
Epigenetic Inactivation of RIPK3-Dependent Necroptosis Augments Cisplatin Chemoresistance in Human Osteosarcoma

 ,
, 
Abstract
:1. Introduction
2. Results
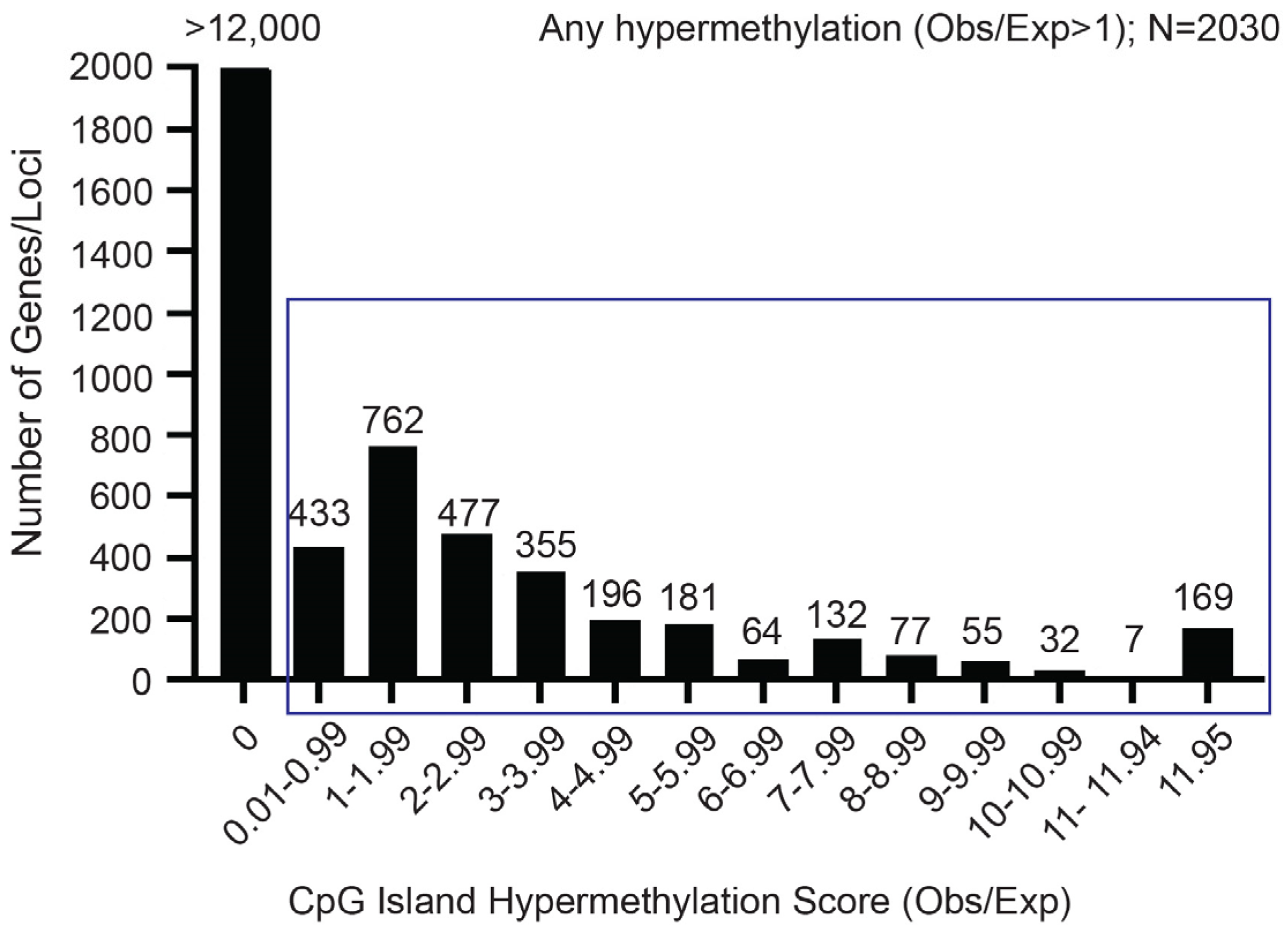
2.1. The RIPK3 CpG Island Exhibits Aberrant Methylation in OS Cell Lines and Tumor Samples
2.2. Aberrant CpG Island Methylation Is Associated with Downregulation of RIPK3 and MLKL Expression in Human OS Cell Lines
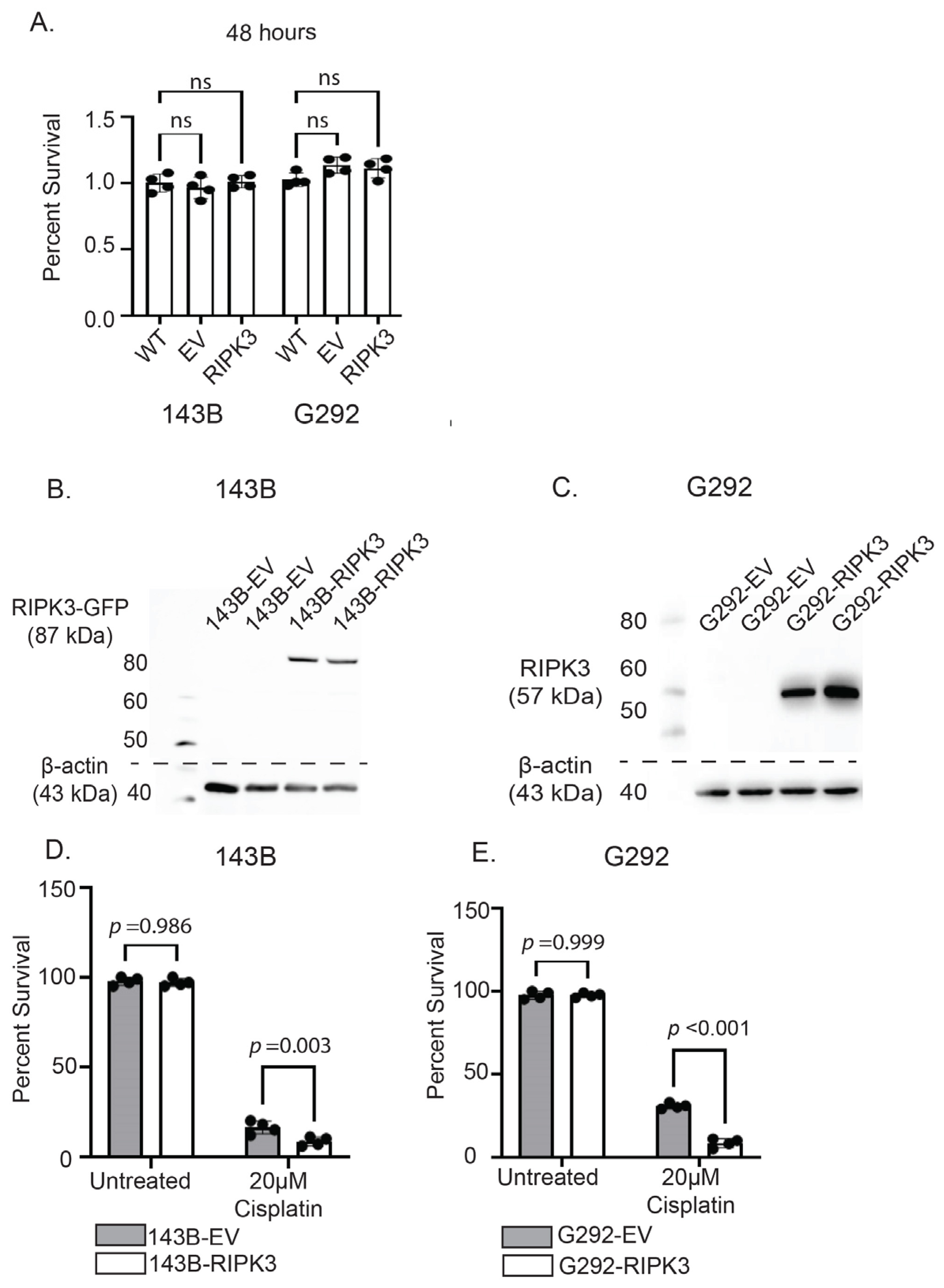
2.3. RIPK3 Expression in OS Cells Lines Does Not Alter Survival Without Therapy but Significantly Increases Susceptibility to Cisplatin Through Activation of Necroptosis
2.4. Knockdown of RIPK3 Expression Desensitizes OS Cells to Cisplatin Cytotoxicity
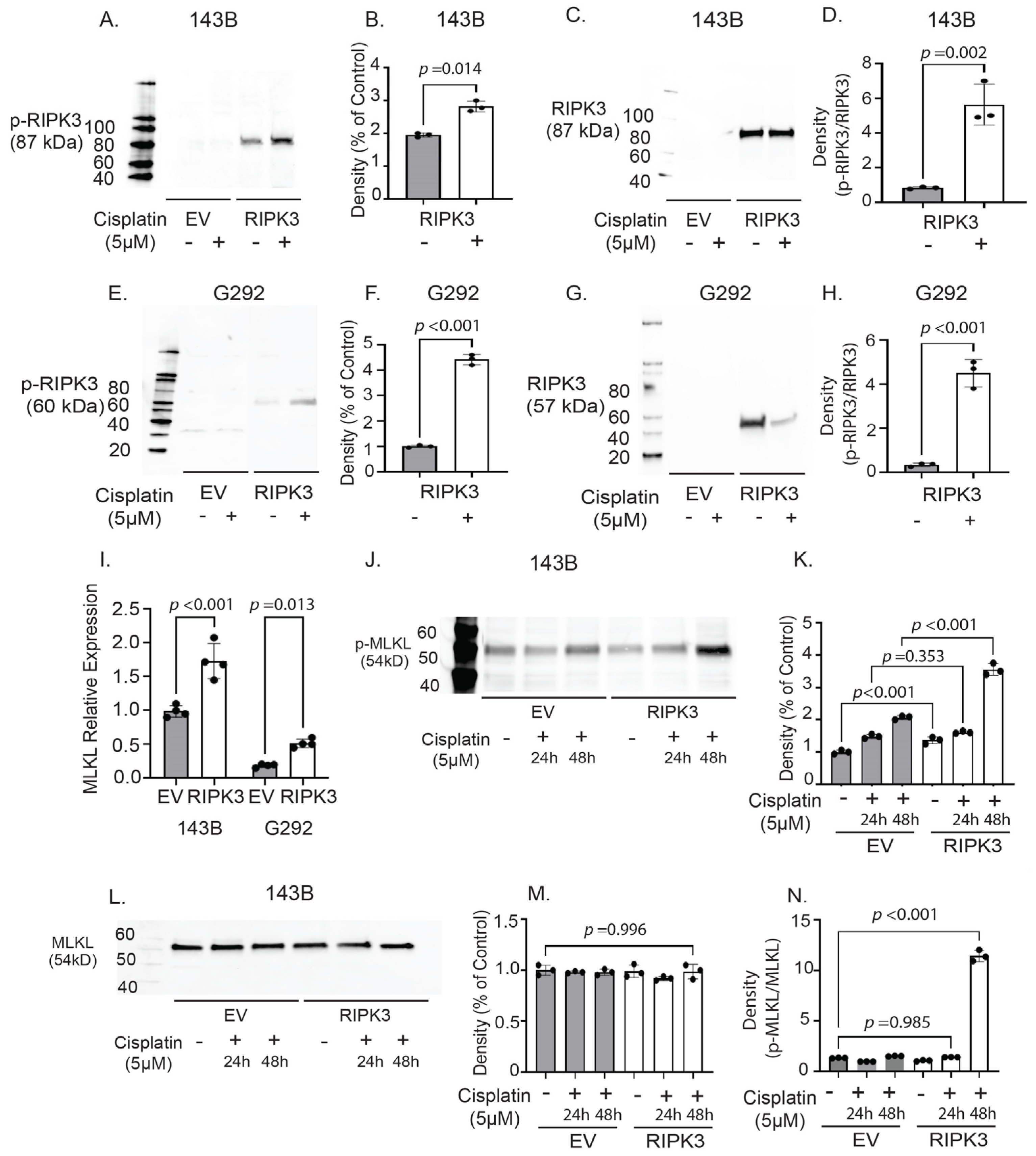
2.5. RIPK3 Expression in OS Cells Is Associated with Activation of Molecular Effectors of Necroptosis Following Cisplatin Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Human Primary OS Tumor Samples
4.3. DNA Isolation
4.4. Methylation Profiling and Analysis
4.5. Transfections
4.6. Transductions
4.7. Pyrosequencing
4.8. Next Generation Sequencing
4.9. Western Blots
4.10. RT-qPCR
4.11. RIPK3 DsiRNA Knockdown
4.12. Cisplatin Sensitivity Assay
4.13. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OS | Osteosarcoma |
| RIPK3 | Receptor interacting protein kinase-3 |
| IPF | Idiopathic pulmonary fibrosis |
| MS | Multiple sclerosis |
| ALS | Amyotrophic lateral sclerosis |
| AD | Alzheimer’s disease |
| DsiRNA | Dicer substrate RNA |
| MLKL | Mixed lineage kinase domain-like |
| hFOB | Human fetal osteoblasts |
| hMSC | Human mesenchymal stem cells |
References
- Damron, T.A.; Ward, W.G.; Stewart, A. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National cancer data base report. Clin. Orthop. Relat. Res. 2007, 459, 40–47. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Gorlick, R. Osteosarcoma. Pediatr. Clin. N. Am. 1997, 44, 973–989. [Google Scholar] [CrossRef]
- Kuerbitz, S.J.; Henderson, M.B. Osteosarcoma: A review with emphasis on pathogenesis and chemoresistance. Med. Res. Arch. 2020, 8, 1–37. [Google Scholar] [CrossRef]
- De Noon, S.; Ijaz, J.; Coorens, T.H.; Amary, F.; Ye, H.; Strobl, A.; Lyskjær, I.; Flanagan, A.M.; Behjati, S. MYC amplifications are common events in childhood osteosarcoma. J. Pathol. Clin. Res. 2021, 7, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Li, H.M.; Huang, Z. Comprehensive analysis of the expression and prognosis for cyclin-dependent protein kinase family in osteosarcoma. Nucleosides Nucleotides Nucleic Acids 2024, 1–24. [Google Scholar] [CrossRef]
- Synoradzki, K.J.; Bartnik, E.; Czarnecka, A.M.; Fiedorowicz, M.; Firlej, W.; Brodziak, A.; Stasinska, A.; Rutkowski, P.; Grieb, P. TP53 in biology and treatment of osteosarcoma. Cancers 2021, 13, 4284. [Google Scholar] [CrossRef]
- Xie, L.; Yang, Y.; Guo, W.; Che, D.; Xu, J.; Sun, X.; Liu, K.; Ren, T.; Liu, X.; Yang, Y.; et al. The clinical implications of tumor mutational burden in osteosarcoma. Front. Oncol. 2021, 10, 595527. [Google Scholar] [CrossRef]
