
Chromoplexy: A Pathway to Genomic Complexity and Cancer Development


Abstract
1. Introduction
2. Definition and Prevalence
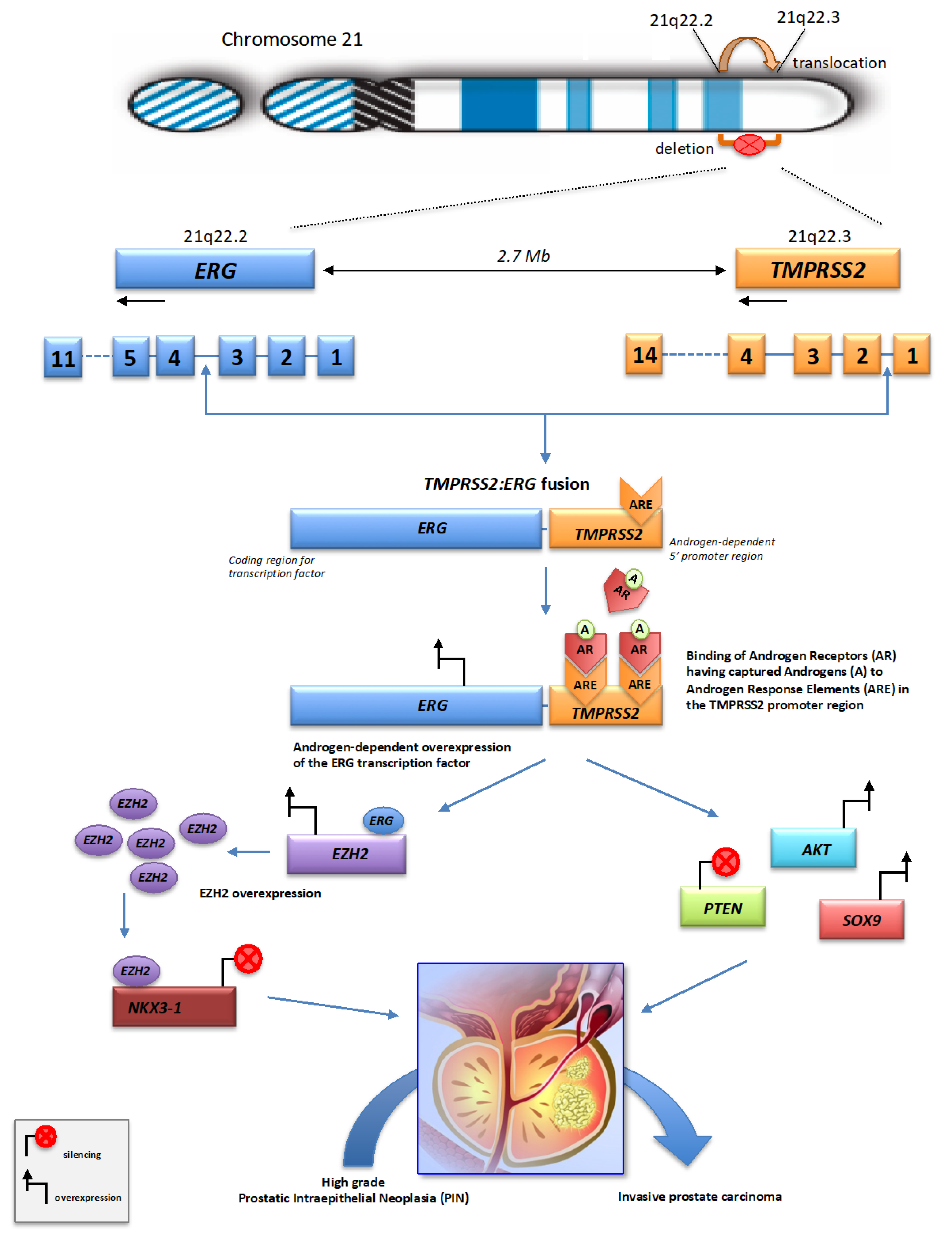
3. Mechanisms of Chromoplexy in Prostate Cancer
4. Involvement of Chromoplexy in Other Cancers
5. Comparison with Chromothripsis
6. Conclusions
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.M.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; et al. Chromothripsis as a Mechanism Driving Complex de Novo Structural Rearrangements in the Germline. Hum. Mol. Genet. 2011, 20, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Wilson, R.K. Chromothripsis and Human Disease: Piecing Together the Shattering Process. Cell 2012, 148, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Erez, A.; Nagamani, S.C.S.; Dhar, S.U.; Kołodziejska, K.E.; Dharmadhikari, A.V.; Cooper, M.L.; Wiszniewska, J.; Zhang, F.; Withers, M.A.; et al. Chromosome Catastrophes Involve Replication Mechanisms Generating Complex Genomic Rearrangements. Cell 2011, 146, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, É.A.; Piazzon, F.B.; Dutra, R.L.; Dias, A.T.; Montenegro, M.M.; Novo-Filho, G.M.; Costa, T.V.M.M.; Nascimento, A.M.; Kim, C.A.; Kulikowski, L.D. Complex Structural Rearrangement Features Suggesting Chromoanagenesis Mechanism in a Case of 1p36 Deletion Syndrome. Mol. Genet. Genomics 2014, 289, 1037–1043. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Collins, C.C. Chromoplexy: A New Paradigm in Genome Remodeling and Evolution. Asian J. Androl. 2013, 15, 711–712. [Google Scholar] [CrossRef]
- Shen, M.M. Chromoplexy: A New Category of Complex Rearrangements in the Cancer Genome. Cancer Cell 2013, 23, 567–569. [Google Scholar] [CrossRef]
- Anderson, N.D.; de Borja, R.; Young, M.D.; Fuligni, F.; Rosic, A.; Roberts, N.D.; Hajjar, S.; Layeghifard, M.; Novokmet, A.; Kowalski, P.E.; et al. Rearrangement Bursts Generate Canonical Gene Fusions in Bone and Soft Tissue Tumors. Science 2018, 361, eaam8419. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The Genomic Complexity of Primary Human Prostate Cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef]
- Dahiya, R.; Hu, Q.; Ly, P. Mechanistic Origins of Diverse Genome Rearrangements in Cancer. Semin. Cell Dev. Biol. 2022, 123, 100–109. [Google Scholar] [CrossRef]
- Dzamba, M.; Ramani, A.K.; Buczkowicz, P.; Jiang, Y.; Yu, M.; Hawkins, C.; Brudno, M. Identification of Complex Genomic Rearrangements in Cancers Using CouGaR. Genome Res. 2017, 27, 107–117. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [CrossRef] [PubMed]
- Baelen, J.; Dewaele, B.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P.; Hompes, D.; Sinnaeve, F.; Wafa, H.; Vanden Bempt, I. Optical Genome Mapping for Comprehensive Cytogenetic Analysis of Soft-Tissue and Bone Tumors for Diagnostic Purposes. J. Mol. Diagn. 2024, 26, 374–386. [Google Scholar] [CrossRef]
- Dardas, Z.; Marafi, D.; Duan, R.; Fatih, J.M.; El-Rashidy, O.F.; Grochowski, C.M.; Carvalho, C.M.B.; Jhangiani, S.N.; Bi, W.; Du, H.; et al. Genomic Balancing Act: Deciphering DNA Rearrangements in the Complex Chromosomal Aberration Involving 5p15.2, 2q31.1, and 18q21.32. Eur. J. Hum. Genet. 2025, 33, 231–238. [Google Scholar] [CrossRef]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-Induced TOP2B-Mediated Double-Strand Breaks and Prostate Cancer Gene Rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef]
