
Isolated and Syndromic Genetic Optic Neuropathies: A Review of Genetic and Phenotypic Heterogeneity

 ,
,  ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Methods
3. Results
3.1. Molecular Causes of Optic Neuropathies
3.1.1. Isolated (Nonsyndromic) Genetic Optic Neuropathies
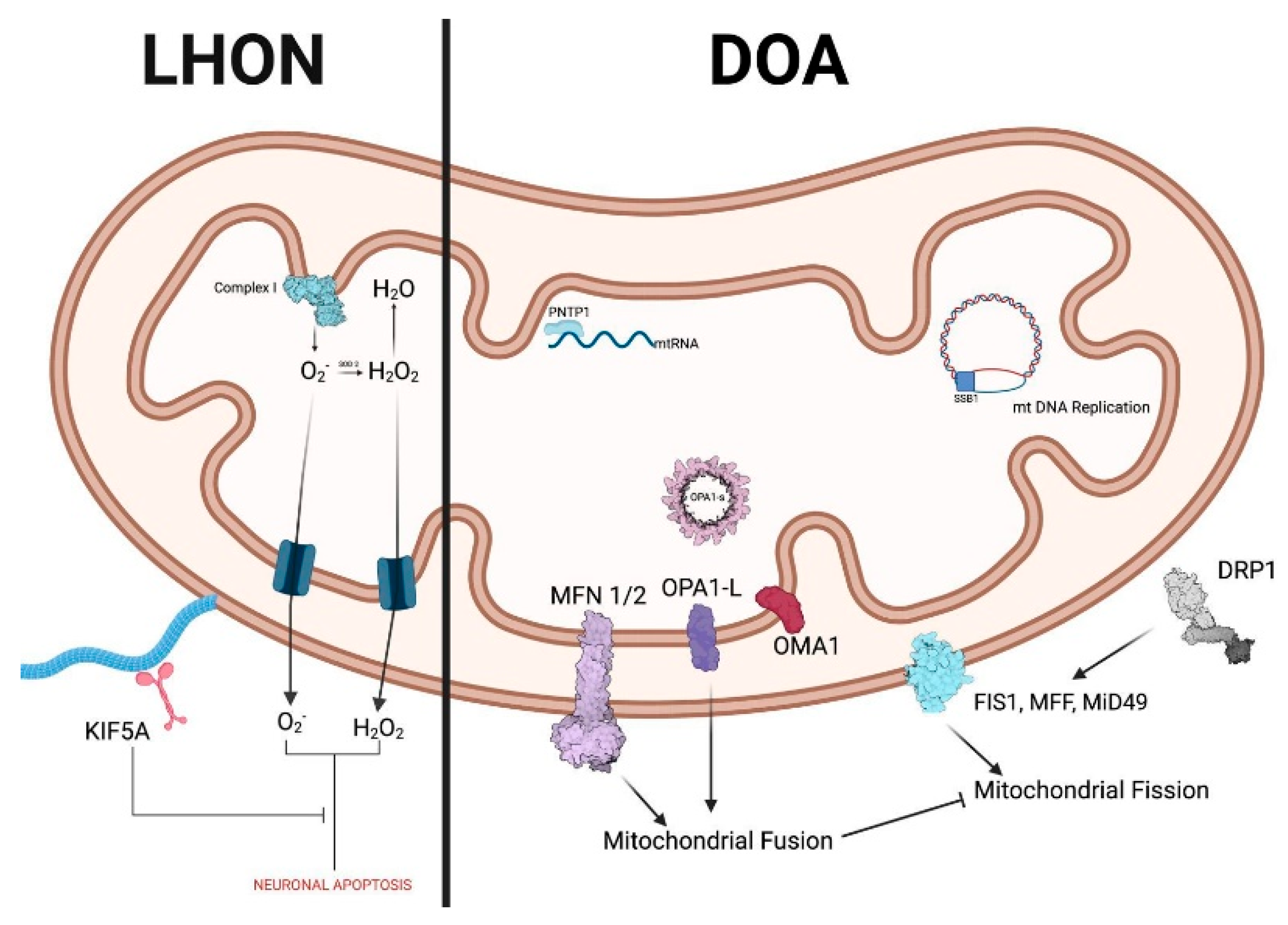
Leber Hereditary Optic Neuropathy (LHON)
Dominant Optic Atrophy (DOA)
Other HONs

{kind=link}
{kind=link}
| Locus (Phenotype MIM No.) | Gene | Inheritance | Phenotype(s) |
|---|---|---|---|
| OPA1 (165500) | OPA1 | AD | Optic atrophy Optic atrophy plus |
| OPA3 (165300) | OPA3 | AD/AR | Optic atrophy + possible cataract |
| OPA5 (610708) | DNM1L | AD | Optic atrophy |
| OPA7 (612988) | TMEM126A | AR | Optic atrophy + possible HI, CD, ND |
| OPA9 (616289) | ACO2 | AD/AR | Optic atrophy + possible ND |
| OPA10 (616732) | RTN4IP1 | AR | Optic atrophy + possible ND |
| OPA11 (617302) | YME1L1 | AR | Optic atrophy + ND |
| OPA12 (618977) | AFG3L2 | AD | Optic atrophy + possible ND |
| OPA13 (165510) | SSBP1 | AD | Optic atrophy and retinal abnormalities + possible HI, PN |
| OPA14 (620550) | MIEF1 | AD | Optic atrophy |
| OPA15 (620583) | MCAT | AR | Optic atrophy |
| OPA16 (620629) | MECR | AR | Optic atrophy + HI |
3.2. Syndromic HONs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu-Wai-Man, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited mitochondrial optic neuropathies. J. Med. Genet. 2009, 46, 145–158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Milea, D.; Amati-Bonneau, P.; Reynier, P.; Bonneau, D. Genetically determined optic neuropathies. Curr. Opin. Neurol. 2010, 23, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.A.; Korsten, A.; Leonhardt, M.; Dimitriadis, K.; De Coo, I.F.; Klopstock, T.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F.; Yu-Wai-Man, P. Quality of life in patients with Leber hereditary optic neuropathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3112–3115. [Google Scholar] [CrossRef] [PubMed]
- Eckmann-Hansen, C.; Bek, T.; Sander, B.; Larsen, M. Vision-related quality of life and visual ability in patients with autosomal dominant optic atrophy. Acta Ophthalmol. 2022, 100, 797–804. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Newman, N.J.; Biousse, V. Hereditary optic neuropathies. Eye 2004, 18, 1144–1160. [Google Scholar] [CrossRef] [PubMed]
- Balducci, N.; Ciardella, A.; Gattegna, R.; Zhou, Q.; Cascavilla, M.L.; La Morgia, C.; Savini, G.; Parisi, V.; Bandello, F.; Carelli, V.; et al. Optical coherence tomography angiography of the peripapillary retina and optic nerve head in dominant optic atrophy. Mitochondrion 2017, 36, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Biousse, V.; Newman, N.J. Neuro-ophthalmology of mitochondrial diseases. Curr. Opin. Neurol. 2003, 16, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Optic nerve degeneration and mitochondrial dysfunction: Genetic and acquired optic neuropathies. Neurochem. Int. 2002, 40, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Jurkute, N.; Majander, A.; Bowman, R.; Votruba, M.; Abbs, S.; Acheson, J.; Lenaers, G.; Amati-Bonneau, P.; Moosajee, M.; Arno, G.; et al. Clinical utility gene card for: Inherited optic neuropathies including next-generation sequencing-based approaches. Eur. J. Hum. Genet. 