- Beird, H.C.; Bielack, S.S.; Flanagan, A.M.; Gill, J.; Heymann, D.; Janeway, K.A.; Livingston, J.A.; Roberts, R.D.; Strauss, S.J.; Gorlick, R. Osteosarcoma. Nat. Rev. Dis. Primers 2022, 8, 77. [Google Scholar] [CrossRef]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol. 2020, 6, 724–734. [Google Scholar] [CrossRef]
- Harrison, D.J.; Geller, D.S.; Gill, J.D.; Lewis, V.O.; Gorlick, R. Current and future therapeutic approaches for osteosarcoma. Expert Rev. Anticancer Ther. 2018, 18, 39–50. [Google Scholar] [CrossRef]
- Tippett, V.L.; Tattersall, L.; Ab Latif, N.B.; Shah, K.M.; Lawson, M.A.; Gartland, A. The strategy and clinical relevance of in vitro models of MAP resistance in osteosarcoma: A systematic review. Oncogene 2023, 42, 259–277. [Google Scholar] [CrossRef]
- Garcia-Ortega, D.Y.; Cabrera-Nieto, S.A.; Caro-Sánchez, H.S.; Cruz-Ramos, M. An overview of resistance to chemotherapy in osteosarcoma and future perspectives. Cancer Drug Resist. 2022, 5, 762. [Google Scholar] [CrossRef] [PubMed]
- Marchandet, L.; Lallier, M.; Charrier, C.; Baud’huin, M.; Ory, B.; Lamoureux, F. Mechanisms of resistance to conventional therapies for osteosarcoma. Cancers 2021, 13, 683. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, J.; Li, Y. Effects of microenvironment in osteosarcoma on chemoresistance and the promise of immunotherapy as an osteosarcoma therapeutic modality. Front. Immunol. 2022, 13, 871076. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef]
- Mella, C.; Tsarouhas, P.; Brockwell, M.; Ball, H.C. The Role of Chronic Inflammation in Pediatric Cancer. Cancers 2025, 17, 154. [Google Scholar] [CrossRef]
- Buckley, D.N.; Tew, B.Y.; Gooden, C.; Salhia, B. A comprehensive analysis of minimally differentially methylated regions common to pediatric and adult solid tumors. npj Precis. Oncol. 2024, 8, 125. [Google Scholar] [CrossRef]
- Cristalli, C.; Scotlandi, K. Targeting DNA Methylation Machinery in Pediatric Solid Tumors. Cells 2024, 13, 1209. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- Lee, J.-M.; Yoshida, M.; Kim, M.-S.; Lee, J.-H.; Baek, A.-R.; Jang, A.S.; Kim, D.J.; Minagawa, S.; Chin, S.S.; Park, C.-S. Involvement of alveolar epithelial cell necroptosis in idiopathic pulmonary fibrosis pathogenesis. Am. J. Respir. Cell Mol. Biol. 2018, 59, 215–224. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jiang, N.; Su, W.; Zhuo, Y. Necroptosis: A novel pathway in neuroinflammation. Front. Pharmacol. 2021, 12, 701564. [Google Scholar] [CrossRef]
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 2015, 62, 592–601. [Google Scholar] [CrossRef]
- Höckendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer Cell 2016, 30, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Koo, G.-B.; Morgan, M.J.; Lee, D.-G.; Kim, W.-J.; Yoon, J.-H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Dai, W.; Cheng, J.; Leng, X.; Hu, X.; Ao, Y. The potential role of necroptosis in clinical diseases. Int. J. Mol. Med. 2021, 47, 89. [Google Scholar] [CrossRef]