- Mani, R.S.; Amin, M.A.; Li, X.; Kalyana-Sundaram, S.; Veeneman, B.A.; Wang, L.; Ghosh, A.; Aslam, A.; Ramanand, S.G.; Rabquer, B.J.; et al. Inflammation-Induced Oxidative Stress Mediates Gene Fusion Formation in Prostate Cancer. Cell Rep. 2016, 17, 2620–2631. [Google Scholar] [CrossRef]
- Mani, R.-S.; Tomlins, S.A.; Callahan, K.; Ghosh, A.; Nyati, M.K.; Varambally, S.; Palanisamy, N.; Chinnaiyan, A.M. Induced Chromosomal Proximity and Gene Fusions in Prostate Cancer. Science 2009, 326, 1230. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of Diet and Nutrition on Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef]
- Mosquera, J.-M.; Mehra, R.; Regan, M.M.; Perner, S.; Genega, E.M.; Bueti, G.; Shah, R.B.; Gaston, S.; Tomlins, S.A.; Wei, J.T.; et al. Prevalence of TMPRSS2-ERG Fusion Prostate Cancer among Men Undergoing Prostate Biopsy in the United States. Clin. Cancer Res. 2009, 15, 4706–4711. [Google Scholar] [CrossRef]
- Charlot, C.; Dubois-Pot, H.; Serchov, T.; Tourrette, Y.; Wasylyk, B. A Review of Post-Translational Modifications and Subcellular Localization of Ets Transcription Factors: Possible Connection with Cancer and Involvement in the Hypoxic Response. Methods Mol. Biol. 2010, 647, 3–30. [Google Scholar] [CrossRef]
- Paoloni-Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the TMPRSS2 Gene, Which Encodes a Novel Serine Protease with Transmembrane, LDLRA, and SRCR Domains and Maps to 21q22.3. Genomics 1997, 44, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Adamo, P.; Ladomery, M.R. The Oncogene ERG: A Key Factor in Prostate Cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef]
- Scaravilli, M.; Koivukoski, S.; Latonen, L. Androgen-Driven Fusion Genes and Chimeric Transcripts in Prostate Cancer. Front. Cell Dev. Biol. 2021, 9, 623809. [Google Scholar] [CrossRef]
- Stopsack, K.H.; Su, X.A.; Vaselkiv, J.B.; Graff, R.E.; Ebot, E.M.; Pettersson, A.; Lis, R.T.; Fiorentino, M.; Loda, M.; Penney, K.L.; et al. Transcriptomes of Prostate Cancer with TMPRSS2:ERG and Other ETS Fusions. Mol. Cancer Res. 2023, 21, 14–23. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Smith, L.R.; Roulston, D.; Helgeson, B.E.; Cao, X.; Wei, J.T.; Rubin, M.A.; Shah, R.B.; et al. TMPRSS2:ETV4 Gene Fusions Define a Third Molecular Subtype of Prostate Cancer. Cancer Res. 2006, 66, 3396–3400. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct Classes of Chromosomal Rearrangements Create Oncogenic ETS Gene Fusions in Prostate Cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef]
- Hermans, K.G.; van Marion, R.; van Dekken, H.; Jenster, G.; van Weerden, W.M.; Trapman, J. TMPRSS2:ERG Fusion by Translocation or Interstitial Deletion Is Highly Relevant in Androgen-Dependent Prostate Cancer, but Is Bypassed in Late-Stage Androgen Receptor-Negative Prostate Cancer. Cancer Res. 2006, 66, 10658–10663. [Google Scholar] [CrossRef]
