

New Amidino-Substituted Benzimidazole Derivatives as Human Dipeptidyl Peptidase III Inhibitors: Synthesis, In Vitro Evaluation, QSAR, and Molecular Docking Studies

 , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussion
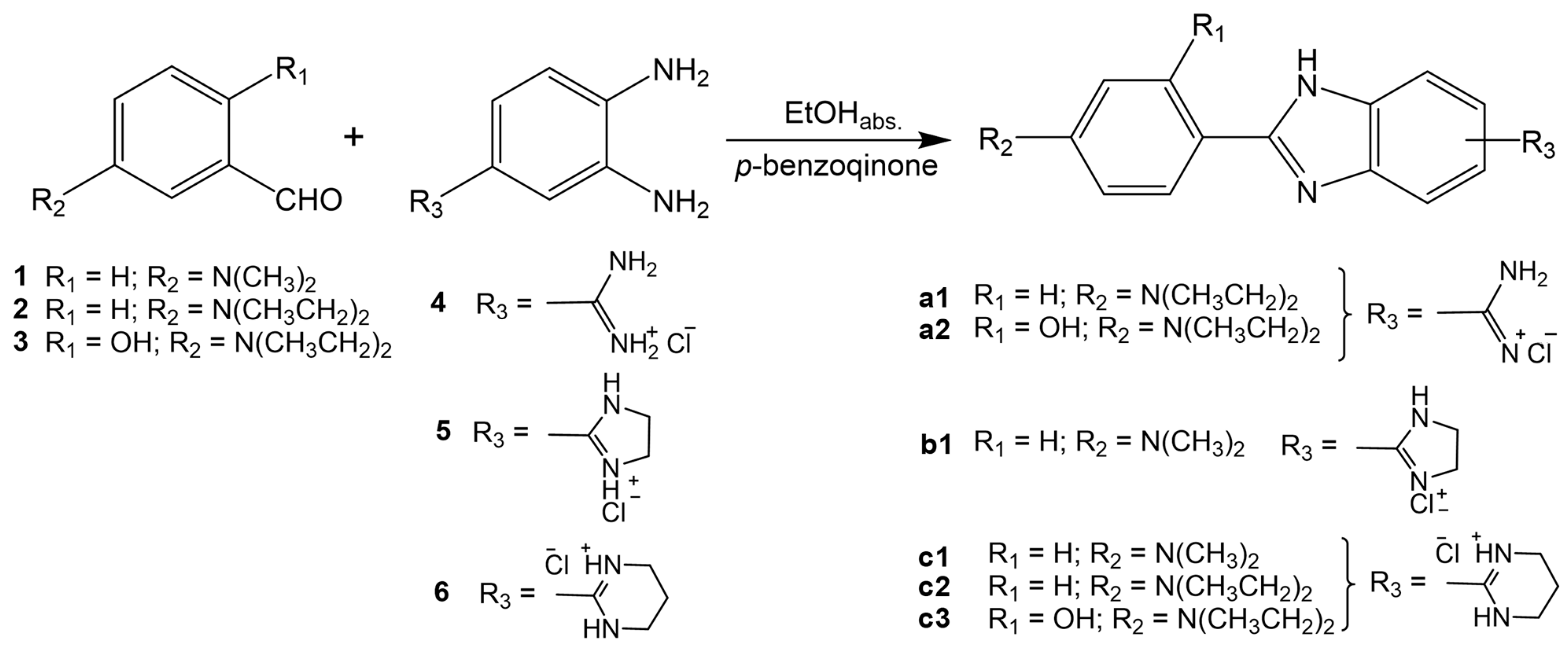