- Laha, D.; Grant, R.; Mishra, P.; Nilubol, N. The role of tumor necrosis factor in manipulating the immunological response of tumor microenvironment. Front. Immunol. 2021, 12, 656908. [Google Scholar] [CrossRef]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Najafov, A.; Chen, H.; Yuan, J. Necroptosis and cancer. Trends Cancer 2017, 3, 294–301. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef]
- Zhan, C.; Huang, M.; Yang, X.; Hou, J. MLKL: Functions beyond serving as the Executioner of Necroptosis. Theranostics 2021, 11, 4759. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Kim, Y.-S. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp. Mol. Med. 2022, 54, 1695–1704. [Google Scholar] [CrossRef]
- Tan, Y.; Sementino, E.; Cheung, M.; Peri, S.; Menges, C.W.; Kukuyan, A.-M.; Zhang, T.; Khazak, V.; Fox, L.A.; Ross, E.A. Somatic epigenetic silencing of RIPK3 inactivates necroptosis and contributes to chemoresistance in malignant mesothelioma. Clin. Cancer Res. 2021, 27, 1200–1213. [Google Scholar] [CrossRef]
- Wu, B.; Li, J.; Wang, H.; Liu, J.; Li, J.; Sun, F.; Feng, D.C. RIPK1 is aberrantly expressed in multiple B-cell cancers and implicated in the underlying pathogenesis. Discov. Oncol. 2023, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Wu, D.; Du, J. RIPK3 modulates sarcoma through immune checkpoint HAVCR2. Oncol. Lett. 2022, 24, 381. [Google Scholar] [CrossRef]
- Tian, W.; Li, Y.; Zhang, J.; Li, J.; Gao, J. Combined analysis of DNA methylation and gene expression profiles of osteosarcoma identified several prognosis signatures. Gene 2018, 650, 7–14. [Google Scholar] [CrossRef]
- An, F.; Chang, W.; Song, J.; Zhang, J.; Li, Z.; Gao, P.; Wang, Y.; Xiao, Z.; Yan, C. Reprogramming of glucose metabolism: Metabolic alterations in the progression of osteosarcoma. J. Bone Oncol. 2024, 44, 100521. [Google Scholar] [CrossRef] [PubMed]
- Mohseny, A.B.; Machado, I.; Cai, Y.; Schaefer, K.-L.; Serra, M.; Hogendoorn, P.C.; Llombart-Bosch, A.; Cleton-Jansen, A.-M. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab. Investig. 2011, 91, 1195–1205. [Google Scholar] [CrossRef]
- Song, M.-S.; Alluin, J.; Rossi, J.J. The effect of dicer knockout on RNA interference using various dicer substrate small interfering RNA (DsiRNA) structures. Genes 2022, 13, 436. [Google Scholar] [CrossRef]
- Martinez-Osorio, V.; Abdelwahab, Y.; Ros, U. The many faces of MLKL, the executor of necroptosis. Int. J. Mol. Sci. 2023, 24, 10108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; He, W.; Sun, L. Necrosome core machinery: MLKL. Cell. Mol. Life Sci. 2016, 73, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.-G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. Sicot-J 2018, 4, 12. [Google Scholar] [CrossRef]