2019, 27, 494–502. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Delatycki, M.B.; Corben, L.A. Clinical features of Friedreich ataxia. J. Child. Neurol. 2012, 27, 1133–1137. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- D’Esposito, F.; Zeppieri, M.; Cordeiro, M.F.; Capobianco, M.; Avitabile, A.; Gagliano, G.; Musa, M.; Barboni, P.; Gagliano, C. Insights on the Genetic and Phenotypic Complexities of Optic Neuropathies. Genes 2024, 15, 1559. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carelli, V.; Carbonelli, M.; de Coo, I.F.; Kawasaki, A.; Klopstock, T.; Lagrèze, W.A.; La Morgia, C.; Newman, N.J.; Orssaud, C.; Pott, J.W.R.; et al. International Consensus Statement on the Clinical and Therapeutic Management of Leber Hereditary Optic Neuropathy. J. Neuroophthalmol. 2017, 37, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mascialino, B.; Leinonen, M.; Meier, T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. Eur. J. Ophthalmol. 2012, 22, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Barboni, P.; Carbonelli, M.; Savini, G.; Ramos Cdo, V.; Carta, A.; Berezovsky, A.; Salomao, S.R.; Carelli, V.; Sadun, A.A. Natural history of Leber’s hereditary optic neuropathy: Longitudinal analysis of the retinal nerve fiber layer by optical coherence tomography. Ophthalmology 2010, 117, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, C.; Van Stavern, G.; McClelland, C. Leber hereditary optic neuropathy: Current perspectives. Clin. Ophthalmol. 2015, 9, 1165–1176. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Balducci, N.; Savini, G.; Cascavilla, M.L.; La Morgia, C.; Triolo, G.; Giglio, R.; Carbonelli, M.; Parisi, V.; Sadun, A.A.; Bandello, F.; et al. Macular nerve fiber and ganglion cell layer changes in acute Leber’s hereditary optic neuropathy. Br. J. Ophthalmol. 2016, 100, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Savini, G.; Barboni, P.; Valentino, M.L.; Montagna, P.; Cortelli, P.; De Negri, A.M.; Sadun, F.; Bianchi, S.; Longanesi, L.; Zanini, M.; et al. Retinal nerve fiber layer evaluation by optical coherence tomography in unaffected carriers with Leber’s hereditary optic neuropathy mutations. Ophthalmology 2005, 112, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Newman, N.J.; Miller, N.R.; Johns, D.R.; Lott, M.T.; Wallace, D.C. Visual recovery in patients with Leber’s hereditary optic neuropathy and the 11778 mutation. J. Clin. Neuroophthalmol. 1992, 12, 10–14. [Google Scholar] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas LJ2nd Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Achilli, A.; Iommarini, L.; Olivieri, A.; Pala, M.; Hooshiar Kashani, B.; Reynier, P.; La Morgia, C.; Valentino, M.L.; Liguori, R.; Pizza, F.; et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber’s hereditary optic neuropathy. PLoS ONE 2012, 7, e42242. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu-Wai-Man, P.; Carelli, V.; Newman, N.J.; Silva, M.J.; Linden, A.; Van Stavern, G.; Szaflik, J.P.; Banik, R.; Lubiński, W.; Pemp, B.; et al. The therapeutic benefit of idebenone in patients with Leber hereditary optic neuropathy: The LEROS nonrandomized controlled trial. Cell Rep. Med. 2024, 5, 101437. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011, 134 Pt 9, 2677–2686. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Rizzo, G.; Carbonelli, M.; De Negri, A.M.; Sadun, F.; Carta, A.; Guerriero, S.; Simonelli, F.; et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain 2011, 134 Pt 9, e188. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Metz, G.; Yu-Wai-Man, P.; Büchner, B.; Gallenmüller, C.; Bailie, M.; Nwali, N.; Griffiths, P.G.; von Livonius, B.; Reznicek, L.; et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain 2013, 136 Pt 2, e230. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Newman, N.J.; Yu-Wai-Man, P.; Subramanian, P.S.; Moster, M.L.; Wang, A.G.; Donahue, S.P.; Leroy, B.P.; Carelli, V.; Biousse, V.; Vignal-Clermont, C.; et al. Randomized trial of bilateral gene therapy injection for m.11778G>A MT-ND4 Leber optic neuropathy. Brain 2023, 146, 1328–1341. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wong, D.C.S.; Harvey, J.P.; Jurkute, N.; Thomas, S.M.; Moosajee, M.; Yu-Wai-Man, P.; Gilhooley, M.J. OPA1 Dominant Optic Atrophy: Pathogenesis and Therapeutic Targets. J. Neuroophthalmol. 2023, 43, 464–474. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Asanad, S.; Tian, J.J.; Frousiakis, S.; Jiang, J.P.; Kogachi, K.; Felix, C.M.; Fatemeh, D.; Irvine, A.G.; Ter-Zakarian, A.; Falavarjani, K.G.; et al. Optical Coherence Tomography of the Retinal Ganglion Cell Complex in Leber’s Hereditary Optic Neuropathy and Dominant Optic Atrophy. Curr. Eye Res. 2019, 44, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Corajevic, N.; Larsen, M.; Rönnbäck, C. Thickness mapping of individual retinal layers and sectors by Spectralis SD-OCT in Autosomal Dominant Optic Atrophy. Acta Ophthalmol. 2018, 96, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133 Pt 3, 771–786. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Whitehead, M.; Harvey, J.P.; Sladen, P.E.; Becchi, G.; Singh, K.; Sun, Y.J.; Burgoyne, T.; Duchen, M.R.; Yu-Wai-Man, P.; Cheetham, M.E. Disruption of mitochondrial homeostasis and permeability transition pore opening in OPA1 iPSC-derived retinal ganglion cells. Acta Neuropathol. Commun. 2025, 13, 28. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jurkute, N.; D’Esposito, F.; Robson, A.G.; Pitceathly, R.D.S.; Cordeiro, F.; Raymond, F.L.; Moore, A.T.; Michaelides, M.; Yu-Wai-Man, P.; Webster, A.R.; et al. SSBP1-Disease Update: Expanding the Genetic and Clinical Spectrum, Reporting Variable Penetrance and Confirming Recessive Inheritance. Invest. Ophthalmol. Vis. Sci. 2021, 62, 12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Thiselton, D.L.; Alexander, C.; Morris, A.; Brooks, S.; Rosenberg, T.; Eiberg, H.; Kjer, B.; Kjer, P.; Bhattacharya, S.S.; Votruba, M. A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: Evidence for a founder effect. Hum. Genet. 