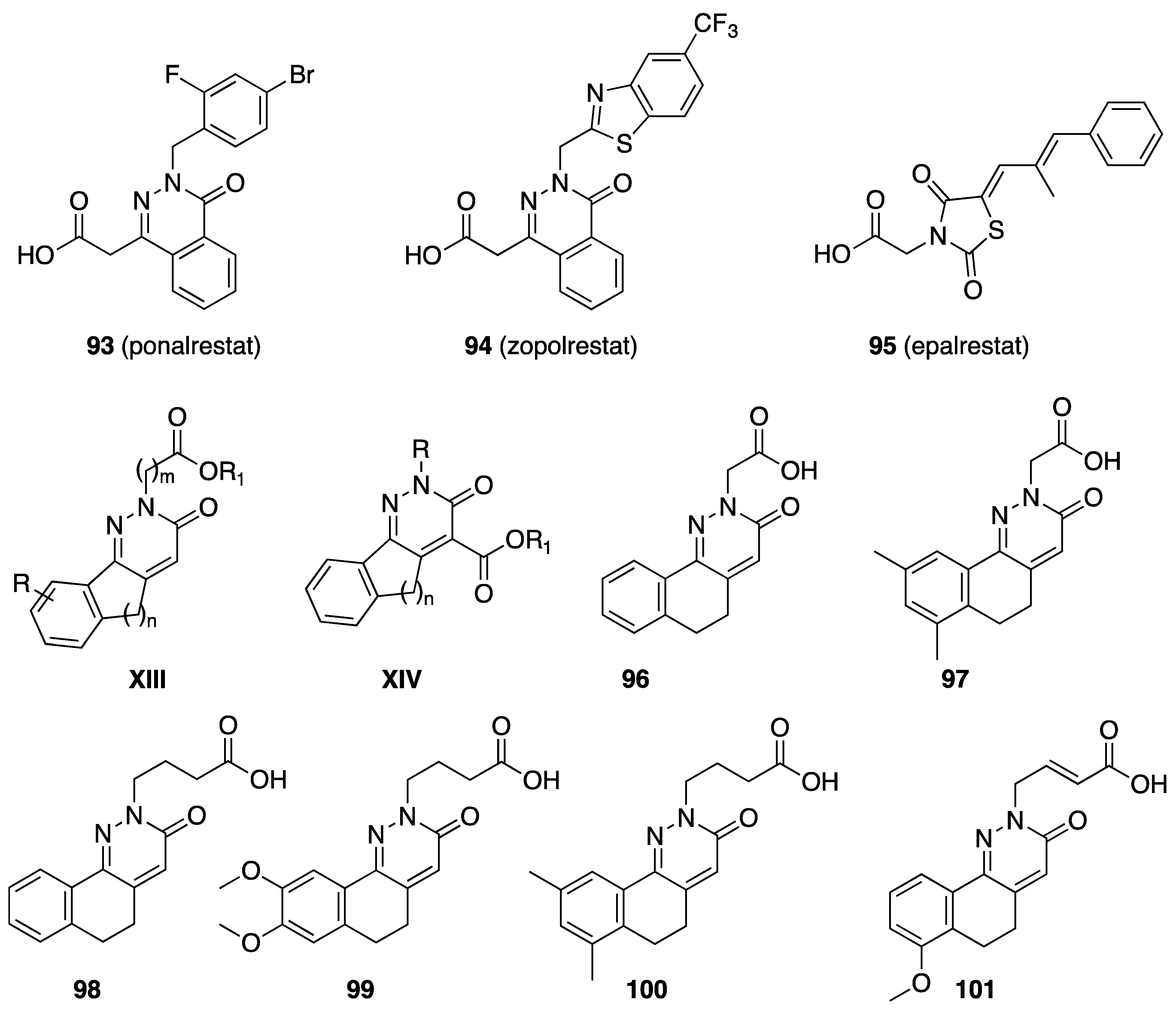
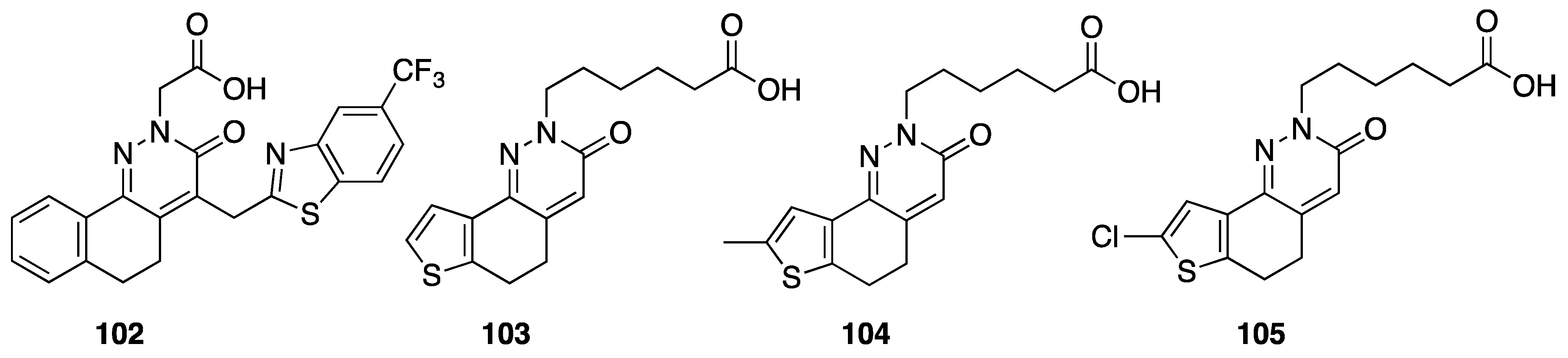
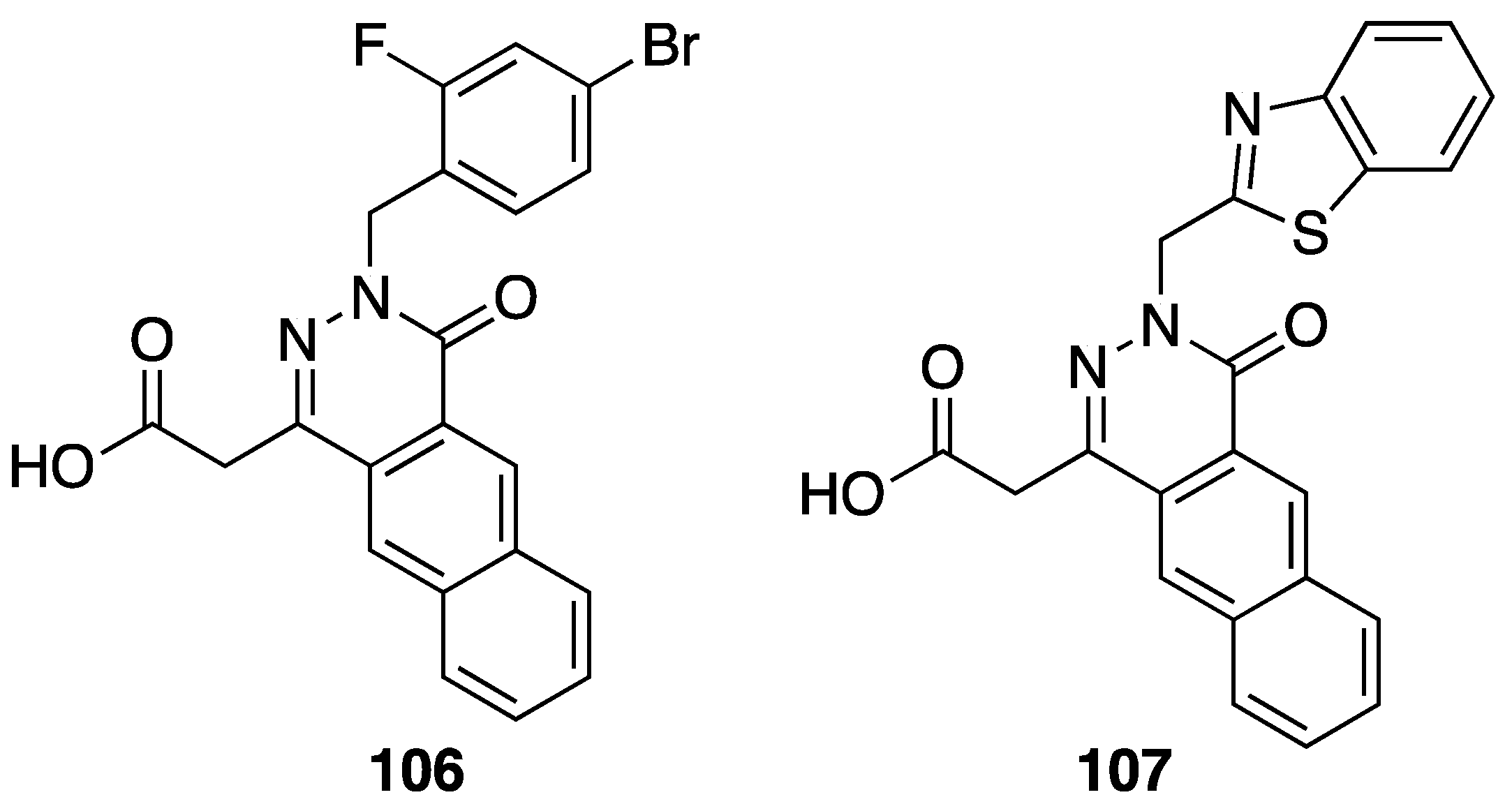
6. Antidiabetic Activity
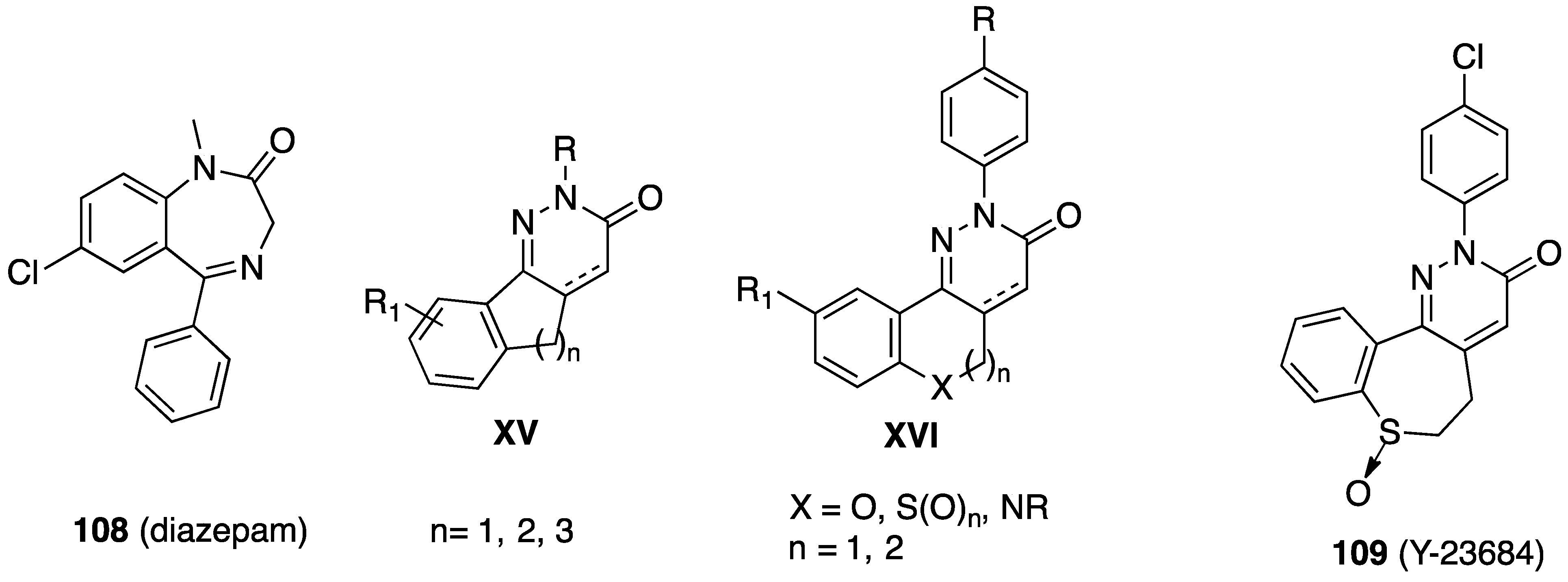
7. Anxiety Disorders
8. Antiviral Activity
9. Other Biological Activities
9.1. Matrix Metalloproteinase Inhibitors
9.2. Phosphodiesterase 5 Inhibitors
9.3. Adenosine Inhibitors
9.4. Translocator Protein 18 kDa (TSPO) Ligands
9.5. Multitarget-Directed Ligands
9.6. PKM2 Activators to Reduce Photoreceptor Apoptosis
10. Conclusions
Funding
Conflicts of Interest
References
- Kabir, E.; Uzzaman, M. A review on biological and medicinal impact of heterocyclic compounds. Results Chem. 2022, 4, 100606. [Google Scholar] [CrossRef]
- Landi, G.; Linciano, P.; Tassone, G.; Costi, M.P.; Mangani, S.; Pozzi, C. High-resolution crystal structure of trypanosoma brucei pteridine reductase 1 in complex with an innovative tricyclic-based inhibitor. Acta Cryst. 2020, D76, 558–564. [Google Scholar] [CrossRef]
- Alsaiari, A.A.; Almehmadi, M.M.; Asif, M. Diverse pharmacological potential of pyridazine analogs against various diseases. Med. Chem. 2024, 20, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, W.; Shaquiquzzaman, M.; Akhter, M.; Verma, G.; Khan, M.F.; Alam, M.M. The therapeutic journey of pyridazinone. Eur. J. Med. Chem. 2016, 123, 256–281. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-X.; Gong, Y.-P.; Zhang, X.; Ma, L.-Y.; Zhao, W. Pyridazine as a privileged structure: An updated review on anticancer activity of pyridazine containing bioactive molecules. Eur. J. Med. Chem. 2021, 209, 112946. [Google Scholar] [CrossRef]
- Zeinab, S.A.-R.; Aya, M.S.; Riham, F.G. An overview of pyridazin-3(2H)-one: A core for developing bioactive agents targeting cardiovascular diseases and cancer. Fut. Med. Chem. 2024, 16, 1685–1703. [Google Scholar] [CrossRef]
- Singh, J.; Kumar, V.; Silakari, P.; Kumar, S. Pyridazinones: A versatile scaffold in the development of potential target-based novel anticancer agents. J. Heterocycl. Chem. 2023, 60, 929–949. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, D.; Bansal, R. Pyridazinone: An attractive lead for anti-inflammatory and analgesic drug discovery. Future Med. Chem. 2017, 9, 95–127. [Google Scholar] [CrossRef]
- Holava, H.M., Jr.; Partyka, R.A. Benzocycloalka[l,2-c]pyridazones. J. Med. Chem. 1971, 14, 262–264. [Google Scholar]
- Campagna, F.; Palluotto, F.; Carotti, A.; Casini, G.; Genchi, G. Synthesis and structure-affinity relationships at the central benzodiazepine receptor of pyridazino[4,3-b]indoles and indeno[1,2-c]pyridazines. Bioorg. Med. Chem. 1999, 7, 1533–1538. [Google Scholar] [CrossRef]
- Bakewell, S.J.; Coates, W.J.; Comer, M.B.; Reeves, M.L.; Warrington, B.H. Inotropic, vasodilator and low Km, cAMP-selective, cGMP-inhibited phosphodiesterase (PDEIII) inhibitory activities of 4a-methyl-4,4a-dihydro-5H-indeno[1,2-c]pyridazine-3(2H)-ones and 4a-methyl-4,4a,5,6-tetrahydrobenzo[h]cinnoline-3(2H)-ones. Eur. J. Med. Chem. 1990, 25, 765–774. [Google Scholar] [CrossRef]
- Attiq, A.; Afzal, S.; Ahmad, W.; Kandeel, M. Hegemony of inflammation in atherosclerosis and coronary artery disease. Eur. J. Pharmacol. 2024, 966, 176338. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, Q.; Inchakalody, V.P.; Bedhiafi, T.; Mestiri, S.; Taib, N.; Uddin, S.; Merhi, M.; Dermine, S. Chronic inflammation and cancer; the two sides of a coin. Life Sci. 2024, 338, 122390. [Google Scholar] [CrossRef] [PubMed]
- British National Formulary, September 2024–March 2025 (BNF 88); Pharmaceutical Press: London, UK, 2024.