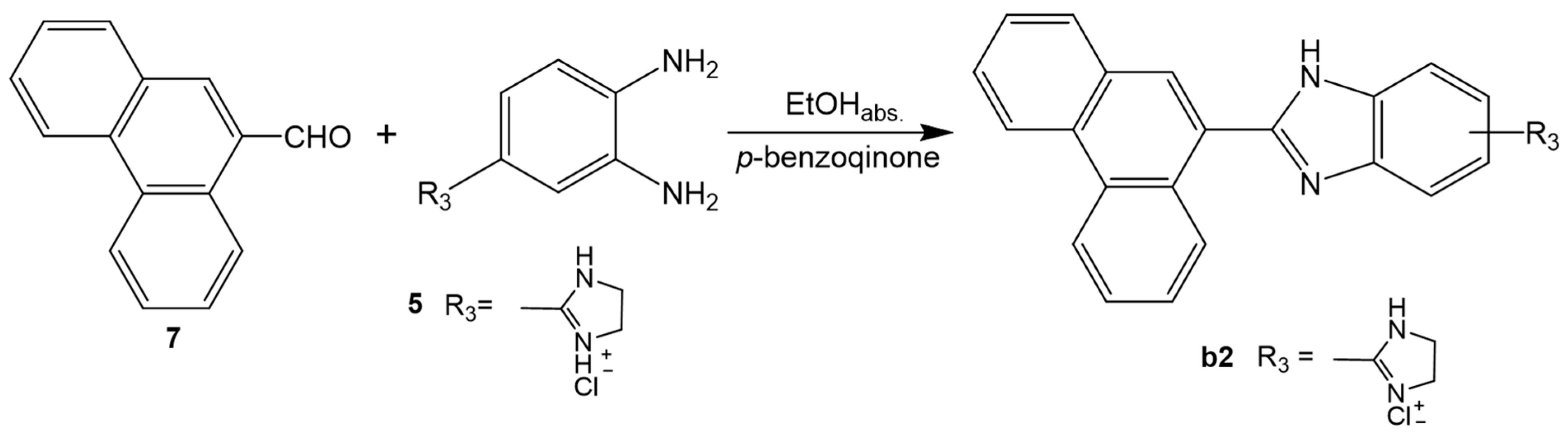
2.1. Chemistry
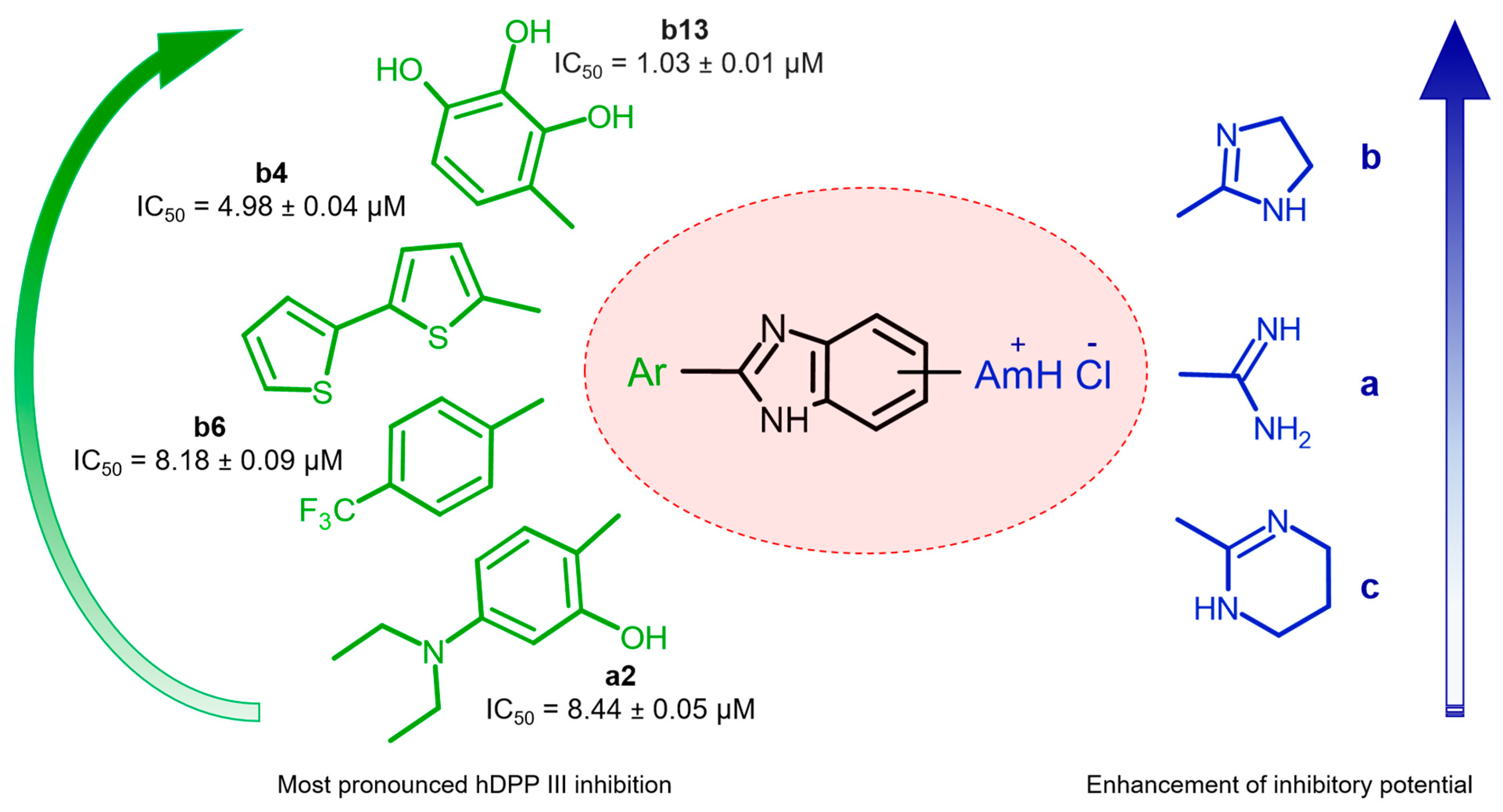