- Boyland, R.; Amin, S.; Shostrom, V.; Zheng, C.; Allison, J.; Lin, C. Comparison of overall survival of adult and pediatric osteosarcoma patients using the national cancer database. BMC Cancer 2025, 25, 290. [Google Scholar] [CrossRef]
- Hong, A.; Millington, S.; Ahern, V.; McCowage, G.; Boyle, R.; Tattersall, M.; Haydu, L.; Stalley, P. Limb preservation surgery with extracorporeal irradiation in the management of malignant bone tumor: The oncological outcomes of 101 patients. Ann. Oncol. 2013, 24, 2676–2680. [Google Scholar] [CrossRef]
- Cripe, T.P.; Yeager, N.D. Malignant Pediatric Bone Tumors-Treatment & Management; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Meazza, C.; Luksch, R.; Luzzati, A. Managing axial bone sarcomas in childhood. Expert Rev. Anticancer Ther. 2021, 21, 747–764. [Google Scholar] [CrossRef]
- Zhou, C.; Li, H.; Zeng, H.; Wang, P. Incidence trends, overall survival, and metastasis prediction using multiple machine learning and deep learning techniques in pediatric and adolescent population with osteosarcoma and Ewing’s sarcoma: Nomogram and webpage. Clin. Transl. Oncol. 2024, 1–12. [Google Scholar] [CrossRef]
- Martins-Neves, S.R.; Sampaio-Ribeiro, G.; Gomes, C.M. Chemoresistance-related stem cell signaling in osteosarcoma and its plausible contribution to poor therapeutic response: A discussion that still matters. Int. J. Mol. Sci. 2022, 23, 11416. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Patrizio, M.P.; Fantoni, L.; Casotti, C.; Riganti, C.; Serra, M. Drug resistance in osteosarcoma: Emerging biomarkers, therapeutic targets and treatment strategies. Cancers 2021, 13, 2878. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G. Relationship between RFC gene expression and intracellular drug concentration in methotrexate-resistant osteosarcoma cells. Genet. Mol. Res. 2014, 13, 5313–5321. [Google Scholar] [CrossRef]
- Malfatti, M.C.; Bellina, A.; Antoniali, G.; Tell, G. Revisiting two decades of research focused on targeting APE1 for cancer therapy: The pros and cons. Cells 2023, 12, 1895. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, D.; Cogdell, D.; Du, X.; Li, H.; Pang, Y.; Sun, Y.; Hu, L.; Sun, B.; Trent, J. APEX1 gene amplification and its protein overexpression in osteosarcoma: Correlation with recurrence, metastasis, and survival. Technol. Cancer Res. Treat. 2010, 9, 161–169. [Google Scholar] [CrossRef]
- Gong, G.; Ganesan, K.; Xiong, Q.; Zheng, Y. Anti-invasive and anti-migratory effects of ononin on human osteosarcoma cells by limiting the MMP2/9 and EGFR-Erk1/2 pathway. Cancers 2023, 15, 758. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-G.; Mo, H.-M.; Liu, X.-Q.; Li, Y.; Du, L.; Qiao, H.; Fan, Q.-M.; Zhao, J.; Zhang, S.-H.; Tang, T.-T. TIMP3 overexpression improves the sensitivity of osteosarcoma to cisplatin by reducing IL-6 production. Front. Genet. 2018, 9, 135. [Google Scholar] [CrossRef]