2001, 109, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Almind, G.J.; Ek, J.; Rosenberg, T.; Eiberg, H.; Larsen, M.; Lucamp, L.; Brøndum-Nielsen, K.; Grønskov, K. Dominant optic atrophy in Denmark—Report of 15 novel mutations in OPA1, using a strategy with a detection rate of 90%. BMC Med. Genet. 2012, 13, 65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferré, M.; Bonneau, D.; Milea, D.; Chevrollier, A.; Verny, C.; Dollfus, H.; Ayuso, C.; Defoort, S.; Vignal, C.; Zanlonghi, X.; et al. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum. Mutat. 2009, 30, E692–E705. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Schimpf-Linzenbold, S.; Mazzola, P.; Kieninger, S.; Xiao, T.; Kellner, U.; Neuhann, T.; Kelbsch, C.; Tonagel, F.; Wilhelm, H.; et al. Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: Identification and classification of 48 novel variants. PLoS ONE 2021, 16, e0253987. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Jüschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye Res. 2021, 83, 100935. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Veitia, R.A.; Caburet, S.; Birchler, J.A. Mechanisms of Mendelian dominance. Clin. Genet. 2018, 93, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog. Retin. Eye Res. 2013, 36, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.A.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Barron, M.J.; Griffiths, P.; Turnbull, D.M.; Bates, D.; Nichols, P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br. J. Ophthalmol. 2004, 88, 286–290. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu Wai Man, C.Y.; Chinnery, P.F.; Griffiths, P.G. Optic neuropathies--importance of spatial distribution of mitochondria as well as function. Med. Hypotheses 2005, 65, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Valentin, K.; Georgi, T.; Riedl, R.; Aminfar, H.; Singer, C.; Klopstock, T.; Wedrich, A.; Schneider, M. Idebenone Treatment in Patients with OPA1-Dominant Optic Atrophy: A Prospective Phase 2 Trial. Neuroophthalmology 2023, 47, 237–247. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sarzi, E.; Seveno, M.; Piro-Mégy, C.; Elzière, L.; Quilès, M.; Péquignot, M.; Müller, A.; Hamel, C.P.; Lenaers, G.; Delettre, C. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci. Rep. 2018, 8, 2468. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Venkatesh, A.; Li, Z.; Christiansen, A.; Lim, K.H.; Kach, J.; Hufnagel, R.; Aznarez, I.; Liau, G. Antisense oligonucleotide mediated increase of OPA1 expression using TANGO technology for the treatment of autosomal dominant optic atrophy. Investig. Ophthalmol. Vis. Sci. 2020, 61, 2755. [Google Scholar]
- Sladen, P.E.; Perdigão, P.R.L.; Salsbury, G.; Novoselova, T.; van der Spuy, J.; Chapple, J.P.; Yu-Wai-Man, P.; Cheetham, M.E. CRISPR-Cas9 correction of OPA1 c.1334G>A: P.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Mol. Ther. Nucleic Acids 2021, 26, 432–443. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- OMIM. Available online: https://www.omim.org.