2.2. Human DPP III Inhibitory Activity
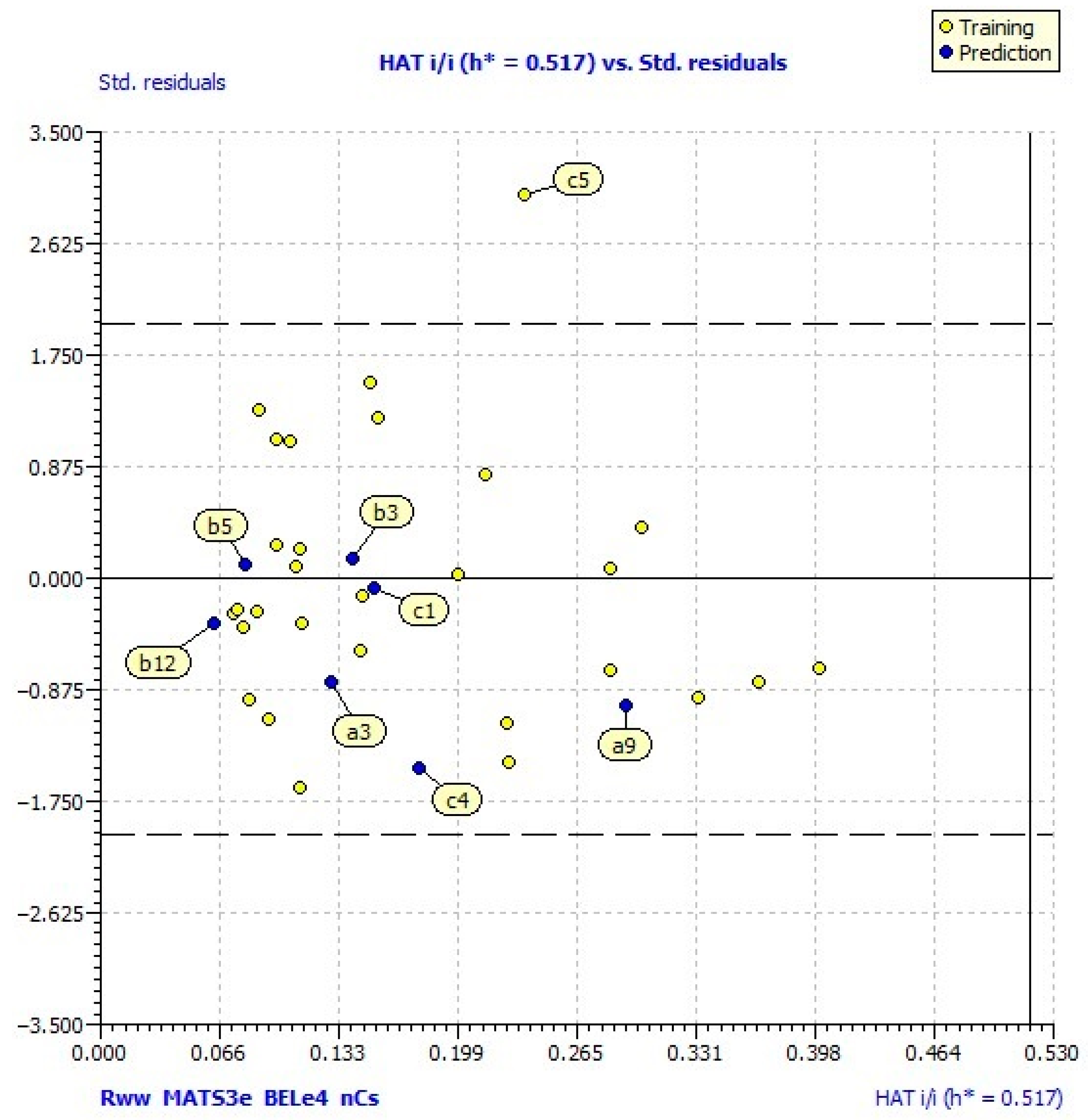