- Rosa, F.A.; Gonçalves, D.S.; Pianoski, K.E.; da Silva, M.J.V.; Ames, F.Q.; Aguitar, R.P.; Volpato, H.; Lazarin-Bidóia, D.; Nakamura, C.V.; Bersani-Amado, C.A. Discovery of a new pyrido[2,3-d]pyridazine-2,8-dione derivative as a potential anti-inflammatory agent through COX-1/COX-2 dual inhibition. RSC Med. Chem. 2024, 15, 1038–1045. [Google Scholar] [CrossRef]
- Cignarela, G.; Grella, G.; Loriga, M.; Curzu, M.M.; Schiatti, G. Inattesa attività antiinfiammatoria di strutture rigide derivate da 6-arilpiridazinoni antiipertensivi. Nota I—Sintesi e attività di 4,4a-diidro-5H-indeno[1,2-c]piridazin-3-oni. Il Farm. 1978, 33, 866–874. [Google Scholar]
- Cignarella, G.; Loriga, M.; Pinna, G.A.; Pirisi, M.A.; Schiatti, P.; Selva, D. Inattesa attività antiinfiammatoria di strutture rigide derivate da 6-arilpiridazinoni antiipertensivi. Nota III—Sintesi e attività di 7-fluoro e 5-cheto-5H-indeno[1,2-c]piridazine. Il Farm. 1982, 37, 133–144. [Google Scholar]
- Pinna, G.A.; Curzu, M.M.; Loriga, M.; Cignarella, G.; Barlocco, D.; Malandrino, S. Rigid congeners of arylpyridazinones IV—Synthesis and activity of derivatives of the new heterocyclic system 9H-indeno[2,1-c]pyridazine. Il Farm. 1985, 40, 979–986. [Google Scholar]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 20 January 2025).
- Chen, X.C.; Yang, L.; Aslam, M.F.; Tao, J.; Zhang, X.; Ren, P.; Wang, Y.; Chao, P. Functional analysis, virtual screening, and molecular dynamics revealed potential novel drug targets and their inhibitors against cardiovascular disease in human. J. Biomol. Struct. Dyn. 2024, 42, 6982–6996. [Google Scholar] [CrossRef]
- Xu, Y.; Ren, Y.; Zhang, J.; Niu, B.; Liu, M.; Xu, T.; Zhang, X.; Shen, J.; Wang, K.; Cao, Z. Discovery of pyridazinone derivatives bearing tetrahydroimidazo[1,2-a]pyrazine scaffold as potent inhibitors of transient receptor potential canonical 5 to ameliorate hypertension-induced renal injury in rats. Eur. J. Med. Chem. 2024, 275, 116565. [Google Scholar] [CrossRef]
- Aziz, M.W.; Mohamed, K.O.; Farag, D.B.; Khalifa, A.K.; Mahmoud, Z. Synthesis of potent vasodilating agents: In silico and in vitro evaluation of 6-(4-substituted phenyl)-3-pyridazinone derivatives as potential hydralazine analogues. Sci. Rep. 2024, 14, 29514. [Google Scholar] [CrossRef]
- Cignarella, G.; Barlocco, D.; Pinna, G.A.; Loriga, M.; Tofanetti, O.; Germini, M.; Sala, F. Conformationally restricted congeners of hypotensive and platelet aggregation inhibitors: 6-aryl-5-methyl-4,5-dihydro-3(2H)-pyridazinones derived from 5H-indeno[1,2-c]pyridazine. J. Med. Chem. 1986, 29, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Cignarella, G.; Barlocco, D.; Landrini, L.; Folloni, M.; Pinna, G.A.; Sala, F.; Germini, M. New congeners of antihypertensive and antithrombotic 7-amino or 7-acetyl-amino substituted 4,4a-dihydro-5H-indeno[1,2-c]pyridazine-3-ones. Il Farm. 1988, 43, 169–179. [Google Scholar]
- Cignarella, G.; Barlocco, D.; Pinna, G.A.; Loriga, M.; Curzu, M.M.; Tofanetti, O.; Germini, M.; Cazzulani, P.; Cavalletti, E. Synthesis and biological evaluation of substituted benzo[h]cinnolinones and 3H-benzo[6,7]cyclohepta[1,2-c]pyridazinones: Higher homologues of the antihypertensive and antithrombotic 5H-indeno[1,2-c]pyridazinones. J. Med. Chem. 1989, 32, 2277–2282. [Google Scholar] [CrossRef]
- Barlocco, D.; Pinna, G.A.; Carboni, L.; Cipolla, P. Synthesis and pharmacological study of 4,4a,5,6-tetrahydro-4a-substituted-benzo[h]cinnolin-3(2H)ones. Il Farm. 1989, 44, 967–974. [Google Scholar] [CrossRef]
- Toma, L.; Cignarella, G.; Barlocco, D.; Ronchetti, F. Conformationally constrained congeners of 6-aryl-5-methyl-4,5-dihydro-3(2H)-pyridazinones active on the cardiovascular system: Conformational studies by molecular mechanics calculations and 1H NMR spectroscopy. J. Med. Chem. 1990, 33, 1591–1594. [Google Scholar] [CrossRef]
- Sircar, I.; Duell, B.L.; Cain, M.H.; Burke, S.E.; Bristol, J.A. Cardiotonic agents. 4. Synthesis and biological evaluation of N-substituted 2,4,4a,5-tetrahydro-3H-indeno[1,2-c]pyridazin-3-ones: Rigid structures derived from CI-930 and analogues. J. Med. Chem. 1986, 29, 2142–2148. [Google Scholar] [CrossRef]
- Pinna, G.A.; Curzu, M.M.; Fraghì, P.; Gavini, E.; D’Amico, M. Synthesis and pharmacological evaluation of 5,6-dihydrobenzo[f]cinnoline-2(3H)ones analogues of antihypertensive and antiaggregating benzo[h]cinnolinones. Il Farm. 1996, 51, 653–658. [Google Scholar]
- Jayashree, B.S.; Nikhil, P.S.; Paul, S. Bioisosterism in drug discovery and development—An overview. Med. Chem. 2022, 18, 915–925. [Google Scholar] [CrossRef]