- Tune, B.X.J.; Sim, M.S.; Poh, C.L.; Guad, R.M.; Woon, C.K.; Hazarika, I.; Das, A.; Gopinath, S.C.; Rajan, M.; Sekar, M. Matrix metalloproteinases in chemoresistance: Regulatory roles, molecular interactions, and potential inhibitors. J. Oncol. 2022, 2022, 3249766. [Google Scholar] [CrossRef]
- Adamopoulos, C.; Gargalionis, A.N.; Basdra, E.K.; Papavassiliou, A.G. Deciphering signaling networks in osteosarcoma pathobiology. Exp. Biol. Med. 2016, 241, 1296–1305. [Google Scholar] [CrossRef]
- Ma, Y.; Ren, Y.; Han, E.Q.; Li, H.; Chen, D.; Jacobs, J.J.; Gitelis, S.; O’Keefe, R.J.; Konttinen, Y.T.; Yin, G. Inhibition of the Wnt-β-catenin and Notch signaling pathways sensitizes osteosarcoma cells to chemotherapy. Biochem. Biophys. Res. Commun. 2013, 431, 274–279. [Google Scholar] [CrossRef]
- Martins-Neves, S.R.; Sampaio-Ribeiro, G.; Gomes, C.M. Self-renewal and pluripotency in osteosarcoma stem cells’ chemoresistance: Notch, hedgehog, and wnt/β-catenin interplay with embryonic markers. Int. J. Mol. Sci. 2023, 24, 8401. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Wang, J.; Wang, S. Role of autophagy in drug resistance and regulation of osteosarcoma. Mol. Clin. Oncol. 2022, 16, 72. [Google Scholar] [CrossRef]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J. Hypoxia-inducible factor (HIF)–mediated effects of the hypoxic niche in bone cancer. In Bone Sarcomas and Bone Metastases-From Bench to Bedside; Elsevier: Amsterdam, The Netherlands, 2022; pp. 321–335. [Google Scholar]
- Zhou, J.; Lan, F.; Liu, M.; Wang, F.; Ning, X.; Yang, H.; Sun, H. Hypoxia inducible factor-1α as a potential therapeutic target for osteosarcoma metastasis. Front. Pharmacol. 2024, 15, 1350187. [Google Scholar] [CrossRef]
- Chen, H.; Gong, Z.; Zhou, H.; Han, Y. Deciphering chemoresistance in osteosarcoma: Unveiling regulatory mechanisms and function through the lens of noncoding RNA. Drug Dev. Res. 2024, 85, e22167. [Google Scholar] [CrossRef]
- Ferretti, V.A.; León, I.E. Long non-coding RNAs in cisplatin resistance in osteosarcoma. Curr. Treat. Options Oncol. 2021, 22, 41. [Google Scholar] [CrossRef]
- Hu, X.; Wen, Y.; Tan, L.-y.; Wang, J.; Tang, F.; Wang, Y.-T.; Zheng, C.-X.; Zhang, Y.-Q.; Gong, T.-J.; Min, L. Exosomal long non-coding RNA ANCR mediates drug resistance in osteosarcoma. Front. Oncol. 2022, 11, 735254. [Google Scholar] [CrossRef]
- Qin, S.; Wang, Y.; Ma, C.; Lv, Q. Competitive endogenous network of circRNA, lncRNA, and miRNA in osteosarcoma chemoresistance. Eur. J. Med. Res. 2023, 28, 354. [Google Scholar] [CrossRef]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brünner, N.; Kristensen, B.W. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neuro-Oncol. 2011, 103, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, J.M.; Wijetunga, N.A.; Fazzari, M.J.; Krailo, M.; Barkauskas, D.A.; Gorlick, R.; Greally, J.M. Predictive properties of DNA methylation patterns in primary tumor samples for osteosarcoma relapse status. Epigenetics 2015, 10, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Bertin, J.; Gough, P.; Orlowski, G.