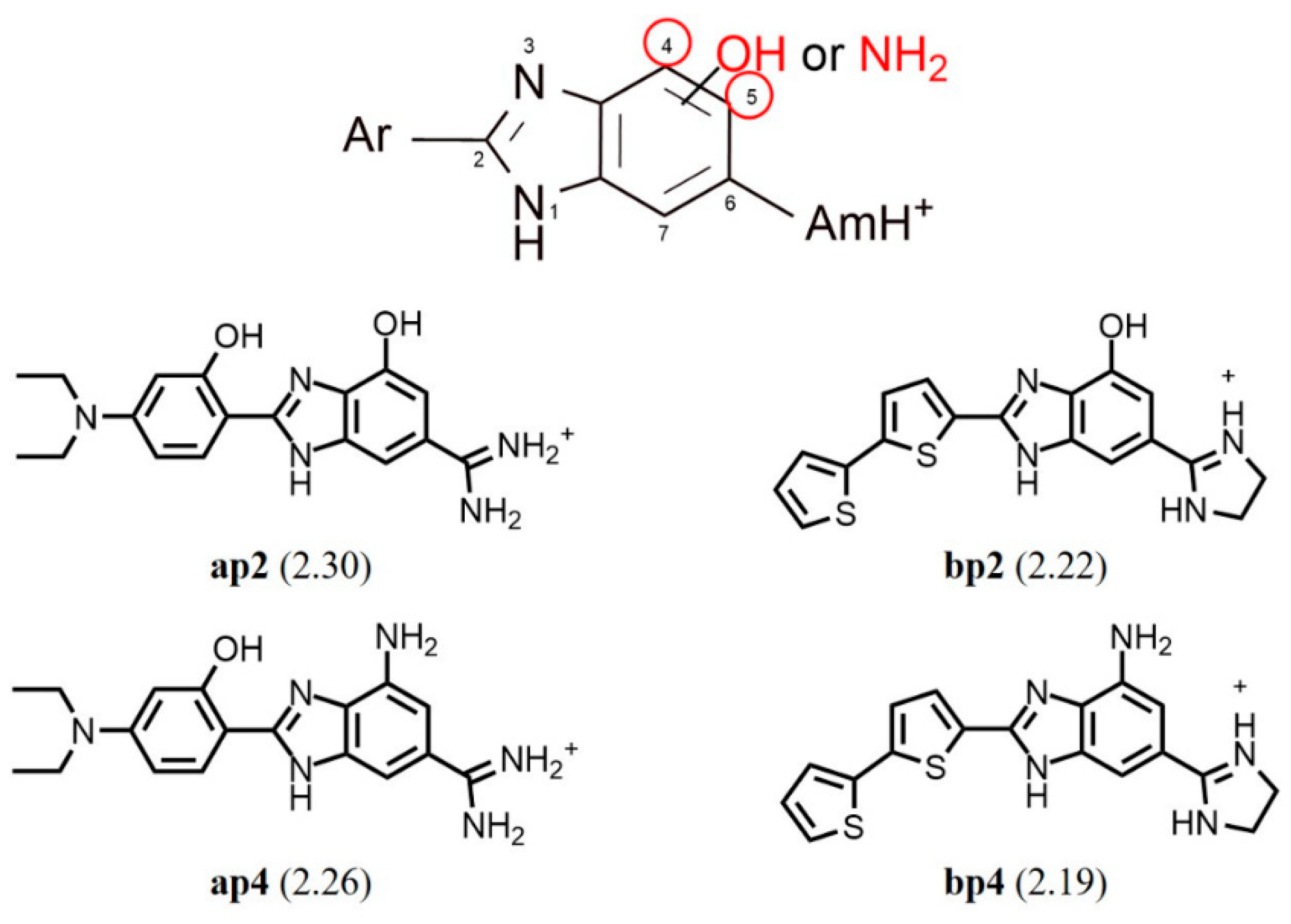
2.3. QSAR Model Analysis
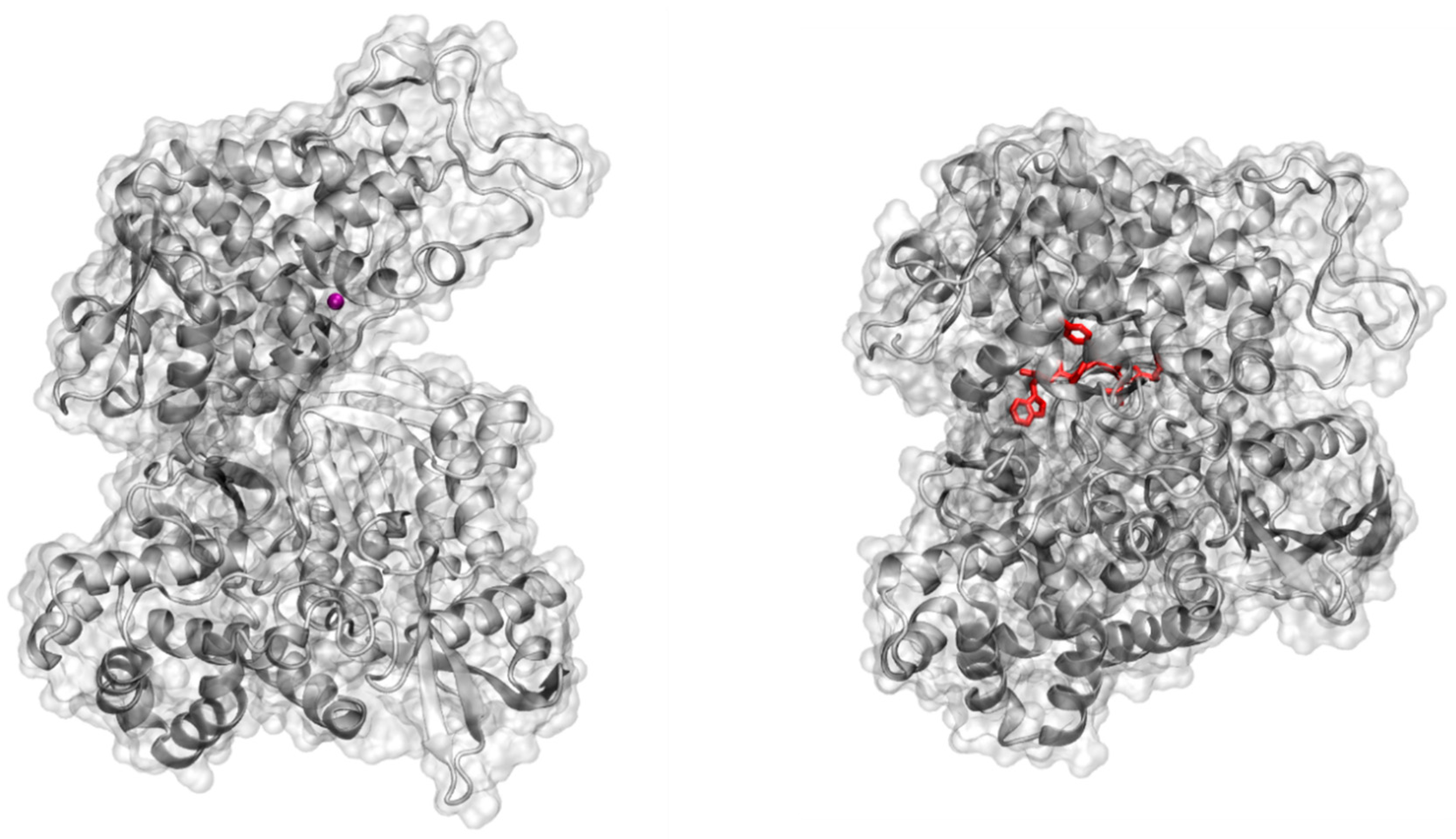