- Subbaiah, M.A.M.; Meanwell, N.A. Bioisosteres of the phenyl ring: Recent strategic applications in lead optimization and drug design. J. Med. Chem. 2021, 64, 14046–14128. [Google Scholar] [CrossRef]
- Cignarella, G.; Barlocco, D.; Curzu, M.M.; Pinna, G.A.; Cazzulani, P.; Cassin, M.; Lumachi, B. Synthesis and pharmacological evaluation of 4,4a-dihydro-5H-[1]benzopyrano[4,3-c]pyridazin-3(2H)-ones bioisosters of antihypertensiove and antithrombotic benzo[h]cinnolinones. Eur. J. Med. Chem. 1990, 25, 749–756. [Google Scholar] [CrossRef]
- Pinna, G.A.; Curzu, M.M.; Cignarella, G.; Barlocco, D.; D’Amico, M.; Filippelli, A.; De Novellis, V.; Rossi, F. Synthesis and pharmacological evaluation of thienocinnolin-3-(2H)-ones, bioisosters of antihypertensive and antithrombotic benzo[h]cinnolinones. Eur. J. Med. Chem. 1994, 29, 447–454. [Google Scholar] [CrossRef]
- Pinna, G.A.; Salis, E.; Berta, D.; Gavini, E. Synthesis and pharmacological evaluation of 4a-methyl-4,4a,5,6-tetrahydrothieno[2,3-h]cinnolin-3(H)-ones. Il Farm. 1997, 52, 29–33. [Google Scholar]
- Monge, A.; Parrado, P.; Font, M.; Fernãndez-Alvarez, E. Selective thromboxane synthetase inhibitors and antihypertensive agents. New derivatives of 4-hydrazino-5H-pyridazino[4,5-b]indole, 4-hydrazinopyridazino[4,5-a]indole, and related compounds. J. Med. Chem. 1987, 30, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Nobuhara, Y.; Shimamura, H.; Tsukamoto, Y.; Yoshihara, K.; Yamaguchi, A.; Ohki, M. Pyridazinones. 2. Synthesis and antisecretory and antiulcer activities of 2-thiourea and 2-cyanoguanidine derivatives. J. Med. Chem. 1983, 26, 373–381. [Google Scholar] [CrossRef]
- Cignarella, G.; Barlocco, D.; Villa, S.; Curzu, M.M.; Pinna, G.A.; Lavezzo, A.; Bestetti, A. Tricyclic 3-(2H)-pyridazinone derivatives. Synthesis and evaluation of their antisecretory and antiulcer activity. Eur. J. Med. Chem. 1992, 27, 819–823. [Google Scholar] [CrossRef]
- Barlocco, D.; Cignarella, G.; Vianello, P.; Dal Piaz, V.; Giovannoni, M.P.; Malandrino, S.; Barocelli, E.; Chiavarini, M.; Impicciatore, M. Indenopyridazinone derivatives as potential antisecretory and antiulcer agents. Drug Des. Discov. 1997, 15, 95–103. [Google Scholar]
- Barocelli, E.; Chiavarini, M.; Ballabeni, V.; Barlocco, D.; Vianello, P.; Dal Piaz, V.; Impicciatore, M. Study of the antisecretory and antiulcer mechanisms of a new indenopirydazinone derivative in rats. Pharm. Res. 1997, 35, 487–492. [Google Scholar] [CrossRef]
- Roche, V.F. Drugs used to treat neoplastic diseases. In Foye’s Principles of Medicinal Chemistry, 8th ed.; Roche, V.F., Zito, W.S., Lemke, T.L., Williams, D.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2020; Volume 33, pp. 1309–1442. [Google Scholar]
- Dobbelstein, M.; Moll, U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat. Rev. Drug Discov. 2014, 13, 179–196. [Google Scholar] [CrossRef]
- Li, Y. DNA adducts in cancer chemotherapy. J. Med. Chem. 2024, 67, 5113–5143. [Google Scholar] [CrossRef]
- Hawash, M. Recent advances of tubulin inhibitors targeting the colchicine binding site for cancer therapy. Biomolecules 2022, 12, 1843. [Google Scholar] [CrossRef]
- Sabnis, R.W. Novel exatecan-derived topoisomerase-1 inhibitors for treating cancer. ACS Med. Chem. Lett. 2024, 15, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Sharma, V.; Mehra, A.; Singh, I. DNA intercalators as anticancer agents. Chem. Biol. Drug Des. 2022, 100, 580–598. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome inhibitor drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Wang, K.; He, Z.; Jin, G.; Jin, S.; Du, Y.; Yuan, S.; Zhang, J. Targeting DNA methyltransferases for cancer therapy. Bioorg. Chem. 2024, 151, 107652. [Google Scholar] [CrossRef]
- Xu, M.; Zhao, C.; Zhu, B.; Wang, L.; Zhou, H.; Yan, D.; Gu, Q.; Xu, J. Discovering high potent Hsp90 inhibitors as antinasopharyngeal carcinoma agents through fragment assembling approach. J. Med. Chem. 2021, 64, 2010–2023. [Google Scholar] [CrossRef]
- Barlocco, D.; Bergomi, M.; Menta, E.; Palumbo, M.; Cignarella, G. Tricyclic indenopyridazinone derivatives: Synthesis, cytotoxic activity, and DNA-binding properties. Rec. Trav. Chim. Pays-Bas 1996, 115, 25–30. [Google Scholar] [CrossRef]
- Murineddu, G.; Cignarella, G.; Chelucci, G.; Loriga, G.; Pinna, G.A. Synthesis and cytotoxic activities of pyrrole[2,3-d]pyridazine-4-one derivatives. Chem. Pharm. Bull. 2002, 50, 754–759. [Google Scholar] [CrossRef]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef] [PubMed]