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.-M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. TMPRSS2:ERG Fusion-Associated Deletions Provide Insight into the Heterogeneity of Prostate Cancer. Cancer Res. 2006, 66, 8337–8341. [Google Scholar] [CrossRef]
- Mehra, R.; Han, B.; Tomlins, S.A.; Wang, L.; Menon, A.; Wasco, M.J.; Shen, R.; Montie, J.E.; Chinnaiyan, A.M.; Shah, R.B. Heterogeneity of TMPRSS2 Gene Rearrangements in Multifocal Prostate Adenocarcinoma: Molecular Evidence for an Independent Group of Diseases. Cancer Res. 2007, 67, 7991–7995. [Google Scholar] [CrossRef]
- Pellestor, F.; Gaillard, J.B.; Schneider, A.; Puechberty, J.; Gatinois, V. Chromoanagenesis, the Mechanisms of a Genomic Chaos. Semin. Cell Dev. Biol. 2022, 123, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Burssed, B.; Zamariolli, M.; Bellucco, F.T.; Melaragno, M.I. Mechanisms of Structural Chromosomal Rearrangement Formation. Mol. Cytogenet. 2022, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, K.A.; Saramäki, O.R.; Furusato, B.; Kimura, T.; Takahashi, H.; Egawa, S.; Suzuki, H.; Keiger, K.; Ho Hahm, S.; Isaacs, W.B.; et al. Loss of PTEN Is Associated with Aggressive Behavior in ERG-Positive Prostate Cancer. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 2333–2344. [Google Scholar] [CrossRef]
- Burkhardt, L.; Fuchs, S.; Krohn, A.; Masser, S.; Mader, M.; Kluth, M.; Bachmann, F.; Huland, H.; Steuber, T.; Graefen, M.; et al. CHD1 Is a 5q21 Tumor Suppressor Required for ERG Rearrangement in Prostate Cancer. Cancer Res. 2013, 73, 2795–2805. [Google Scholar] [CrossRef]
- Melling, N.; Thomsen, E.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Minner, S.; Koop, C.; Graefen, M.; Heinzer, H.; Wittmer, C.; et al. Overexpression of Enhancer of Zeste Homolog 2 (EZH2) Characterizes an Aggressive Subset of Prostate Cancers and Predicts Patient Prognosis Independently from Pre- and Postoperatively Assessed Clinicopathological Parameters. Carcinogenesis 2015, 36, 1333–1340. [Google Scholar] [CrossRef]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased Mobility of Double-Strand Breaks Requires Mec1, Rad9 and the Homologous Recombination Machinery. Nat. Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef]
- García Fernández, F.; Fabre, E. The Dynamic Behavior of Chromatin in Response to DNA Double-Strand Breaks. Genes 2022, 13, 215. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Spielmann, M.; Mundlos, S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 2016, 32, 225–237. [Google Scholar] [CrossRef]
- Rajderkar, S.; Barozzi, I.; Zhu, Y.; Hu, R.; Zhang, Y.; Li, B.; Alcaina Caro, A.; Fukuda-Yuzawa, Y.; Kelman, G.; Akeza, A.; et al. Topologically Associating Domain Boundaries Are Required for Normal Genome Function. Commun. Biol. 2023, 6, 435. [Google Scholar] [CrossRef]