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hong, M.; Li, Y.; Chen, D.; Wu, Y.; Hu, Y. Programmed cell death tunes tumor immunity. Front. Immunol. 2022, 13, 847345. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Bridelance, J.; Roelandt, R.; Vandenabeele, P.; Takahashi, N. MLKL in cancer: More than a necroptosis regulator. Cell Death Differ. 2021, 28, 1757–1772. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Jiang, X.; Deng, W.; Tao, S.; Tang, Z.; Chen, Y.; Tian, M.; Wang, T.; Tao, C.; Li, Y.; Fang, Y. A RIPK3-independent role of MLKL in suppressing parthanatos promotes immune evasion in hepatocellular carcinoma. Cell Discov. 2023, 9, 7. [Google Scholar] [CrossRef]
- Liccardi, G.; Annibaldi, A. MLKL post-translational modifications: Road signs to infection, inflammation and unknown destinations. Cell Death Differ. 2023, 30, 269–278. [Google Scholar] [CrossRef]
- Herold, N. A guardian turned rogue: TP53 promoter translocations rewire stress responses to oncogenic effectors in osteosarcoma. Cancer Gene Ther. 2024, 31, 805–806. [Google Scholar] [CrossRef]
- Tian, H.; Cao, J.; Li, B.; Nice, E.C.; Mao, H.; Zhang, Y.; Huang, C. Managing the immune microenvironment of osteosarcoma: The outlook for osteosarcoma treatment. Bone Res. 2023, 11, 11. [Google Scholar] [PubMed]
- Yong, L.; Shi, Y.; Wu, H.-L.; Dong, Q.-Y.; Guo, J.; Hu, L.-S.; Wang, W.-H.; Guan, Z.-P.; Yu, B.-S. p53 inhibits CTR1-mediated cisplatin absorption by suppressing SP1 nuclear translocation in osteosarcoma. Front. Oncol. 2023, 12, 1047194. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Baldari, C.; Maiorano, G.; Mastronuzzi, A.; Carai, A.; Quintarelli, C.; De Angelis, B.; Cortese, B.; Gigli, G.; Palamà, I.E. Nanoparticles for diagnosis and target therapy in pediatric brain cancers. Diagnostics 2022, 12, 173. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wallach, M.; Krishna, A.; Kurmasheva, R.; Sridhar, S. Recent developments in nanomedicine for pediatric cancer. J. Clin. Med. 2021, 10, 1437. [Google Scholar] [CrossRef]
- McEachron, T.A.; Helman, L.J. Recent advances in pediatric cancer research. Cancer Res. 2021, 81, 5783–5799. [Google Scholar]
- Chang, W.-I.; Zhou, L.; Seyhan, A.A.; Zhang, Y.; El-Deiry, W.S. Novel therapeutic targeting of epigenetic aberrations in pediatric sarcomas through combination of ONC201 and HDAC inhibitors. Cancer Res. 2020, 80 (Suppl. S16), 3902. [Google Scholar] [CrossRef]
- Chilamakuri, R.; Agarwal, S. Dual targeting of PI3K and HDAC by CUDC-907 inhibits pediatric neuroblastoma growth. Cancers 2022, 14, 1067. [Google Scholar] [CrossRef]
- Pommert, L.; Schafer, E.S.; Malvar, J.; Gossai, N.; Florendo, E.; Pulakanti, K.; Heimbruch, K.; Stelloh, C.; Chi, Y.Y.; Sposto, R. Decitabine and vorinostat with FLAG chemotherapy in pediatric relapsed/refractory AML: Report from the therapeutic advances in childhood leukemia and lymphoma (TACL) consortium. Am. J. Hematol. 2022, 97, 613–622. [Google Scholar]