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diab Rep. 2016, 16, 6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takeda, K.; Inoue, H.; Tanizawa, Y.; Matsuzaki, Y.; Oba, J.; Watanabe, Y.; Shinoda, K.; Oka, Y. WFS1 (Wolfram syndrome 1) gene product: Predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum. Mol. Genet. 2001, 10, 477–484. [Google Scholar] [CrossRef] [PubMed]
- de Muijnck, C.; Brink, J.B.T.; Bergen, A.A.; Boon, C.J.F.; van Genderen, M.M. Delineating Wolfram-like syndrome: A systematic review and discussion of the WFS1-associated disease spectrum. Surv. Ophthalmol. 2023, 68, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Hofmann, S.; Hamasaki, D.I.; Yamamoto, H.; Kreczmanski, P.; Schmitz, C.; Parel, J.M.; Schmidt-Kastner, R. Wolf ram syndrome 1 (WFS1) protein expression in retinal ganglion cells and optic nerve glia of the cynomolgus monkey. Exp. Eye Res. 2006, 83, 1303–1306. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Tanabe, K.; Kaneko, N.; Hishimura, N.; Nakamura, A. Comprehensive overview of disease models for Wolfram syndrome: Toward effective treatments. Mamm. Genome 2024, 35, 1–12. [Google Scholar] [CrossRef] [PubMed]
- El-Shanti, H.; Lidral, A.C.; Jarrah, N.; Druhan, L.; Ajlouni, K. Homozygosity mapping identifies an additional locus for Wolfram syndrome on chromosome 4q. Am. J. Hum. Genet. 2000, 66, 1229–1236. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mozzillo, E.; Delvecchio, M.; Carella, M.; Grandone, E.; Palumbo, P.; Salina, A.; Aloi, C.; Buono, P.; Izzo, A.; D’Annunzio, G.; et al. A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2. BMC Med. Genet. 2014, 15, 88. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rondinelli, M.; Novara, F.; Calcaterra, V.; Zuffardi, O.; Genovese, S. Wolfram syndrome 2: A novel CISD2 mutation identified in Italian siblings. Acta Diabetol. 2015, 52, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, F.; Barboni, P.; Liguori, R.; Valentino, M.L.; Savini, G.; Gellera, C.; Mariotti, C.; Rizzo, G.; Tonon, C.; Manners, D.; et al. Visual system involvement in patients with Friedreich’s ataxia. Brain 2009, 132 Pt 1, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Reetz, K.; Dogan, I.; Hohenfeld, C.; Didszun, C.; Giunti, P.; Mariotti, C.; Durr, A.; Boesch, S.; Klopstock, T.; Rodríguez de Rivera Garrido, F.J.; et al. Nonataxia symptoms in Friedreich Ataxia: Report from the Registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS). Neurology 2018, 91, e917–e930. [Google Scholar] [CrossRef] [PubMed]
- Buesch, K.; Zhang, R. A systematic review of disease prevalence, health-related quality of life, and economic outcomes associated with Friedreich’s Ataxia. Curr. Med. Res. Opin. 2022, 38, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Argentini, M.; Carelle-Calmels, N.; Martelli, A.; Puccio, H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008, 17, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Bidichandani, S.I.; Delatycki, M.B.; Napierala, M.; Duquette, A. Friedreich Ataxia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1998; pp. 1993–2025. [Google Scholar] [PubMed]