- Pau, A.; Murineddu, G.; Asporoni, B.; Murruzzu, C.; Grella, G.E.; Pinna, G.A.; Curzu, M.M.; Marchesi, I.; Bagella, L. Synthesis and cytotoxicity of novel hexahydrothienocycloheptapyridazinone derivatives. Molecules 2009, 14, 3494–3508. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. Recent update on development of small-molecule STAT3 inhibitors for cancer therapy: From phosphorylation inhibition to protein degradation. J. Med. Chem. 2021, 64, 8884–8915. [Google Scholar] [CrossRef] [PubMed]
- Masciocchi, D.; Gelain, A.; Porta, F.; Meneghetti, F.; Pedretti, A.; Celentano, G.; Barlocco, D.; Legnani, L.; Toma, L.; Kwon, B.M.; et al. Synthesis, structure–activity relationships and stereochemical investigations of new tricyclic pyridazinone derivatives as potential STAT3 inhibitors. Med. Chem. Commun. 2013, 4, 1181–1188. [Google Scholar] [CrossRef]
- Wu, B.; Liang, Z.; Lan, H.; Teng, X.; Wang, C. The role of PKM2 in cancer progression and its structural and biological basis. J. Physiol. Biochem. 2024, 80, 261–275. [Google Scholar] [CrossRef]
- Jiang, J.-K.; Boxer, M.B.; Heiden, M.G.V.; Shen, M.; Skoumbourdis, A.P.; Southall, N.; Veith, H.; Leister, W.; Austin, C.P.; Park, H.W.; et al. Evaluation of thieno[3,2-b]pyrrole[3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg. Med. Chem. Lett. 2010, 20, 3387–3393. [Google Scholar] [CrossRef]
- Liu, T.; Padyana, A.K.; Judd, E.T.; Jin, L.; Hammoudeh, D.; Kung, C.; Dang, L. Structure-based design of AG-946, a pyruvate kinase activator. ChemMedChem 2024, 19, e202300559. [Google Scholar] [CrossRef]
- Bruel, A.; Logé, C.; de Tauzia, M.-L.; Ravache, M.; Le Guevel, R.; Guillouzo, C.; Lohier, J.-F.; Sopkova-de Oliveira Santos, J.; Lozach, O.; Meijer, L.; et al. Synthesis and biological evaluation of new 5-benzylated 4-oxo-3,4-dihydro-5H-pyridazino[4,5-b]indoles as PI3Ka inhibitors. Eur. J. Med. Chem. 2012, 57, 225–233. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, J.; Ren, W.C.; Chen, H.; Zhang, J. DYRK1A inhibitors for disease therapy: Current status and perspectives. Eur. J. Med. Chem. 2022, 229, 114062. [Google Scholar] [CrossRef]
- Zheng, X.-F.; Sarkar, A.; Lotana, H.; Syed, A.; Nguyen, H.; Ivey, R.G.; Kennedy, J.J.; Whiteaker, J.R.; Tomasik, B.; Huang, K.; et al. CDK5-cyclin B1 regulates mitotic fidelity. Nature 2024, 633, 932–940. [Google Scholar] [CrossRef]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K inhibitors in cancer: Clinical implications and adverse effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef] [PubMed]
- Bruel, A.; Bénéteau, R.; Chabanne, M.; Lozach, O.; Le Guevel, R.; Ravache, M.; Bénédetti, H.; Meijer, L.; Logé, C.; Robert, J.-M. Synthesis of new pyridazino[4,5-b]indol-4-ones and pyridazin-3(2H)-one analogs as DYRK1A inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 5037–5040. [Google Scholar] [CrossRef] [PubMed]
- Panathur, N.; Gokhale, N.; Dalimba, U.; Koushik, P.V.; Yogeeswari, P.; Sriram, D. Synthesis of novel 5-[(1,2,3-triazol-4-yl)methyl]-1-methyl-3H-pyridazino[4,5-b]indol-4-one derivatives by click reaction and exploration of their anticancer activity. Med. Chem. Res. 2016, 25, 135–148. [Google Scholar] [CrossRef]
- Sarhan, A.A.M.; Boraei, A.T.A.; Barakat, A.; Nafie, M.S. Discovery of hydrazide-based pyridazino[4,5-b]indole scaffold as a new phosphoinositide 3-kinase (PI3K) inhibitor for breast cancer therapy. RSC Adv. 2020, 10, 19534–19541. [Google Scholar] [CrossRef]
- Zeng, Y.; Arisa, O.; Peer, C.J.; Fojo, A.; Figg, W.D. PARP inhibitors: A review of the pharmacology, pharmacokinetics, and pharmacogenetics. Semin. Oncol. 2024, 51, 19–24. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Y.; Zhang, Z.; Zhao, J.; Jia, W.; Xia, C.; Wang, F.; Liu, T. Multi-therapies based on PARP inhibition: Potential therapeutic approaches for cancer treatment. J. Med. Chem. 2022, 65, 16099–16127. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First global approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/search?term=E-7016 (accessed on 20 January 2025).
- Wang, J.; Tan, H.; Sun, Q.; Ge, Z.; Wang, X.; Wang, Y.; Li, R. Design, synthesis and biological evaluation of pyridazino[3,4,5-de]quinazolin-3(2H)-one as a new class of PARP-1 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 2340–2344. [Google Scholar] [CrossRef]
- Renu, S.; Priyanka, R.; Sarita, K.; EsraKüpeli, A.; Khayatkashani, M.; Seyed, M.N.; Anurag, K. Dihydrofolate reductase (DHFR) inhibitors: A comprehensive review. Curr. Med. Chem. 2024, 31, 799–824. [Google Scholar] [CrossRef]
- Ewida, M.A.; Abou El Ella, D.A.; Lasheen, D.S.; Ewida, H.A.; El-Gazzar, Y.I.; El-Subbagh, H.I. Imidazo[2′,1′:2,3]thiazolo[4,5-d]pyridazinone as a new scaffold of DHFR inhibitors: Synthesis, biological evaluation and molecular modeling study. Bioorg. Chem. 2018, 80, 11–23. [Google Scholar] [CrossRef]