- Fatma, H.; Maurya, S.K.; Siddique, H.R. Epigenetic modifications of c-MYC: Role in cancer cell reprogramming, progression and chemoresistance. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 166–176. [Google Scholar]
- Li, W.; Wang, Y.; Liu, R.; Kasinski, A.L.; Shen, H.; Slack, F.J.; Tang, D.G. MicroRNA-34a: Potent tumor suppressor, cancer stem cell inhibitor, and potential anticancer therapeutic. Front. Cell Dev. Biol. 2021, 9, 640587. [Google Scholar]
- Wang, D.; Yi, H.; Geng, S.; Jiang, C.; Liu, J.; Duan, J.; Zhang, Z.; Shi, J.; Song, H.; Guo, Z. Photoactivated DNA nanodrugs damage mitochondria to improve gene therapy for reversing chemoresistance. ACS Nano 2023, 17, 16923–16934. [Google Scholar] [CrossRef]
- Saeed, H.; Sinha, S.; Mella, C.; Kuerbitz, J.S.; Cales, M.L.; Steele, M.A.; Stanke, J.; Damron, D.; Safadi, F.; Kuerbitz, S.J. Aberrant epigenetic silencing of neuronatin is a frequent event in human osteosarcoma. Oncotarget 2020, 11, 1876. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.; Moussa, F.; Mbimba, T.; Orman, R.; Safadi, F.; Cooper, L. Methods and insights from the characterization of osteoprogenitor cells of bats (Mammalia: Chiroptera). Stem Cell Res. 2016, 17, 54–61. [Google Scholar]
- Ball, H.C.; Holmes, R.K.; Londraville, R.L.; Thewissen, J.G.; Duff, R.J. Leptin in whales: Validation and measurement of mRNA expression by absolute quantitative real-time PCR. PLoS ONE 2013, 8, e54277. [Google Scholar]
- Ball, H.C.; Alejo, A.L.; Samson, T.K.; Alejo, A.M.; Safadi, F.F. Epigenetic regulation of chondrocytes and subchondral bone in osteoarthritis. Life 2022, 12, 582. [Google Scholar] [CrossRef]
- Gilda, J.E.; Gomes, A.V. Stain-Free total protein staining is a superior loading control to β-actin for Western blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Gürtler, A.; Kunz, N.; Gomolka, M.; Hornhardt, S.; Friedl, A.A.; McDonald, K.; Kohn, J.E.; Posch, A. Stain-Free technology as a normalization tool in Western blot analysis. Anal. Biochem. 2013, 433, 105–111. [Google Scholar]
- Sule, R.; Rivera, G.; Gomes, A.V. Western blotting (immunoblotting): History, theory, uses, protocol and problems. Biotechniques 2023, 75, 99–114. [Google Scholar]
- Valente, V.; Teixeira, S.A.; Neder, L.; Okamoto, O.K.; Oba-Shinjo, S.M.; Marie, S.K.; Scrideli, C.A.; Paco-Larson, M.L.; Carlotti, C.G. Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. BMC Mol. Biol. 2009, 10, 17. [Google Scholar]
- Kang, T.H.; Park, Y.; Bader, J.S.; Friedmann, T. The housekeeping gene hypoxanthine guanine phosphoribosyltransferase (HPRT) regulates multiple developmental and metabolic pathways of murine embryonic stem cell neuronal differentiation. PLoS ONE 2013, 8, e74967. [Google Scholar] [CrossRef]
- Chim, L.K.; Williams, I.L.; Bashor, C.J.; Mikos, A.G. Tumor-associated macrophages induce inflammation and drug resistance in a mechanically tunable engineered model of osteosarcoma. Biomaterials 2023, 296, 122076. [Google Scholar]