- Rojas, P.; de Hoz, R.; Cadena, M.; Salobrar-García, E.; Fernández-Albarral, J.A.; López-Cuenca, I.; Elvira-Hurtado, L.; Urcelay-Segura, J.L.; Salazar, J.J.; Ramírez, J.M.; et al. Neuro-Ophthalmological Findings in Friedreich’s Ataxia. J. Pers. Med. 2021, 11, 708. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bombelli, F.; Stojkovic, T.; Dubourg, O.; Echaniz-Laguna, A.; Tardieu, S.; Larcher, K.; Amati-Bonneau, P.; Latour, P.; Vignal, O.; Cazeneuve, C.; et al. Charcot-Marie-Tooth disease type 2A: From typical to rare phenotypic and genotypic features. JAMA Neurol. 2014, 71, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Komen, J.; Ferdinandusse, S. Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta 2011, 1811, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Barboni, P.; Savini, G.; Valentino, M.L.; Montagna, P.; Cortelli, P.; De Negri, A.M.; Sadun, F.; Bianchi, S.; Longanesi, L.; Zanini, M.; et al. Retinal nerve fiber layer evaluation by optical coherence tomography in Leber’s hereditary optic neuropathy. Ophthalmology 2005, 112, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Sadun, F.; De Negri, A.M.; Carelli, V.; Salomao, S.R.; Berezovsky, A.; Andrade, R.; Moraes, M.; Passos, A.; Belfort, R.; da Rosa, A.B.; et al. Ophthalmologic findings in a large pedigree of 11778/Haplogroup J Leber hereditary optic neuropathy. Am. J. Ophthalmol. 2004, 137, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Machado, T.; Cortinhal, T.; Carvalho, A.L.; Teixeira-Marques, F.; Silva, R.; Murta, J.; Marques, J.P. Unraveling the genetic spectrum of inherited deaf-blindness in Portugal. Orphanet J. Rare Dis. 2025, 20, 22. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, M.; Vieta-Ferrer, E.R.; Bakdalieh, A.; Tsai, T. The Role of Visual Electrophysiology in Systemic Hereditary Syndromes. Int. J. Mol. Sci. 2025, 26, 957. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guy, J.; Feuer, W.J.; Davis, J.L.; Porciatti, V.; Gonzalez, P.J.; Koilkonda, R.D.; Yuan, H.; Hauswirth, W.W.; Lam, B.L. Gene Therapy for Leber Hereditary Optic Neuropathy: Low- and Medium-Dose Visual Results. Ophthalmology 2017, 124, 1621–1634. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Klopstock, T.; Zeng, L.H.; Priglinger, C. Leber’s hereditary optic neuropathy—Current status of idebenone and gene replacement therapies. Med. Genet. 2025, 37, 57–63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, X.; Yuan, J.; Qi, J.; Ruan, K.; Li, B.; Dan, Y.; Zhang, Y. The rAAV2-ND1 Gene Therapy for Leber Hereditary Optic Neuropathy. Arch. Clin. Exp. Ophthalmol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Ferro Desideri, L.; Traverso, C.E.; Iester, M. Current treatment options for treating OPA1-mutant dominant optic atrophy. Drugs Today 2022, 58, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, V.W. Precision Medicine for Continuing Phenotype Expansion of Human Genetic Diseases. BioMed Res. Int. 2015, 2015, 745043. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rabenstein, A.; Catarino, C.B.; Rampeltshammer, V.; Schindler, D.; Gallenmüller, C.; Priglinger, C.; Pogarell, O.; Rüther, T.; Klopstock, T. Smoking and alcohol, health-related quality of life and psychiatric comorbidities in Leber’s Hereditary Optic Neuropathy mutation carriers: A prospective cohort study. Orphanet J. Rare Dis. 2021, 16, 127. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ezan, P.; Hardy, E.; Bemelmans, A.; Taiel, M.; Dossi, E.; Rouach, N. Retinal damage promotes mitochondrial transfer in the visual system of a mouse model of Leber hereditary optic neuropathy. Neurobiol. Dis. 2024, 201, 106681. [Google Scholar] [CrossRef] [PubMed]
- Grier, J.; Hirano, M.; Karaa, A.; Shepard, E.; Thompson, J.L.P. Diagnostic odyssey of patients with mitochondrial disease: Results of a survey. Neurol. Genet. 2018, 4, e230. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tung, C.; Varzideh, F.; Farroni, E.; Mone, P.; Kansakar, U.; Jankauskas, S.S.; Santulli, G. Elamipretide: A Review of Its Structure, Mechanism of Action, and Therapeutic Potential. Int. J. Mol. Sci. 2025, 26, 944. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, K.Y.; Osman, R.M.; Esomchukwu, O.; Marchand, M.; Nguyen, B.H.; Tran, S.D. Advances in Regenerative Medicine, Cell Therapy, and 3D Bioprinting for Glaucoma and Retinal Diseases. Adv. Exp. Med. Biol. 2025. [Google Scholar] [CrossRef] [PubMed]

| Trial (ID) | Intervention | Phase | Design | Population | Key Outcomes | Status |
|---|---|---|---|---|---|---|
| RHODOS (NCT00747487) | Idebenone 900 mg/day | II | RCT, placebo-controlled (24 wks) | LHON (<5 yrs onset, n = 85) | No significant primary outcome; secondary VA endpoints improved. | Completed (2011) |
| LEROS (NCT02774005) | Idebenone 900 mg/day (post-approval) | IV | Observational, uncontrolled | LHON (<5 yrs onset, n = 199) | Clinically relevant visual improvement compared with natural history. | Completed (2020) |
| RESCUE/REVERSE (NCT02652780) | Lenadogene nolparvovec (AAV2-ND4, unilateral) | III | Sham-controlled, double-masked RCT | LHON (ND4), n = 76 | Bilateral visual improvement superior to natural history. | Completed (2020–21) |
| REFLECT (NCT03293524) | Lenadogene nolparvovec (bilateral injections) | III | Sham-controlled, double-masked RCT | LHON (ND4), n = 98 | Significant bilateral VA improvement (+22 ETDRS letters). | Completed (2022) |
| Idebenone in DOA (no NCT ID) | Idebenone 900 mg/day (compassionate use) | II | Prospective open-label (12 months) | DOA (OPA1; n = 10) | Modest visual acuity improvement after 1 year. | Completed (2023) |
| NICODA (NCT06007391) | Nicotinamide (vitamin B3, oral) | II/III | Randomized pilot (12 months) | DOA/DOA+ (OPA1; adults > 18, n = 30) | Results pending; ongoing. | Recruiting (2025) |
| DAN-WS (NCT02829268) | Dantrolene sodium (oral) | Ib/IIa | Open-label dose escalation (6 months) | Wolfram syndrome (n = 24) | Well tolerated, no significant visual benefit. | Completed (2021) |
| TREATWOLFRAM (NCT03717909) | Sodium valproate vs. placebo (oral) | II | Double-blind placebo-controlled RCT | Wolfram syndrome (>5 yrs, n = 70) | Primary outcomes pending; ongoing. | Ongoing |
| Molecular Target | Therapeutic Strategy (Type) | Evidence (Stage) |