- Zito, S.W. Drugs used to treat diabetic disorders. In Foye’s Principles of Medicinal Chemistry, 8th ed.; Roche, V.F., Zito, W.S., Lemke, T.L., Williams, D.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2020; Volume 20, pp. 791–823. [Google Scholar]
- Cole, J.B.; Florez, J.C. Genetics of diabetes and diabetes complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Niimi, N.; Yako, H.; Takaku, S.; Chung, S.K.; Sango, K. Aldose reductase and the polyol pathway in schwann cells: Old and new problems. Int. J. Mol. Sci. 2021, 22, 1031. [Google Scholar] [CrossRef] [PubMed]
- Quattrini, L.; La Motta, C. Aldose reductase inhibitors: 2013-present. Expert Opin. Ther. Pat. 2019, 29, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Imran, A.; Shehzad, M.T.; Shah, S.J.A.; Laws, M.; al-Adhami, T.; Rahman, K.M.; Khan, I.A.; Shafiq, Z.; Iqbal, J. Development, molecular docking, and in silico ADME evaluation of selective ALR2 inhibitors for the treatment of diabetic complications via suppression of the polyol pathway. ACS Omega 2022, 7, 26425–26436. [Google Scholar] [CrossRef]
- Sangshetti, J.K.; Pathan, S.K.; Patil, R.; Ansari, S.A.; Chhajed, S.; Arote, R.; Shinde, D.B. Synthesis and biological activity of structurally diverse phthalazine derivatives: A systematic review. Bioorg. Med. Chem. 2019, 27, 3979–3997. [Google Scholar] [CrossRef]
- Costantino, L.; Rastelli, G.; Vescovini, K.; Cignarella, G.; Vianello, P.; Del Corso, A.; Cappiello, M.; Mura, U.; Barlocco, D. Synthesis, activity, and molecular modeling of a new series of tricyclic pyridazinones as selective aldose reductase inhibitors. J. Med. Chem. 1996, 39, 4396–4405. [Google Scholar] [CrossRef]
- Costantino, L.; Rastelli, G.; Cignarella, G.; Barlocco, D. Synthesis and aldose reductase inhibitory activity of a new series of benzo[h]cinnolinone derivatives. Il Farm. 2000, 55, 544–552. [Google Scholar] [CrossRef]
- Rastelli, G.; Vianello, P.; Barlocco, D.; Costantino, L.; Del Corso, D.; Mura, U. Structure-based design of an inhibitor modeled at the substrate active site of aldose reductase. Bioorg. Med. Chem. Lett. 1997, 7, 1897–1902. [Google Scholar] [CrossRef]
- Pau, A.; Asproni, B.; Murineddu, G.; Boatto, G.P.; Grella, G.E.; Rakowitz, D.; Costantino, L.; Pinna, G.A. Thienocinnolinone alkanoic acid derivatives as aldose reductase inhibitors. Med. Chem. 2006, 2, 39–45. [Google Scholar] [CrossRef]
- Mylari, B.L.; Zembrowski, W.J.; Beyer, T.A.; Aldinger, C.E.; Siege, T.W. Orally active aldose reductase inhibitors: Indazoleacetic, oxopyridazineacetic, and oxopyridopyridazineacetic acid derivatives. J. Med. Chem. 1992, 35, 2155–2162. [Google Scholar] [CrossRef]
- Canal, C.E.; Booth, R.G.; Williams, D.A. Drugs used to treat mental, behavioral, and cognitive disorders. In Foye’s Principles of Medicinal Chemistry, 8th ed.; Roche, V.F., Zito, W.S., Lemke, T.L., Williams, D.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2020; Volume 11, pp. 313–421. [Google Scholar]
- Tanaka, H.; Kirihara, S.; Yasumatsu, H.; Yakushiji, T.; Nakao, T. Synthesis and structure-activity analysis of 2-(4-chlorophenyl)-5,6-dihydrothieno[2’,3’:2,3]thiepino[4,5-c]pyridazin-3(2H)-ones as ligands for benzodiazepine receptors. Eur. J. Med. Chem. 1995, 30, 859–868. [Google Scholar] [CrossRef]
- Tanaka, H.; Kirihara, S.; Yasumatsu, H.; Yakushiji, T.; Nakao, T. Synthesis and evaluation of novel 2-aryl-2,5,6,7-tetrahydro-3H-thieno[2’,3’:6,7]cyclohepta[1,2-c]lpyridazin-3-ones and 2-aryl-5,6-dihydrothieno[2,3-h]cinnolin3(2H)-ones as anxiolytics. Eur. J. Med. Chem. 1997, 32, 607–615. [Google Scholar] [CrossRef]
- Primofiore, G.; Da Settimo, F.; Marini, A.M.; Simorini, F.; La Motta, C.; Taliani, S.; Laneri, S.; Trincavelli, L.; Martini, C. Synthesis and benzodiazepine receptor affinity of derivatives of the new tricyclic heteroaromatic system pyrido[3’,2’:5,6]thiopyrano[4,3-c]pyridazin-3(2H,5H)-one. Arch. Pharm. Chem. Life Sci. 2005, 338, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef]
- Jóźwik, I.; Passos, D.O.; Lyumkis, D. Structural biology of HIV integrase strand transfer inhibitors. Trends Pharmacol. Sci. 2020, 41, 611–626. [Google Scholar] [CrossRef]
- Wiscount, C.M.; Williams, P.D.; Tran, L.O.; Embrey, M.W.; Fisher, T.E.; Sherman, V.; Homnick, C.F.; Staas, D.D.; Lyle, T.A.; Wai, J.S.; et al. 10-Hydroxy-7,8-dihydropyrazino[1’,2’:1,5]pyrrolo[2,3-d]pyridazine-1,9(2H,6H)-diones: Potent, orally bioavailable HIV-1 integrase strand-transfer inhibitors with activity against integrase mutants. Bioorg. Med. Chem. Lett. 2008, 18, 4581–4583. [Google Scholar] [CrossRef]