- Osborne, C.S.; Chakalova, L.; Brown, K.E.; Carter, D.; Horton, A.; Debrand, E.; Goyenechea, B.; Mitchell, J.A.; Lopes, S.; Reik, W.; et al. Active Genes Dynamically Colocalize to Shared Sites of Ongoing Transcription. Nat. Genet. 2004, 36, 1065–1071. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Mani, R.-S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Clinckemalie, L.; Spans, L.; Dubois, V.; Laurent, M.; Helsen, C.; Joniau, S.; Claessens, F. Androgen Regulation of the TMPRSS2 Gene and the Effect of a SNP in an Androgen Response Element. Mol. Endocrinol. 2013, 27, 2028–2040. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and Temporal Recruitment of Androgen Receptor and Its Coactivators Involves Chromosomal Looping and Polymerase Tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef]
- Metzger, E.; Willmann, D.; McMillan, J.; Forne, I.; Metzger, P.; Gerhardt, S.; Petroll, K.; von Maessenhausen, A.; Urban, S.; Schott, A.-K.; et al. Assembly of Methylated KDM1A and CHD1 Drives Androgen Receptor-Dependent Transcription and Translocation. Nat. Struct. Mol. Biol. 2016, 23, 132–139. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjöström, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA Methylation Landscape of Advanced Prostate Cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef]
- Kunderfranco, P.; Mello-Grand, M.; Cangemi, R.; Pellini, S.; Mensah, A.; Albertini, V.; Malek, A.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. ETS Transcription Factors Control Transcription of EZH2 and Epigenetic Silencing of the Tumor Suppressor Gene Nkx3.1 in Prostate Cancer. PLoS ONE 2010, 5, e10547. [Google Scholar] [CrossRef]
- Thangapazham, R.; Saenz, F.; Katta, S.; Mohamed, A.A.; Tan, S.-H.; Petrovics, G.; Srivastava, S.; Dobi, A. Loss of the NKX3.1 Tumorsuppressor Promotes the TMPRSS2-ERG Fusion Gene Expression in Prostate Cancer. BMC Cancer 2014, 14, 16. [Google Scholar] [CrossRef]
- Zoma, M.; Curti, L.; Shinde, D.; Albino, D.; Mitra, A.; Sgrignani, J.; Mapelli, S.N.; Sandrini, G.; Civenni, G.; Merulla, J.; et al. EZH2-Induced Lysine K362 Methylation Enhances TMPRSS2-ERG Oncogenic Activity in Prostate Cancer. Nat. Commun. 2021, 12, 4147. [Google Scholar] [CrossRef]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG Expression Cooperates with Loss of PTEN to Promote Cancer Progression in the Prostate. Nat. Genet. 2009, 41, 619–624. [Google Scholar] [CrossRef]
- Squire, J.A. TMPRSS2-ERG and PTEN Loss in Prostate Cancer. Nat. Genet. 2009, 41, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Martens-Uzunova, E.S.; Böttcher, R.; Croce, C.M.; Jenster, G.; Visakorpi, T.; Calin, G.A. Long Noncoding RNA in Prostate, Bladder, and Kidney Cancer. Eur. Urol. 2014, 65, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Taheri, M.; Safarzadeh, A.; Hussen, B.M.; Ghafouri-Fard, S.; Baniahmad, A. LncRNA/miRNA/mRNA Network Introduces Novel Biomarkers in Prostate Cancer. Cells 2022, 11, 3776. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Shin, S.; Kim, M.S.; Baek, I.-P.; Lee, J.Y.; Lee, S.H.; Kim, T.-M.; Lee, S.H.; Chung, Y.-J. Genetic Progression of High Grade Prostatic Intraepithelial Neoplasia to Prostate Cancer. Eur. Urol. 2016, 69, 823–830. [Google Scholar] [CrossRef]
- Segura-Moreno, Y.Y.; Sanabria-Salas, M.C.; Varela, R.; Mesa, J.A.; Serrano, M.L. Decoding the Heterogeneous Landscape in the Development Prostate Cancer. Oncol. Lett. 2021, 21, 376. [Google Scholar] [CrossRef]
- Dunn, T.; Praissman, L.; Hagag, N.; Viola, M.V. ERG Gene Is Translocated in an Ewing’s Sarcoma Cell Line. Cancer Genet. Cytogenet. 1994, 76, 19–22. [Google Scholar] [CrossRef]