- De Luca, A.; Bellavia, D.; Raimondi, L.; Carina, V.; Costa, V.; Fini, M.; Giavaresi, G. Multiple effects of resveratrol on osteosarcoma cell lines. Pharmaceuticals 2022, 15, 342. [Google Scholar] [CrossRef] [PubMed]
- Sunjic, S.B.; Gasparovic, A.C.; Jaganjac, M.; Rechberger, G.; Meinitzer, A.; Grune, T.; Kohlwein, S.D.; Mihaljevic, B.; Zarkovic, N. Sensitivity of osteosarcoma cells to concentration-dependent bioactivities of lipid peroxidation product 4-hydroxynonenal depend on their level of differentiation. Cells 2021, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Thongkumkoon, P.; Sangphukieo, A.; Tongjai, S.; Noisagul, P.; Sangkhathat, S.; Laochareonsuk, W.; Kamolphiwong, R.; Budprom, P.; Teeyakasem, P.; Yongpitakwattana, P. Establishment, characterization, and genetic profiling of patient-derived osteosarcoma cells from a patient with retinoblastoma. Sci. Rep. 2024, 14, 11056. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Percent Methylation—RIPK3 | ||
|---|---|---|
| Sample | Pyrosequencing | NGS |
| hMSC | 10.6 | 12.1 |
| hFOB | 18.9 | 20.1 |
| HOS | 62.9 | 78.7 |
| G292 | 87.9 | 95.7 |
| MNNGHOS | 88.7 | 94.9 |
| MG63 | 91.0 | 96.2 |
| 143B | 88.9 | 94.7 |
| Non-cancerous bone 1 | 3.6 | 1.4 |
| Non-cancerous bone 2 | 1.0 | 0.7 |
| OS 1 | 3.2 | 2.6 |
| OS 2 | 4.6 | 4.4 |
| OS 3 | 0.6 | 4.5 |
| OS 4 | 31.4 | 46.0 |
| OS 5 | 34.4 | 54.5 |
| OS 6 | 1.5 | - |
| OS 7 | 44.4 | 55.6 |
| OS 8 | 6.7 | 8.0 |
| OS 9 | 45.6 | 56.7 |
| OS 10 | 26.4 | 30.4 |
| OS 11 | 33.1 | 44.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Pettee, D.; Mella, C.; Hord, C.; Brockwell, M.; Hardy, S.; Ball, H.C.; Safadi, F.F.; Kuerbitz, S.J. Epigenetic Inactivation of RIPK3-Dependent Necroptosis Augments Cisplatin Chemoresistance in Human Osteosarcoma. Int. J. Mol. Sci. 2025, 26, 3863. https://doi.org/10.3390/ijms26083863
Sharma A, Pettee D, Mella C, Hord C, Brockwell M, Hardy S, Ball HC, Safadi FF, Kuerbitz SJ. Epigenetic Inactivation of RIPK3-Dependent Necroptosis Augments Cisplatin Chemoresistance in Human Osteosarcoma. International Journal of Molecular Sciences. 2025; 26(8):3863. https://doi.org/10.3390/ijms26083863
Chicago/Turabian StyleSharma, Aditya, Daniel Pettee, Christine Mella, Catherine Hord, Maximilian Brockwell, Samantha Hardy, Hope C. Ball, Fayez F. Safadi, and Steven J. Kuerbitz. 2025. "Epigenetic Inactivation of RIPK3-Dependent Necroptosis Augments Cisplatin Chemoresistance in Human Osteosarcoma" International Journal of Molecular Sciences 26, no. 8: 3863. https://doi.org/10.3390/ijms26083863
APA StyleSharma, A., Pettee, D., Mella, C., Hord, C., Brockwell, M., Hardy, S., Ball, H. C., Safadi, F. F., & Kuerbitz, S. J. (2025). Epigenetic Inactivation of RIPK3-Dependent Necroptosis Augments Cisplatin Chemoresistance in Human Osteosarcoma. International Journal of Molecular Sciences, 26(8), 3863. https://doi.org/10.3390/ijms26083863