- Abdel-Hamid, N.M.; Abass, S.A. Matrix metalloproteinase contribution in management of cancer proliferation, metastasis and drug targeting. Mol. Biol. Rep. 2021, 48, 6525–6538. [Google Scholar] [CrossRef]
- de Morais, E.F.; Pinheiro, J.C.; Leite, R.B.; Santos, P.P.A.; Barboza, C.A.G.; Freitas, R.A. Matrix metalloproteinase-8 levels in periodontal disease patients: A systematic review. J. Periodontal. Res. 2018, 53, 156–163. [Google Scholar] [CrossRef]
- Pinna, G.A.; Curzu, M.M.; Murineddu, G.; Chelucci, G.; Cignarella, G.; Menta, E.; Krell, H.W.; Rastelli, G.; Ferrari, A.M. Preparation of thieno[3,2-h]cinnolinones as matrix metalloproteinase inhibitors. Arch. Pharm. Med. Chem. 2000, 333, 37–47. [Google Scholar] [CrossRef]
- Samidurai, A.; Xi, L.; Das, A.; Kukreja, R.C. Beyond erectile dysfunction: cGMP-specific phosphodiesterase 5 inhibitors for other clinical disorders. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 585–615. [Google Scholar] [CrossRef]
- Feixas, J.; Giovannoni, M.P.; Vergelli, C.; Gavaldà, A.; Cesari, N.; Graziano, A.; Dal Piaz, V. New pyrazolo[1’,5’:1,6]pyrimido[4,5-d]pyridazin-4(3H)-ones as potent and selective PDE5 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2381–2384. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, M.P.; Vergelli, C.; Biancalani, C.; Cesari, N.; Graziano, A.; Biagini, P.; Gracia, J.; Gavaldà, A.; Dal Piaz, V. Novel pyrazolopyrimidopyridazinones with potent and selective phosphodiesterase 5 (PDE5) inhibitory activity as potential agents for treatment of erectile dysfunction. J. Med. Chem. 2006, 49, 5363–5371. [Google Scholar] [CrossRef] [PubMed]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on adenosine receptors as a potential targeted therapy in human diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, M.P.; Claudia Vergelli, V.; Cilibrizzi, A.; Crocetti, L.; Biancalani, C.; Graziano, A.; Dal Piaz, V.; Loza, M.I.; Cadavid, M.I.; Díaz, J.L.; et al. Pyrazolo[1’,5’:1,6]pyrimido[4,5-d]pyridazin-4(3H)-ones as selective human A1 adenosine receptor ligands. Bioorg. Med. Chem. 2010, 18, 7890–7899. [Google Scholar] [CrossRef]
- Giovannoni, M.P.; Ciciani, G.; Cilibrizzi, A.; Crocetti, L.; Daniele, S.; Di Cesare Mannelli, L.; Ghelardini, C.; Giacomelli, C.; Guerrini, G.; Martini, C.; et al. Further studies on pyrazolo[1’,5’:1,6]pyrimido[4,5-d]pyridazin-4(3H)-ones as potent and selective human A1 adenosine receptor antagonists. Eur. J. Med. Chem. 2015, 89, 32–41. [Google Scholar] [CrossRef]
- Barresi, E.; Robello, M.; Costa, B.; Da Pozzo, E.; Baglini, E.; Salerno, S.; Da Settimo, F.; Martini, C.; Taliani, S. An update into the medicinal chemistry of translocator protein (TSPO) ligands. Eur. J. Med. Chem. 2021, 209, 112924. [Google Scholar] [CrossRef]
- Salerno, S.; Viviano, M.; Baglini, E.; Poggetti, V.; Giorgini, D.; Castagnoli, J.; Barresi, E.; Castellano, S.; Da Settimo, F.; Taliani, S. TSPO radioligands for neuroinflammation: An overview. Molecules 2024, 29, 4212. [Google Scholar] [CrossRef]
- Singh, P.; Singh, V.K.; Gond, C.; Singh, D.; Tiwari, A.K. Current advances in the structure-activity relationship (SAR) analysis of the old/new 18-kDa translocator protein ligands. Mol. Div. 2024. [Google Scholar] [CrossRef]
- Pathak, C.; Kabra, U.D. A comprehensive review of multi-target directed ligands in the treatment of Alzheimer’s disease. Bioorg. Chem. 2024, 144, 107152. [Google Scholar] [CrossRef]
- Asproni, B.; Catto, M.; Loriga, G.; Murineddu, G.; Corona, P.; Purgatorio, R.; Cichero, E.; Fossa, P.; Scarano, N.; Martínez, A.L.; et al. Novel thienocycloalkylpyridazinones as useful scaffolds for acetylcholinesterase inhibition and serotonin 5-HT6 receptor interaction. Bioorg. Med. Chem. 2023, 84, 117256. [Google Scholar] [CrossRef]
- Karamali, F.; Behtaj, S.; Babaei-Abraki, S.; Hadady, H.; Atefi, A.; Savoj, S.; Soroushzadeh, S.; Najafian, S.; Mohammad Hossein Nasr Esfahani, M.H.N.; Klassen, H. Potential therapeutic strategies for photoreceptor degeneration: The path to restore vision. J. Transl. Med. 2022, 20, 572. [Google Scholar] [CrossRef]
- Wubben, T.J.; Chaudhury, S.; Watch, B.T.; Stuckey, J.A.; Weh, E.; Fernando, R.; Goswami, M.; Pawar, M.; Rech, J.C.; Besirli, C.G. Development of novel small-molecule activators of pyruvate kinase muscle isozyme 2, PKM2, to reduce photoreceptor apoptosis. Pharmaceuticals 2023, 16, 705. [Google Scholar] [CrossRef]



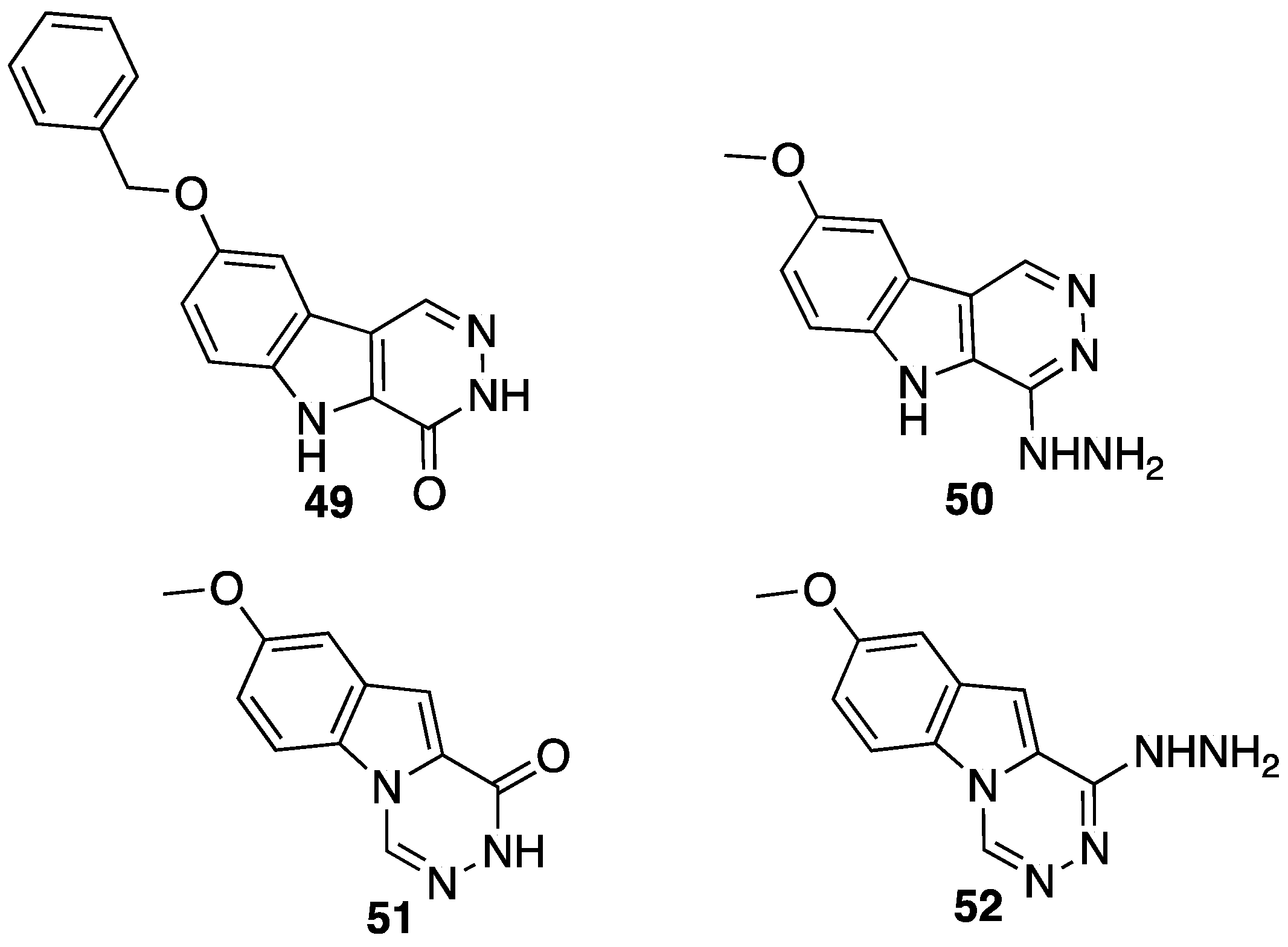

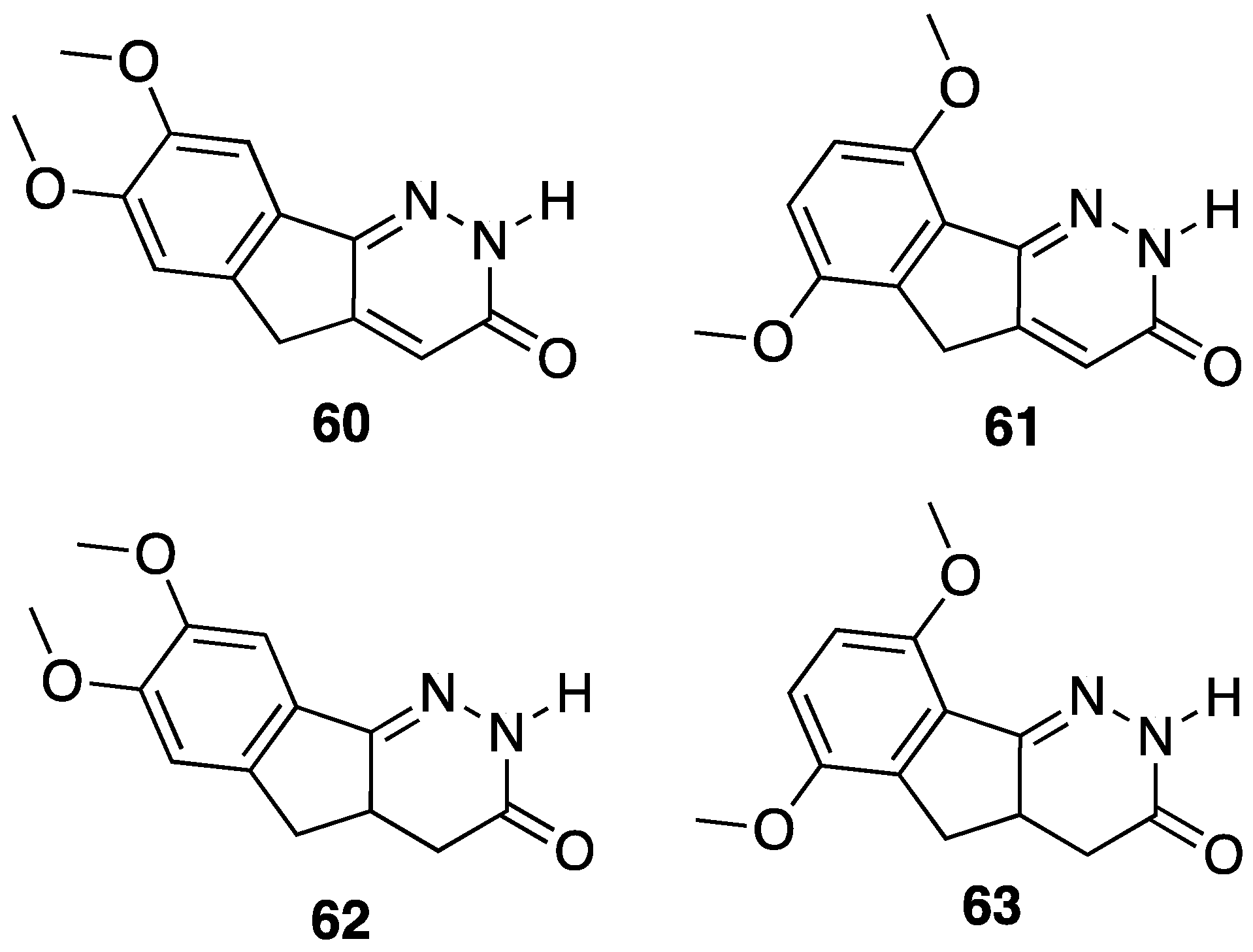
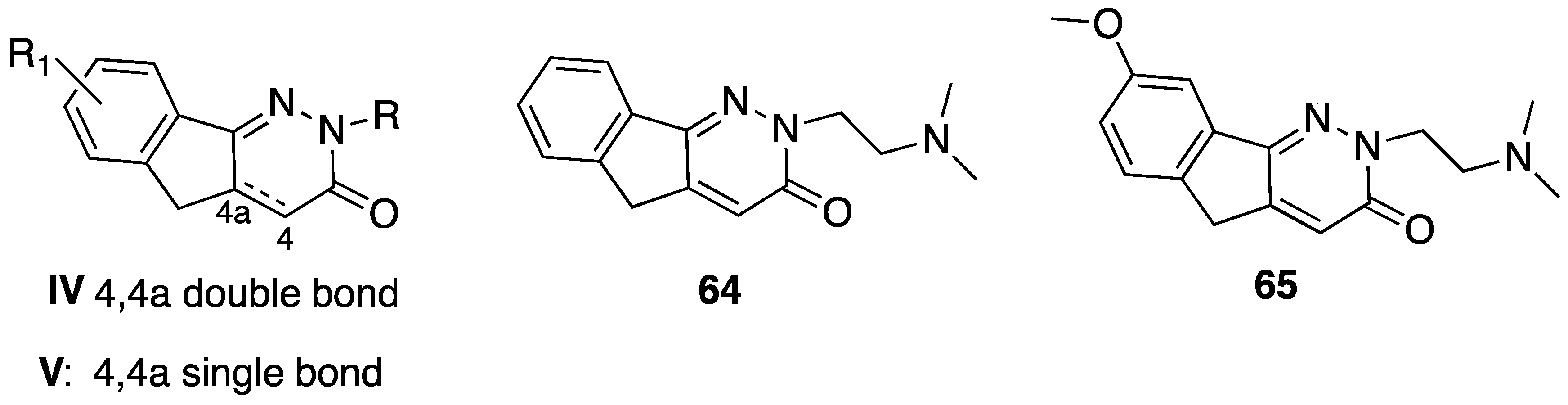
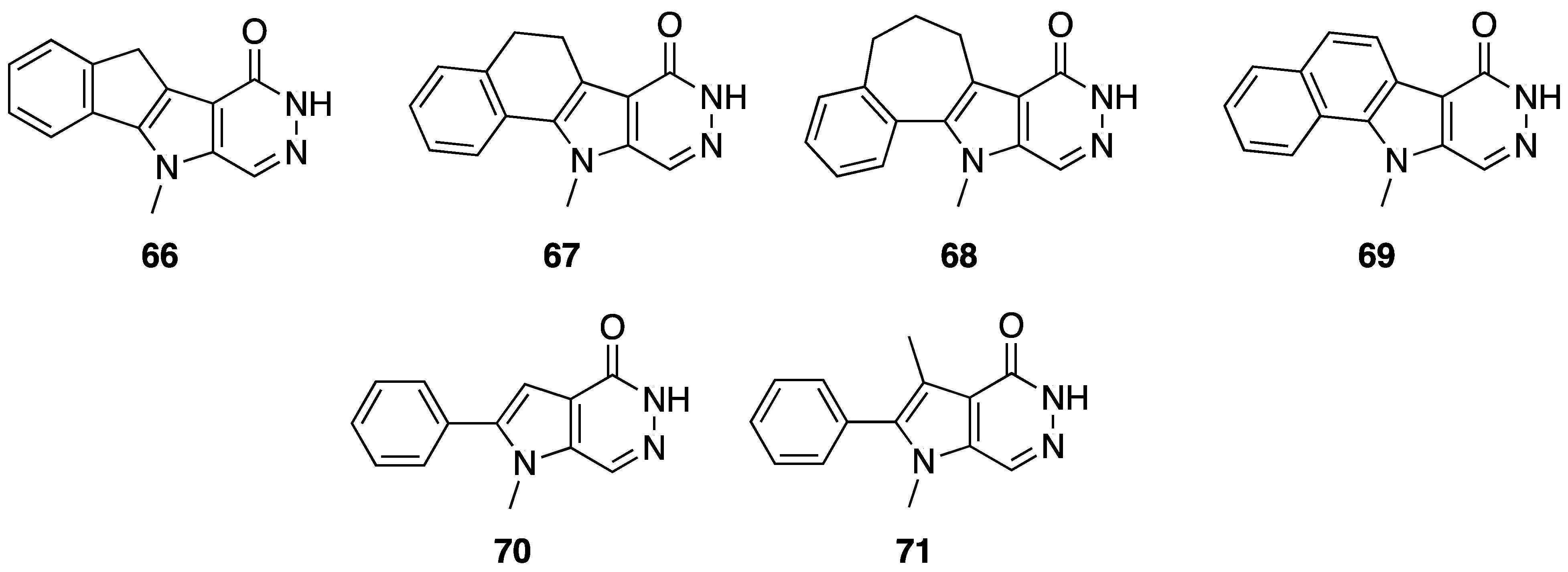

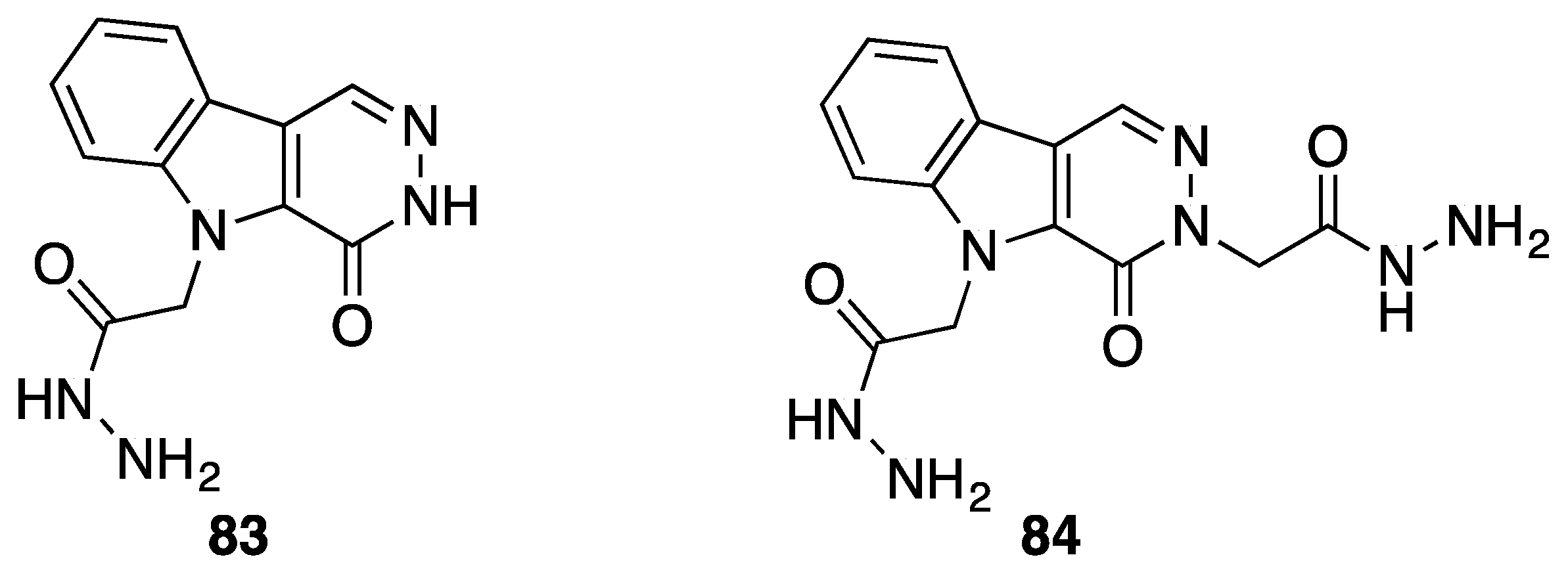
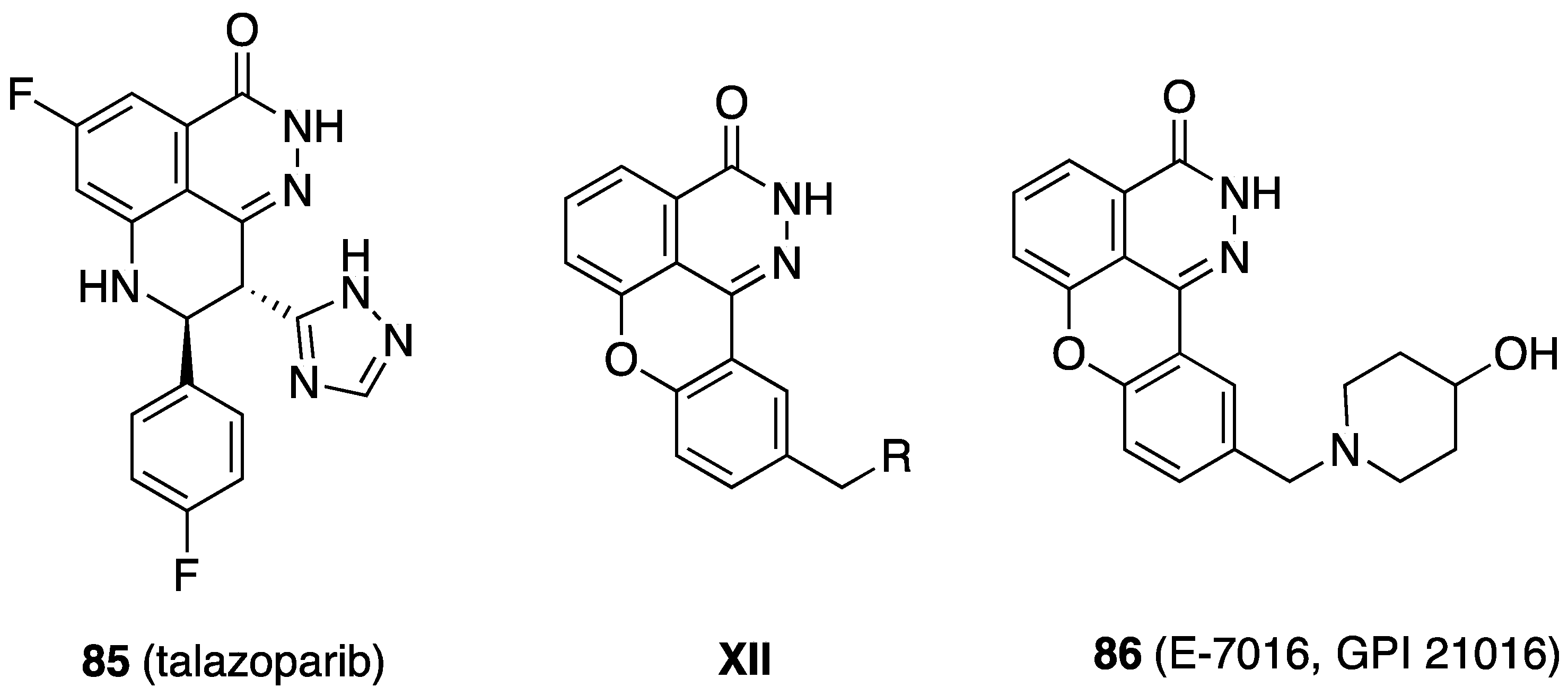
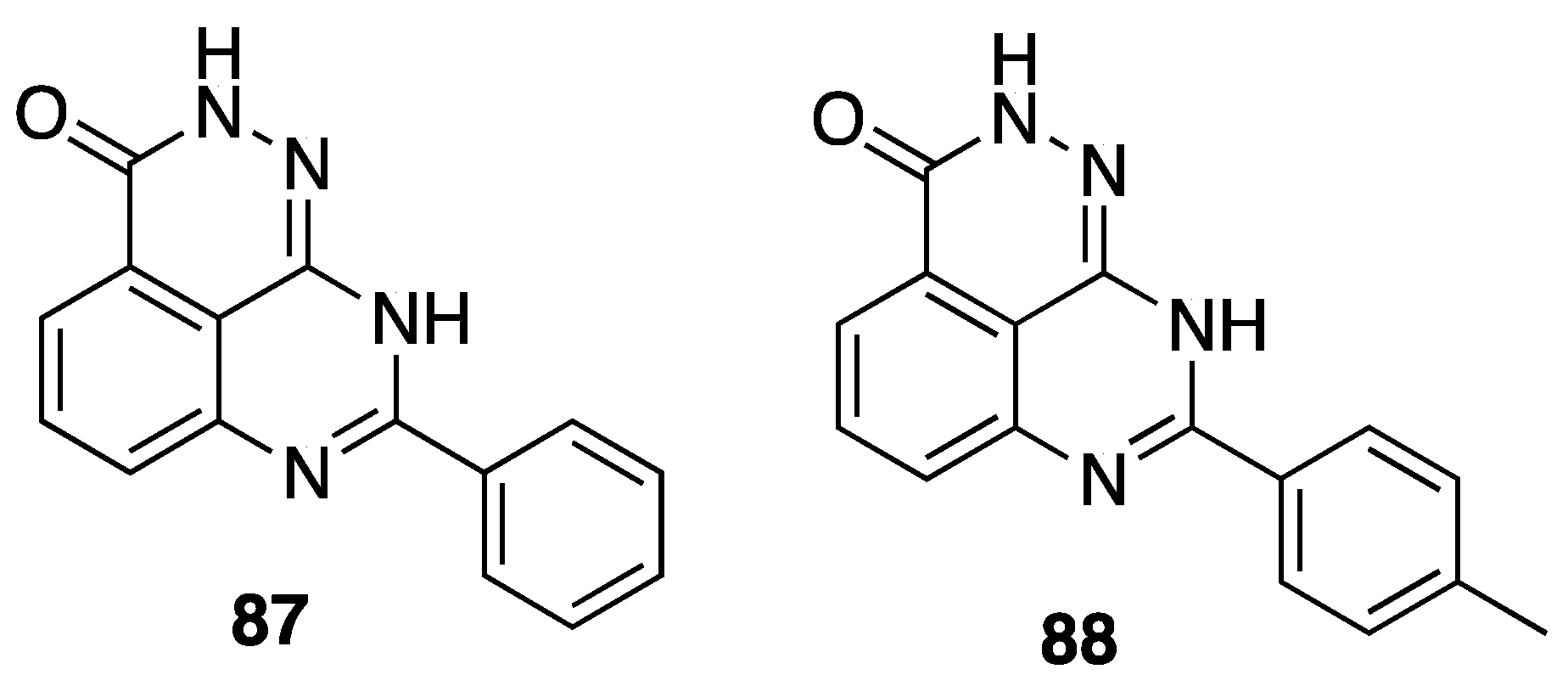