- Zucman, J.; Melot, T.; Desmaze, C.; Ghysdael, J.; Plougastel, B.; Peter, M.; Zucker, J.M.; Triche, T.J.; Sheer, D.; Turc-Carel, C. Combinatorial Generation of Variable Fusion Proteins in the Ewing Family of Tumours. EMBO J. 1993, 12, 4481–4487. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Slotkin, E.; Tap, W.D.; Meyers, P.; Wexler, L.; Healey, J.; Vanoli, F.; Vanderbilt, C.M.; Antonescu, C.R. Chromoplexy Is a Frequent Early Clonal Event in EWSR1-Rearranged Round Cell Sarcomas That Can Be Detected Using Clinically Validated Targeted Sequencing Panels. Cancer Res. 2024, 84, 1504–1516. [Google Scholar] [CrossRef]
- Sole, A.; Grossetête, S.; Heintzé, M.; Babin, L.; Zaïdi, S.; Revy, P.; Renouf, B.; De Cian, A.; Giovannangeli, C.; Pierre-Eugène, C.; et al. Unraveling Ewing Sarcoma Tumorigenesis Originating from Patient-Derived Mesenchymal Stem Cells. Cancer Res. 2021, 81, 4994–5006. [Google Scholar] [CrossRef]
- Moncunill, V.; Gonzalez, S.; Beà, S.; Andrieux, L.O.; Salaverria, I.; Royo, C.; Martinez, L.; Puiggròs, M.; Segura-Wang, M.; Stütz, A.M.; et al. Comprehensive Characterization of Complex Structural Variations in Cancer by Directly Comparing Genome Sequence Reads. Nat. Biotechnol. 2014, 32, 1106–1112. [Google Scholar] [CrossRef]
- Lee, K.J.; Lee, K.H.; Yoon, K.-A.; Sohn, J.Y.; Lee, E.; Lee, H.; Eom, H.-S.; Kong, S.-Y. Chromothripsis in Treatment Resistance in Multiple Myeloma. Genomics Inform. 2017, 15, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.-K.; Park, S.; Park, H.; Kim, S.; Lee, J.; Lee, J.; Youk, J.; Yi, K.; An, Y.; Park, I.K.; et al. Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell 2019, 177, 1842–1857.e21. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, C.; Xue, R.; Liu, M.; Bai, J.; Bao, J.; Wang, Y.; Jiang, N.; Li, Z.; Wang, W.; et al. Deep Whole-Genome Analysis of 494 Hepatocellular Carcinomas. Nature 2024, 627, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Parry, E.M.; Ten Hacken, E.; Wu, C.J. Richter Syndrome: Novel Insights into the Biology of Transformation. Blood 2023, 142, 11–22. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Koster, J.; Molenaar, J.J. Prevalence and Clinical Implications of Chromothripsis in Cancer Genomes. Curr. Opin. Oncol. 2014, 26, 64–72. [Google Scholar] [CrossRef]
- Redin, C.; Brand, H.; Collins, R.L.; Kammin, T.; Mitchell, E.; Hodge, J.C.; Hanscom, C.; Pillalamarri, V.; Seabra, C.M.; Abbott, M.-A.; et al. The Genomic Landscape of Balanced Cytogenetic Abnormalities Associated with Human Congenital Anomalies. Nat. Genet. 2017, 49, 36–45. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Redin, C.E.; Hanscom, C.; Antolik, C.; Stone, M.R.; Glessner, J.T.; Mason, T.; Pregno, G.; Dorrani, N.; et al. Defining the Diverse Spectrum of Inversions, Complex Structural Variation, and Chromothripsis in the Morbid Human Genome. Genome Biol. 2017, 18, 36. [Google Scholar] [CrossRef]