|---|---|---|
| Mitochondrial complex I (LHON mtDNA mutations, e.g., MT-ND4, ND1, ND6) | Idebenone (pharmacological antioxidant, bypassing complex I) | Clinical: EMA-approved; RHODOS trial showed visual acuity improvements in secondary endpoints. |
| Mitochondrial ND4 gene (m.11778G>A mutation in LHON) | Gene therapy (Allotopic expression)—lenadogene nolparvovec (AAV2-ND4) | Clinical: Significant visual acuity improvement in phase III trials (RESCUE, REVERSE, REFLECT). |
| OPA1 (haploinsufficiency) (DOA) | Gene therapy (gene augmentation)—AAV vector carrying wild-type OPA1 | Preclinical: Effective RGC protection in DOA mouse models; no clinical trials yet. |
| OPA1 (haploinsufficiency) (DOA) | Antisense oligonucleotide (ASO)—TANGO technology to increase OPA1 expression | Preclinical: Effective in cell models; under investigation. |
| Mitochondrial biogenesis/NAD+ metabolism (energetic deficit in DOA, LHON) | Nicotinamide (Vitamin B3) (nutraceutical therapy) | Preclinical/Clinical: Improved mitochondrial function in cell models; ongoing phase II/III trials in DOA patients. |
| Retinal ganglion cell (RGC) neuroprotection | Stem cell therapy—intravitreal injection of autologous MSC | Clinical (early): SCOTS trial (phase I/II) reported visual improvements; further studies needed. |
| ER Ca2⁺ homeostasis (ER stress in Wolfram syndrome, WFS1) | Dantrolene sodium—ER stress reduction | Clinical: Phase Ib/IIa trial showed good tolerability; no significant vision improvement at 6 months. |
| ER stress/UPR response (WFS1, Wolfram syndrome) | Sodium valproate—ER stress modulation | Clinical: Ongoing phase II trial (TREATWOLFRAM) assessing long-term efficacy. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeppieri, M.; Gagliano, C.; Di Maita, M.; Avitabile, A.; Gagliano, G.; Dammino, E.; Tognetto, D.; Cordeiro, M.F.; D’Esposito, F. Isolated and Syndromic Genetic Optic Neuropathies: A Review of Genetic and Phenotypic Heterogeneity. Int. J. Mol. Sci. 2025, 26, 3892. https://doi.org/10.3390/ijms26083892
Zeppieri M, Gagliano C, Di Maita M, Avitabile A, Gagliano G, Dammino E, Tognetto D, Cordeiro MF, D’Esposito F. Isolated and Syndromic Genetic Optic Neuropathies: A Review of Genetic and Phenotypic Heterogeneity. International Journal of Molecular Sciences. 2025; 26(8):3892. https://doi.org/10.3390/ijms26083892
Chicago/Turabian StyleZeppieri, Marco, Caterina Gagliano, Marco Di Maita, Alessandro Avitabile, Giuseppe Gagliano, Edoardo Dammino, Daniele Tognetto, Maria Francesca Cordeiro, and Fabiana D’Esposito. 2025. "Isolated and Syndromic Genetic Optic Neuropathies: A Review of Genetic and Phenotypic Heterogeneity" International Journal of Molecular Sciences 26, no. 8: 3892. https://doi.org/10.3390/ijms26083892
APA StyleZeppieri, M., Gagliano, C., Di Maita, M., Avitabile, A., Gagliano, G., Dammino, E., Tognetto, D., Cordeiro, M. F., & D’Esposito, F. (2025). Isolated and Syndromic Genetic Optic Neuropathies: A Review of Genetic and Phenotypic Heterogeneity. International Journal of Molecular Sciences, 26(8), 3892. https://doi.org/10.3390/ijms26083892