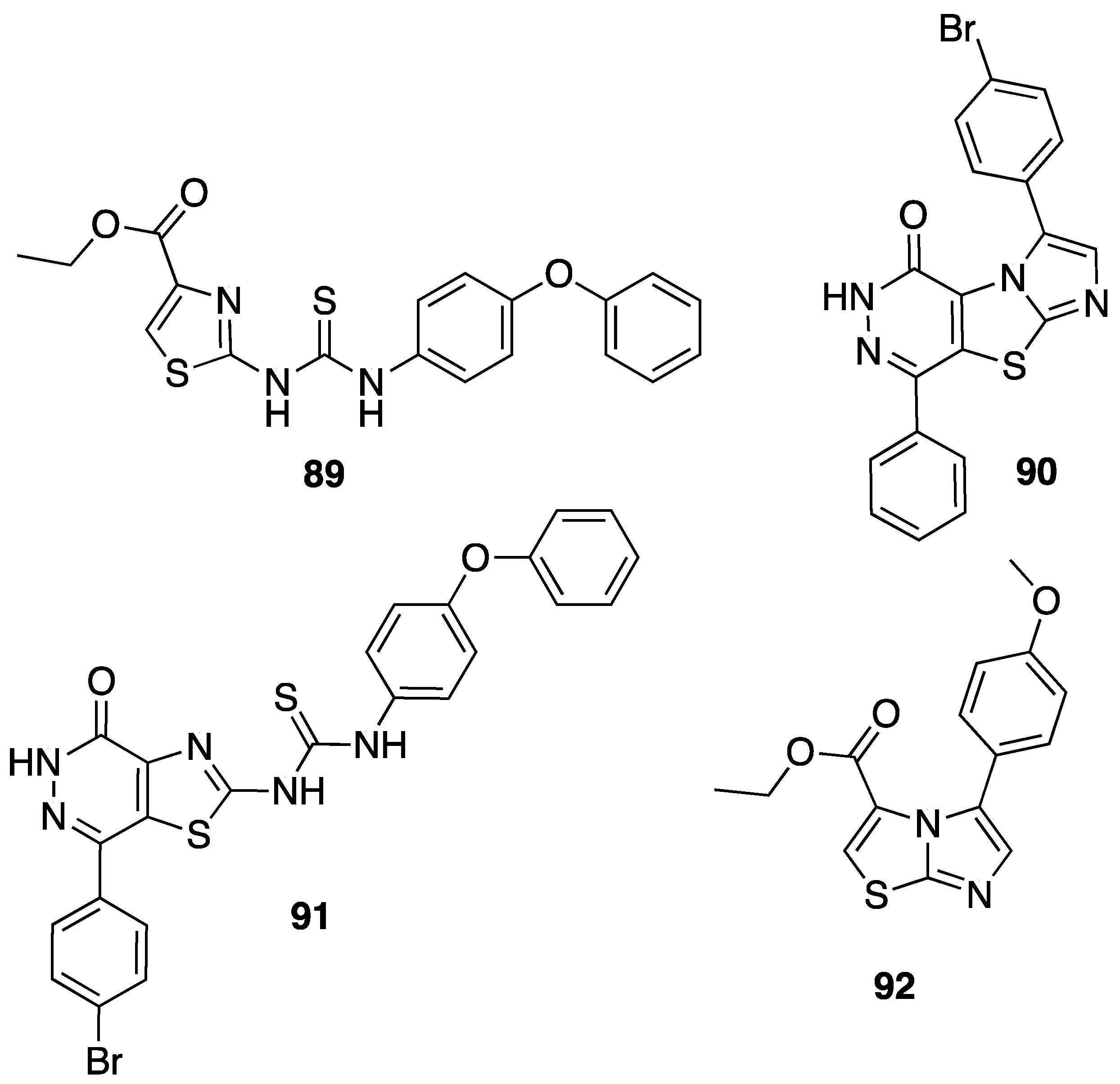


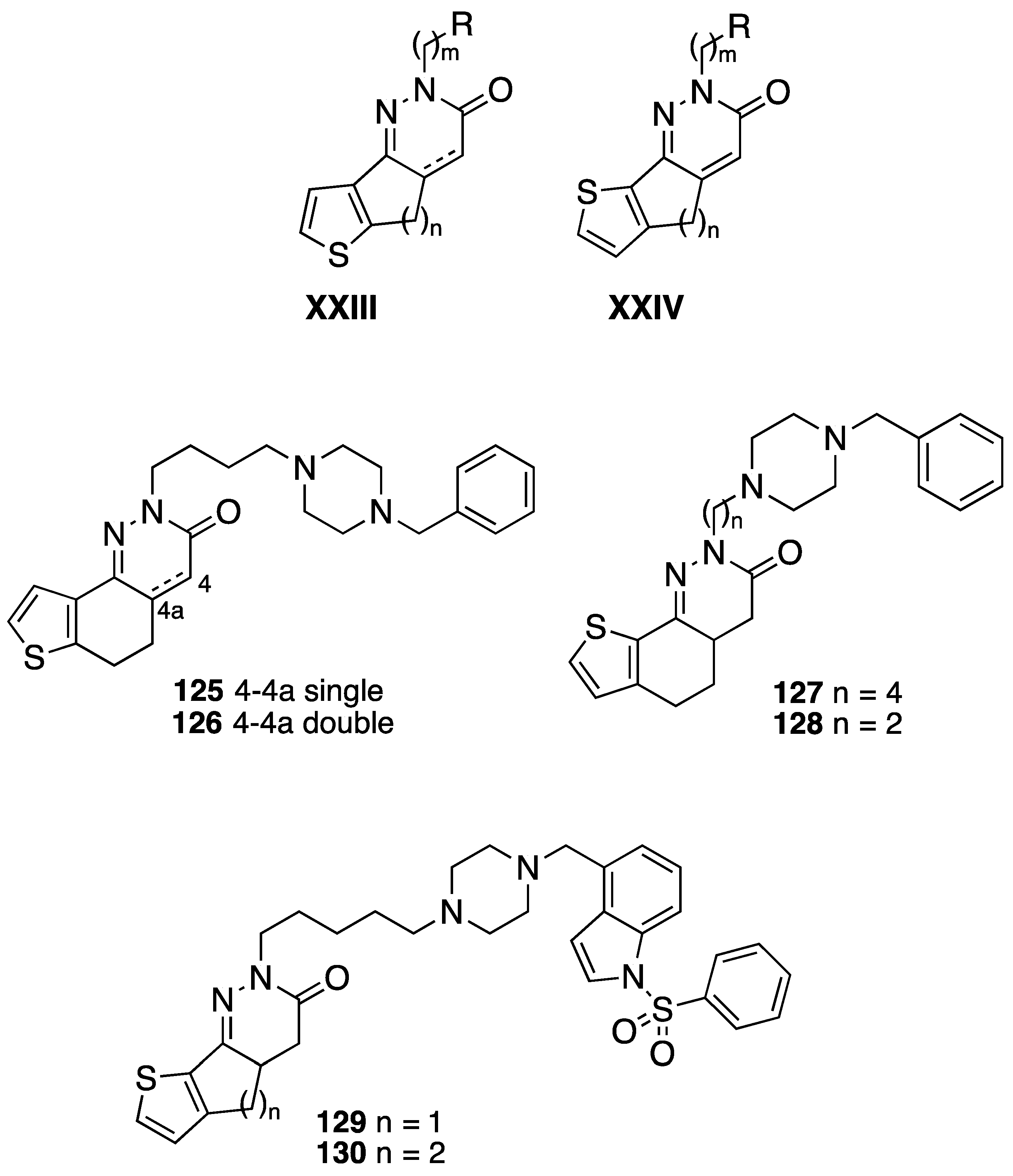
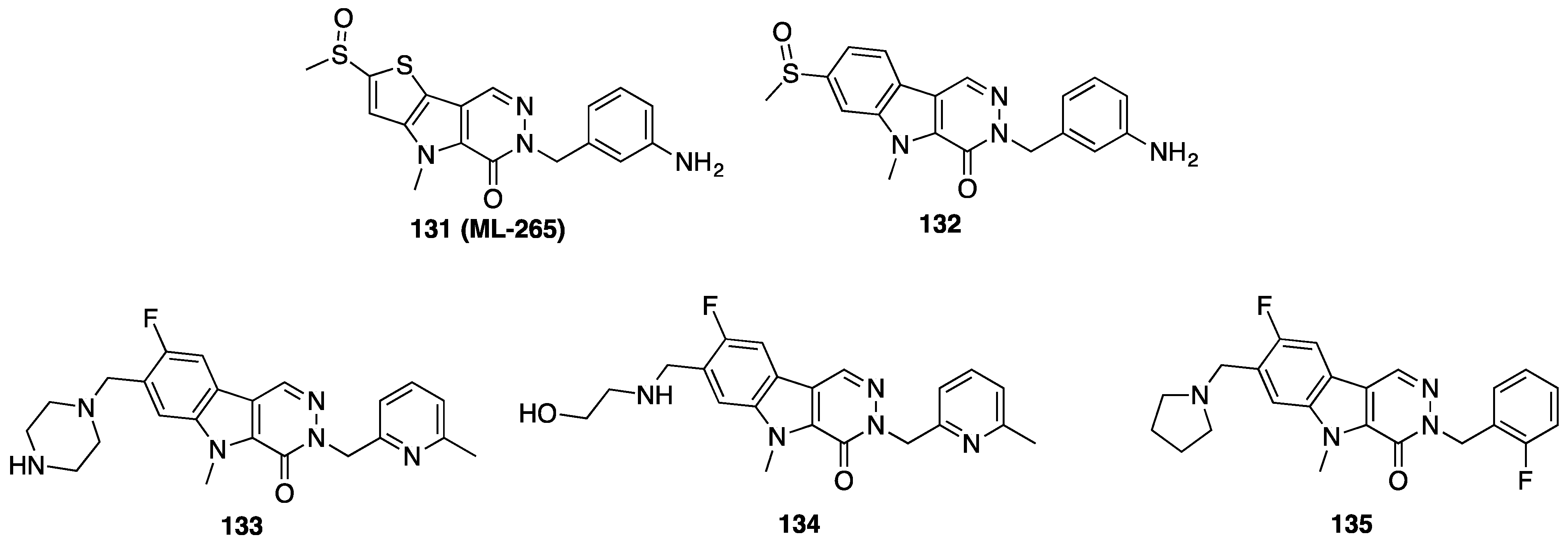

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | mg/Kg | Carrageenin Rat Paw Edema Inhibition % | Compd. | mg/Kg | Carrageenin Rat Paw Edema Inhibition % |
|---|---|---|---|---|---|
| 2 | 50 100 | 22 36 | 6 | 50 100 | 38 49 |
| 3 | 50 100 | 34 42 | 7 | - 100 | - 41 |
| 4 | 50 100 | 20 33 | 8 | - 100 | - 39 |
| 5 | 50 100 | 41 49 | ASA | 50 100 | 31 43 |
| Gen. Formula/Ref. | Compd. | R, R1, n, 4-4a Bond | Biological Activity (In Vivo/In Vitro) |
|---|---|---|---|
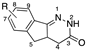 [23,24] | 9 | R = 7-NH2 | antihypertensive, antithrombotic, antiaggregating, anti-inflammatory activity |
| 10 | R = 7-NHCOCH3 | antihypertensive, antithrombotic, antiaggregating, anti-inflammatory, antiulcer activity | |
| 11 | 8-NO2 | antithrombotic activity | |
| 12 | 8-NH2 | antithrombotic activity | |
| 13 | 8-NHCOCH3 | antithrombotic activity | |
| 14 | 9-NHCOCH3 | antithrombotic activity | |
| 15 | 9-NH2 | data not available | |
| 16 | 7-OCH3 | data not available | |
| 17 | 7-OH | antithrombotic activity | |
| 18 | 7-NHCH3 | antihypertensive, antithrombotic activity | |
| 19 | 7-N(CH3)COCH3 | antihypertensive, antithrombotic activity | |
| 20 | 7-N(CH3)COCH2CH3 | antihypertensive, antithrombotic activity | |
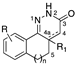 [25,26] | 21 | n = 1, R = 8-NH2, R1 = H, 4-4a single bond | antihypertensive, inotropic, antithrombotic activity |
| 22 | n = 1, R = 8-NHCOCH3, R1 = H, 4-4a single bond | antihypertensive, inotropic, antithrombotic activity | |
| 23 | n = 1, R = 8-NHCOCH3, 4-4a double bond | antihypertensive, inotropic, antithrombotic activity | |
| 24 | n =2, R = 9-NH2, R1 = H, 4-4a single bond | antithrombotic activity | |
| 25 | n = 2, R = 9-NHCOCH3, R1 = H, 4-4a single bond | antithrombotic activity | |
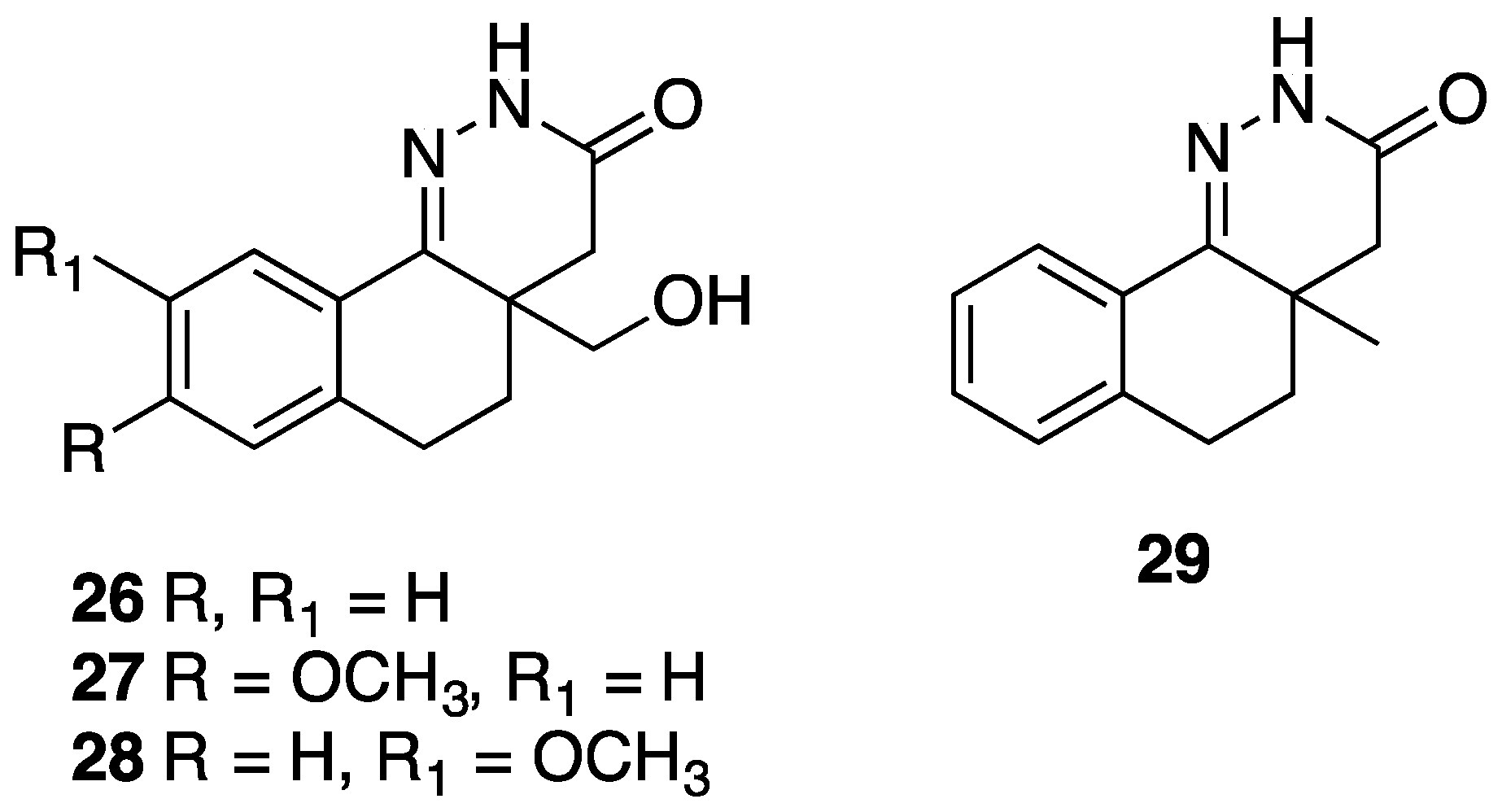
| 26 | n =1, R = H, R1 = CH2OH, 4-4a single bond | antihypertensive activity | |
| 27 | n = 1, R = 8-OCH3, R1 = CH2OH, 4-4a single bond | antihypertensive, antithrombotic activity | |
| 28 | n = 1, R = 9-OCH3, R1 = CH2OH, 4-4a single bond | antithrombotic activity | |
| 29 | n = 1, R = H, R1 = CH3, 4-4a single bond | antithrombotic activity | |
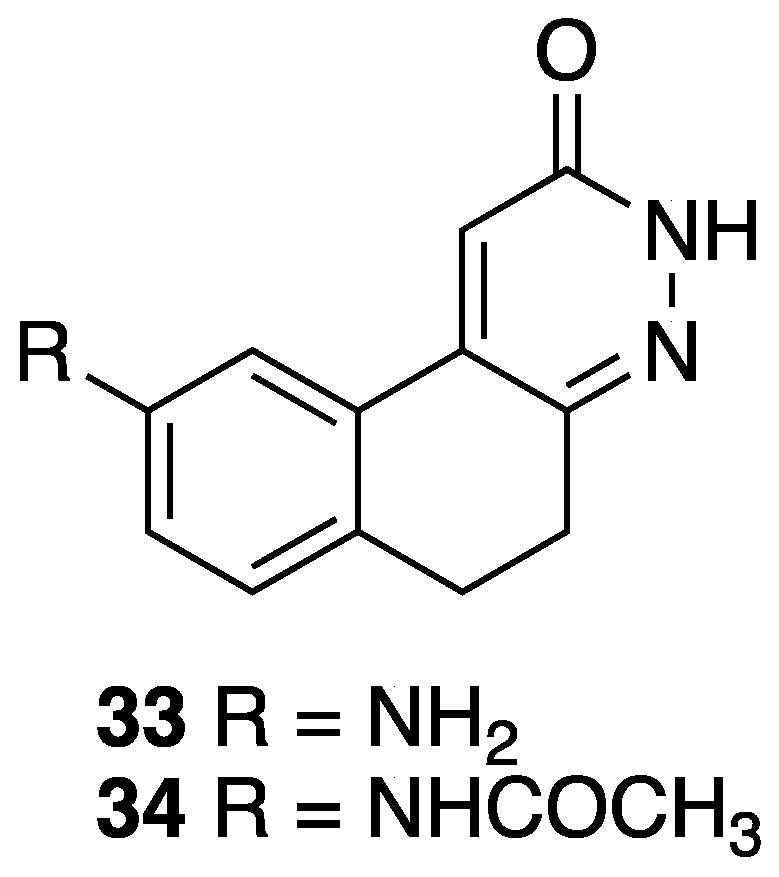
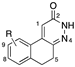 [29] | 33 | R = 9-NH2 | antiaggregating, hypotensive, antihypertensive activity |
| 34 | R = 9-NHCOCH3 | antiaggregating, hypotensive, antihypertensive activity | |
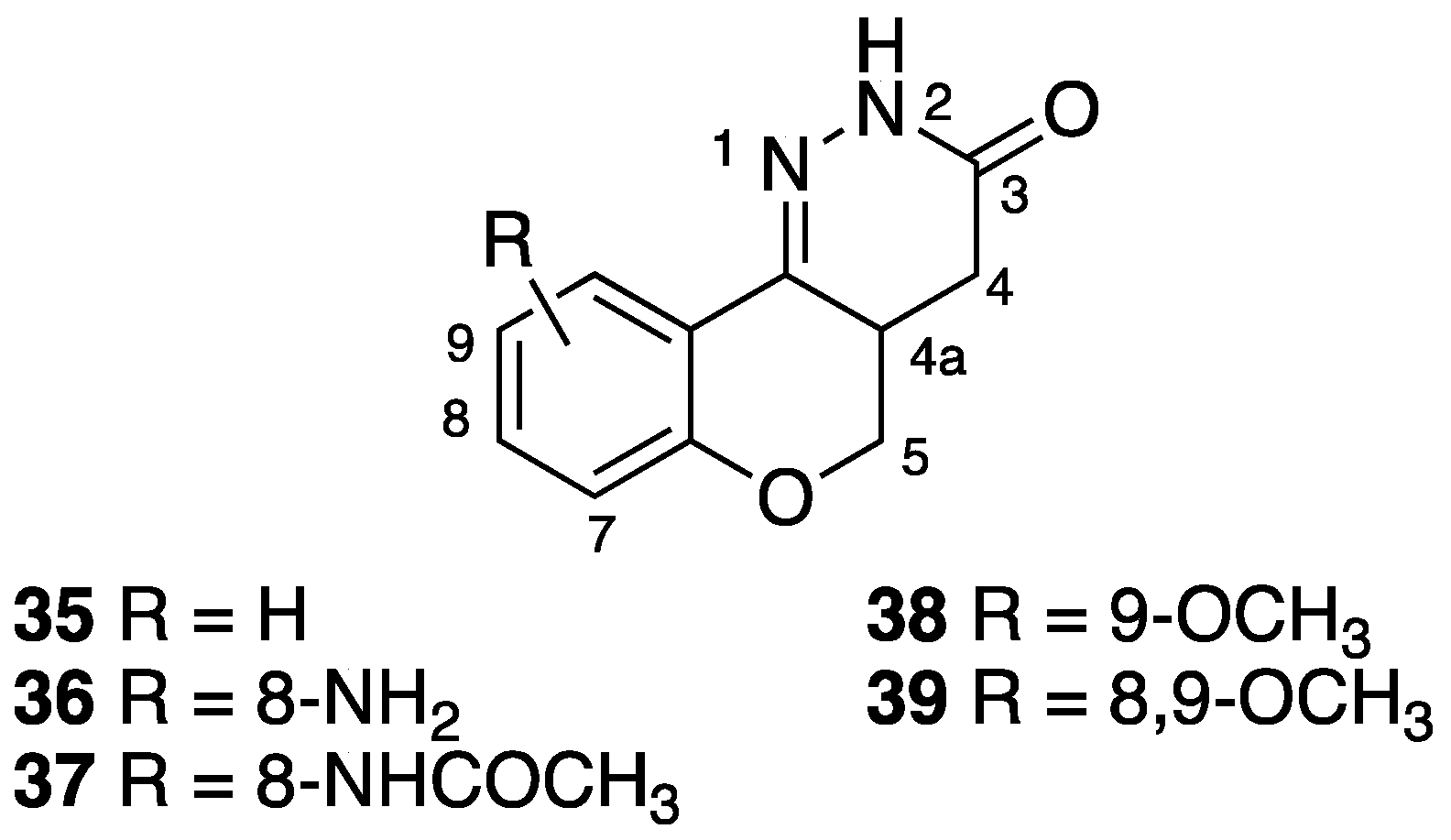
 [32] | 35 | R = H | antiulcer activity |
| 36 | R = 8-NH2 | antithrombotic activity | |
| 37 | R = 8-NHCOCH3 | antihypertensive, antithrombotic activity | |
| 38 | R = 9-OCH3 | antiulcer activity | |
| 39 | R = 8,9-OCH3 | antiulcer activity | |
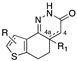 [33,34] | 40 | R = R1 = H, 4-4a single bond | antithrombotic activity |
| 41 | R = 8-NHCOCH3, R1 = H, 4-4a single bond | antithrombotic activity | |
| 42 | R = 9-NHCOCH3, R1 = H, 4-4a single bond | hypotensive activity | |
| 43 | R = 9-NHCOCH3, 4-4a double bond | hypotensive activity, antiaggregating activity | |
| 46 | R = H, R1 = CH3 4-4a single bond | antihypertensive, antiaggregating activity | |
| 47 | R = 8-NHCOCH3, R1 = CH3, 4-4a single bond | hypotensive, antiaggregating activity | |
| 48 | R = 9-NHCOCH3, R1 = CH3, 4-4a single bond | hypotensive, antiaggregating activity | |
 [33] | 44 | R = 7-NHCOCH3 | hypotensive, antiaggregating activity |
| 45 | R = 8-NHCOCH3 | hypotensive, antihypertensive activity, antiaggregating activity |
| Gen. Formula/Ref. | Compd. | X, R1, 4-4a Bond | R | Biological Activity (In Vivo) |
|---|---|---|---|---|
 [37] | 55 | X = O, 4-4a double bond | H | antiulcer activity (ethanol and ASA model), antisecretory activity |
| 56 | X = CH2, 4-4a double bond | H | antiulcer activity (ethanol model) | |
| 57 | X = CH2, R1 = CH3, 4-4a single bond | H | antiulcer activity (ethanol model) | |
| 58 | X = O, R1 = H, 4-4a single bond | 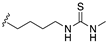 | antiulcer activity (ethanol model) | |
| 59 | X = O, 4-4a double bond | 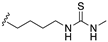 | antiulcer activity (ethanol and ASA model) | |
 [38] | 60 | 4-4a double bond | 7,8-(OCH3)2 | antiulcer activity (ethanol and indomethacine model), antisecretory activity |
| 61 | 4-4a double bond | 6,9-(OCH3)2 | antiulcer activity (ethanol and indomethacine model) | |
| 62 | R1 = H, 4-4a single bond | 7,8-(OCH3)2 | antiulcer activity (ethanol and indomethacine model) | |