- de Pagter, M.S.; van Roosmalen, M.J.; Baas, A.F.; Renkens, I.; Duran, K.J.; van Binsbergen, E.; Tavakoli-Yaraki, M.; Hochstenbach, R.; van der Veken, L.T.; Cuppen, E.; et al. Chromothripsis in Healthy Individuals Affects Multiple Protein-Coding Genes and Can Result in Severe Congenital Abnormalities in Offspring. Am. J. Hum. Genet. 2015, 96, 651–656. [Google Scholar] [CrossRef]
- Bertelsen, B.; Nazaryan-Petersen, L.; Sun, W.; Mehrjouy, M.M.; Xie, G.; Chen, W.; Hjermind, L.E.; Taschner, P.E.M.; Tümer, Z. A Germline Chromothripsis Event Stably Segregating in 11 Individuals through Three Generations. Genet. Med. 2016, 18, 494–500. [Google Scholar] [CrossRef]
- Macera, M.J.; Sobrino, A.; Levy, B.; Jobanputra, V.; Aggarwal, V.; Mills, A.; Esteves, C.; Hanscom, C.; Pereira, S.; Pillalamarri, V.; et al. Prenatal Diagnosis of Chromothripsis, with Nine Breaks Characterized by Karyotyping, FISH, Microarray and Whole-Genome Sequencing. Prenat. Diagn. 2015, 35, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Marcozzi, A.; Pellestor, F.; Kloosterman, W.P. The Genomic Characteristics and Origin of Chromothripsis. Methods Mol. Biol. 2018, 1769, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Mendoza, C.J.; Morton, C.C. The Iceberg under Water: Unexplored Complexity of Chromoanagenesis in Congenital Disorders. Am. J. Hum. Genet. 2019, 104, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA Damage in Micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of Resistance to Systemic Therapy in Metastatic Castration-Resistant Prostate Cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef]
- Pellestor, F.; Gatinois, V. Chromoanagenesis: A Piece of the Macroevolution Scenario. Mol. Cytogenet. 2020, 13, 3. [Google Scholar] [CrossRef]
- Lu, B.; Liu, Y.; Yao, Y.; Yang, T.; Zhang, H.; Yang, X.; Huang, R.; Zhou, W.; Pan, X.; Cui, X. Advances in Sequencing and Omics Studies in Prostate Cancer: Unveiling Molecular Pathogenesis and Clinical Applications. Front. Oncol. 2024, 14, 1355551. [Google Scholar] [CrossRef]
- Warburton, P.E.; Sebra, R.P. Long-Read DNA Sequencing: Recent Advances and Remaining Challenges. Annu. Rev. Genomics Hum. Genet. 2023, 24, 109–132. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Paigin, S.; Afflerbach, A.-K.; Lobermeyer, A.; Werner, S.; Schüller, U.; Bokemeyer, C.; Schuh, A.H.; Bergmann, L.; von Amsberg, G.; et al. Applications of Nanopore Sequencing in Precision Cancer Medicine. Int. J. Cancer 2024, 155, 2129–2140. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J. Natl. Cancer Inst. 2017, 109, djx118. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Chen, G.; Jia, G.; Chao, F.; Xie, F.; Zhang, Y.; Hou, C.; Huang, Y.; Tang, H.; Yu, J.; Zhang, J.; et al. Urine- and Blood-Based Molecular Profiling of Human Prostate Cancer. Front. Oncol. 2022, 12, 759791. [Google Scholar] [CrossRef] [PubMed]
- Militaru, F.C.; Militaru, V.; Crisan, N.; Bocsan, I.C.; Udrea, A.A.; Catana, A.; Kutasi, E.; Militaru, M.S. Molecular Basis and Therapeutic Targets in Prostate Cancer: A Comprehensive Review. Biomol. Biomed. 2023, 23, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Bian, X.; Zhou, J.; Zhang, M. From Microscopes to Molecules: The Evolution of Prostate Cancer Diagnostics. Cytojournal 2024, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, T.S.; Lørup, A.N.; Kongsted, P.; Eefsen, R.L.; Højgaard, M.; Høgdall, E.V. TMPRSS2:ERG Gene Fusion Might Predict Resistance to PARP Inhibitors in Metastatic Castration-Resistant Prostate Cancer. Anticancer Res. 2024, 44, 4203–4211. [Google Scholar] [CrossRef]
- Lalonde, E.; Ishkanian, A.S.; Sykes, J.; Fraser, M.; Ross-Adams, H.; Erho, N.; Dunning, M.J.; Halim, S.; Lamb, A.D.; Moon, N.C.; et al. Tumour Genomic and Microenvironmental Heterogeneity for Integrated Prediction of 5-Year Biochemical Recurrence of Prostate Cancer: A Retrospective Cohort Study. Lancet Oncol. 2014, 15, 1521–1532. [Google Scholar] [CrossRef]
- Ballas, L.K.; Hu, B.R.; Quinn, D.I. Chromoplexy and Hypoxic Microenvironment Drives Prostate Cancer. Lancet Oncol. 2014, 15, 1419–1421. [Google Scholar] [CrossRef]
- Strauss, S.J.; Berlanga, P.; McCabe, M.G. Emerging Therapies in Ewing Sarcoma. Curr. Opin. Oncol. 2024, 36, 297–304. [Google Scholar] [CrossRef]
- Paul, A.K.; Melson, J.W.; Hirani, S.; Muthusamy, S. Systemic Therapy Landscape of Advanced Prostate Cancer. Adv. Cancer Res. 2024, 161, 367–402. [Google Scholar] [CrossRef]
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and Consequences of Chromosomal Instability: From Cellular Adaptation to Genome Chaos-Mediated System Survival. Genes 2020, 11, 1162. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Key-Features | Chromothripsis | Chromoplexy |
|---|---|---|
| Number of events | Single | single or sequential |
| Number of chromosomes involved | Usually 1 or 2 (up to 4) | Multiple (3 to 7) |
| Rearrangements | Balanced rearrangements, deletions, duplications, insertions… ± extra-chromosomal circular elements | Inter- or intra-chromosomal balanced translocations, ± deletions |
| Breakpoints | Numerous (up to 100) Clustering of breakpoints | On average from 5 to 40 |
| Breakpoint signature | Blunt ends (possibly small insertions) | Blunt ends (possibly small insertions) |
| Repair mechanisms | NHEJ | NHEJ/alt-EJ |
| Junction and order of chromosomal fragments | Random order and random orientation | Conservation of the original chromosome orientation |
| Copy number state | Oscillating pattern between 2 copy number states | No copy number alterations |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellestor, F.; Ganne, B.; Gaillard, J.B.; Gatinois, V. Chromoplexy: A Pathway to Genomic Complexity and Cancer Development. Int. J. Mol. Sci. 2025, 26, 3826. https://doi.org/10.3390/ijms26083826
Pellestor F, Ganne B, Gaillard JB, Gatinois V. Chromoplexy: A Pathway to Genomic Complexity and Cancer Development. International Journal of Molecular Sciences. 2025; 26(8):3826. https://doi.org/10.3390/ijms26083826
Chicago/Turabian StylePellestor, Franck, Benjamin Ganne, Jean Baptiste Gaillard, and Vincent Gatinois. 2025. "Chromoplexy: A Pathway to Genomic Complexity and Cancer Development" International Journal of Molecular Sciences 26, no. 8: 3826. https://doi.org/10.3390/ijms26083826
APA StylePellestor, F., Ganne, B., Gaillard, J. B., & Gatinois, V. (2025). Chromoplexy: A Pathway to Genomic Complexity and Cancer Development. International Journal of Molecular Sciences, 26(8), 3826. https://doi.org/10.3390/ijms26083826