| 63 | R1 = H, 4-4a single bond | 6,9-(OCH3)2 | antiulcer activity (ethanol and indomethacine model) |
| Compd./Ref. | Human Cancer Cell Line/Target | Compd./Ref. | Human Cancer Cell Line/Target |
|---|---|---|---|
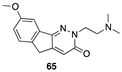 [52] | colon (LoVo and LoVo/DX), murine leukemia (L1210 and L1210/CDDP) | 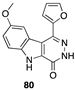 [65] | hepatocellular (Hub-7), colorectal (Caco2), breast (MDA-MB-231) DYRKIA inhibitor |
 [53] | renal (ACHN), leukemia (MOLT-4), non-small-cell lung (NCI-H460), colon (HCT-116), CNS (SF-295) | 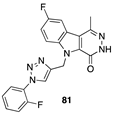 [66] | breast (MDA-MB-231, MCF-7), CNS (U-87, IMR-32) PI3K inhibitor (in silico study). |
 [55] | non-small-cell lung (EKVX, HOP-95), CNS (SNB-75) | 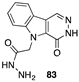 [67] | breast (MCF-7) PI3K inhibitor (in silico study). |
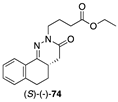 [57] | STAT3 inhibitor | 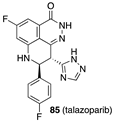 [70] | PARP-1 inhibitor clinical use: breast cancer |
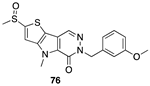 [59] | PKM2 activator | 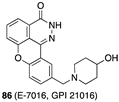 [71] | PARP-1 inhibitor clinical use: melanoma |
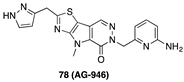 [60] | PKR activator | 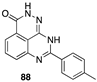 [72] | PARP-1 inhibitor |
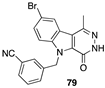 [61] | hepatocellular (Hub-7), colorectal (Caco2, HCT-116), breast (MDA-MB-231), prostate (PC3), lung (NCI-H727). PI3Kα inhibitor | 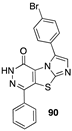 [74] | leukemia (HL-60TB, K-562, RPMI-8226), colon (HCT-116, HT29), melanoma (MDA-MB-435), ovarian (OVCAR-3), breast (MDA-MB468) DHFR inhibitor |
| (Gen.) Formula/Ref. | Compd. | R | R1 | ALR2 IC50 μM or (% Inhibn.) |
|---|---|---|---|---|
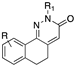 [81,82] | 96 | H | 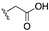 | 12.6 |
| 97 | 7,9-(CH3)2 |  | (31, 48 μM) | |
| 98 | H |  | 11.4 | |
| 99 | 8,9-(OCH3)2 | 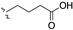 | (35, 79 μM) | |
| 100 | 7,9-(CH3)2 | 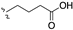 | 17.4 | |
| 101 | 7-OCH3 | 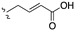 | 4.25 | |
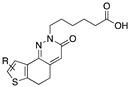 [84] | 103 | H | 7.6 | |
| 104 | 8-CH3 | 18.0 | ||
| 105 | 8-Cl | 31.4 | ||
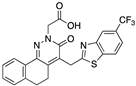 [83] | 102 | 0.15 | ||
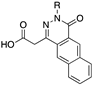 [85] | 106 | 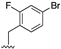 | 0.42 | |
| 107 | 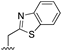 | 0.046 |
| (Gen.) Formula/Ref. | Compd. | X, n, R, R1 | [3H]Diazepam Ki (nM) |
|---|---|---|---|
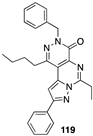
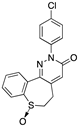 [87] | 109 (Y-23684) | 42.0 | |
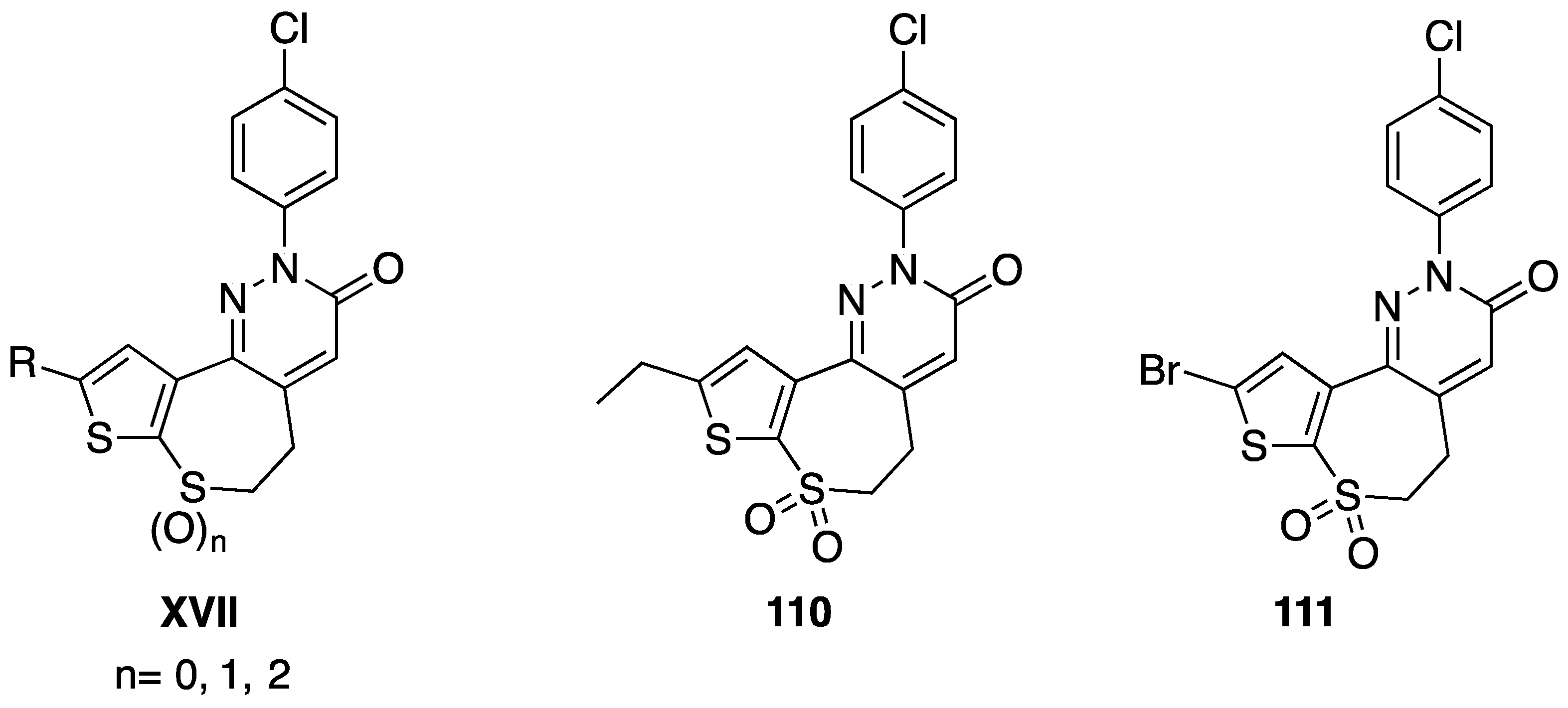
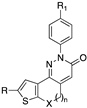 [87,88] | 110 | X = SO2, n = 2, R = CH2CH3, R1 = Cl | 1.1 |
| 111 | X = SO2, n = 2, R = Br, R1 = Cl | 0.61 | |
| 112 | X = CH2, n = 1, R = CH(OH)CH3, R1 = CH3 | 6.4 | |
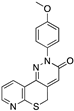 [89] | 113 | 695.0 ([3H]Flumazenil) |
| Compd./Ref. | Target | Compd./Ref. | Target |
|---|---|---|---|
 [95] | MMP-8 inhibitor | 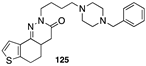 [106] | cholinesterase (AChE, BChE) inhibitor |
 [98] | PDE5 inhibitor | 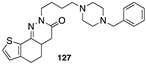 [106] | cholinesterase (AChE, BChE) inhibitor |
 [101] | hA1 antagonist | 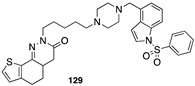 [106] | AChE inhibitor, 5-HT6 |
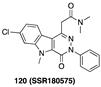 [103] | TSPO | 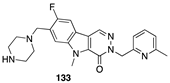 [108] | PKM2 activator |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asproni, B.; Pinna, G.A.; Corona, P.; Coinu, S.; Piras, S.; Carta, A.; Murineddu, G. Therapeutic Potential of Tricyclic Pyridazinone-Based Molecules: An Overview. Int. J. Mol. Sci. 2025, 26, 3806. https://doi.org/10.3390/ijms26083806
Asproni B, Pinna GA, Corona P, Coinu S, Piras S, Carta A, Murineddu G. Therapeutic Potential of Tricyclic Pyridazinone-Based Molecules: An Overview. International Journal of Molecular Sciences. 2025; 26(8):3806. https://doi.org/10.3390/ijms26083806
Chicago/Turabian StyleAsproni, Battistina, Gérard A. Pinna, Paola Corona, Silvia Coinu, Sandra Piras, Antonio Carta, and Gabriele Murineddu. 2025. "Therapeutic Potential of Tricyclic Pyridazinone-Based Molecules: An Overview" International Journal of Molecular Sciences 26, no. 8: 3806. https://doi.org/10.3390/ijms26083806
APA StyleAsproni, B., Pinna, G. A., Corona, P., Coinu, S., Piras, S., Carta, A., & Murineddu, G. (2025). Therapeutic Potential of Tricyclic Pyridazinone-Based Molecules: An Overview. International Journal of Molecular Sciences, 26(8), 3806. https://doi.org/10.3390/ijms26083806