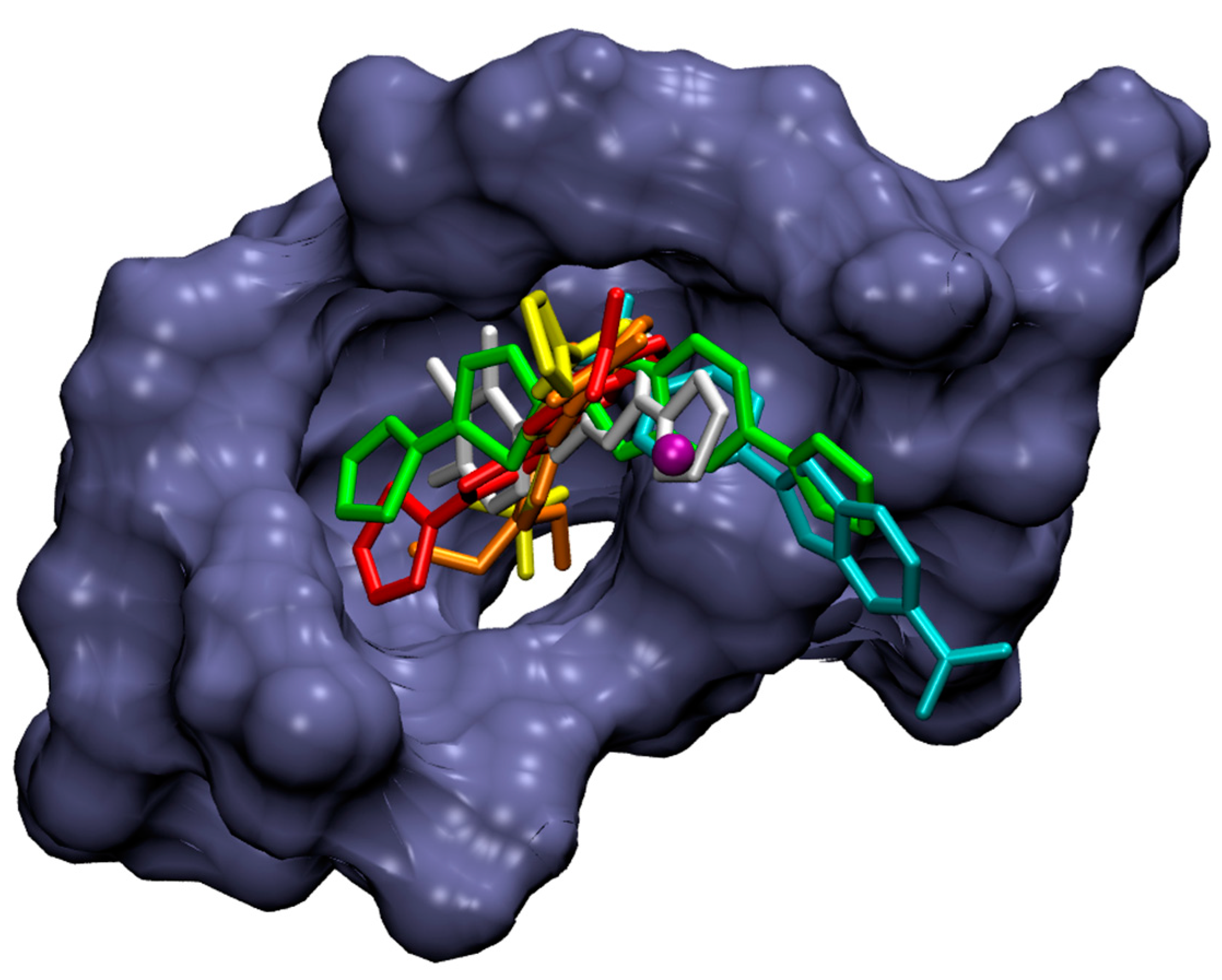
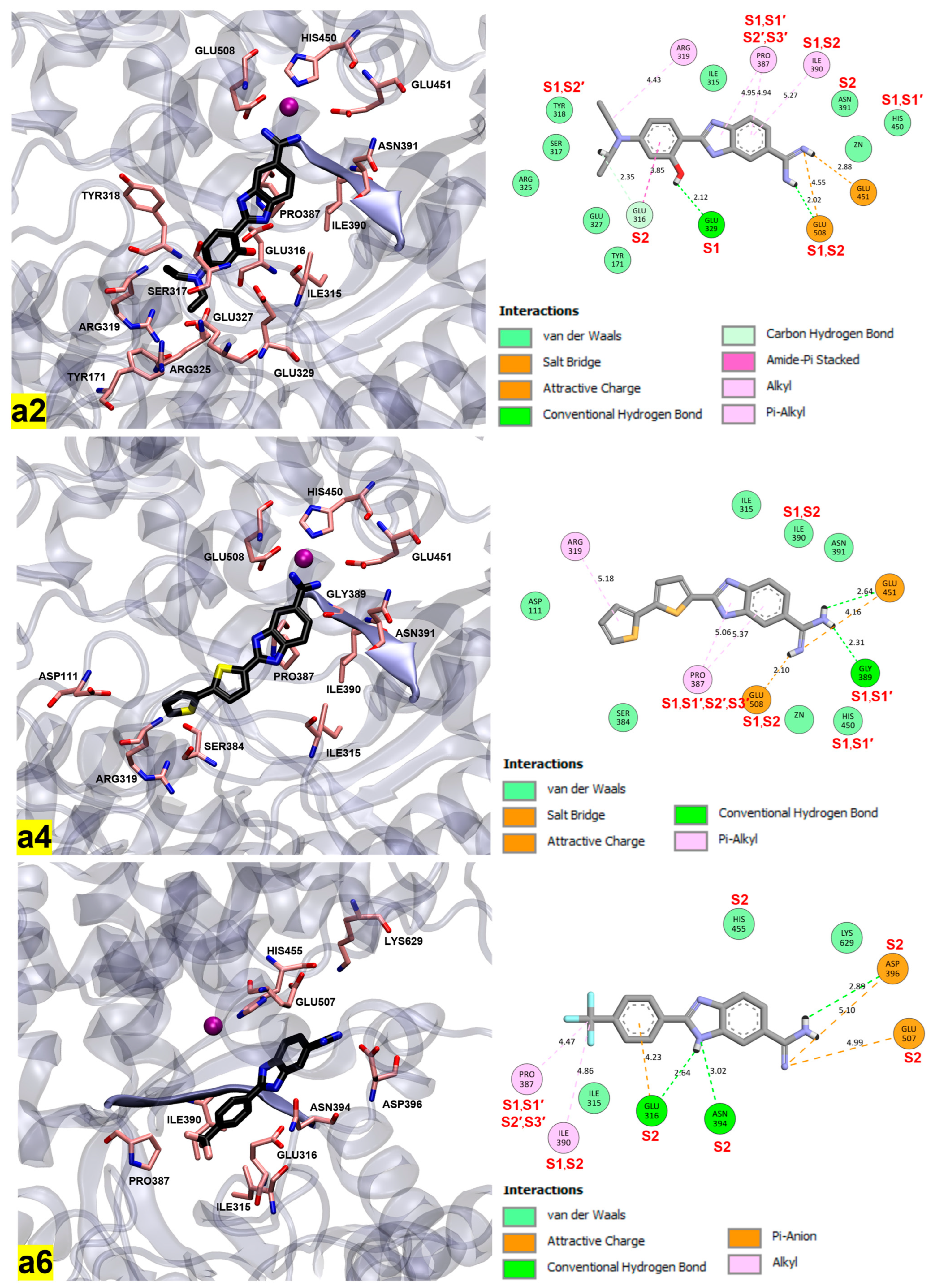
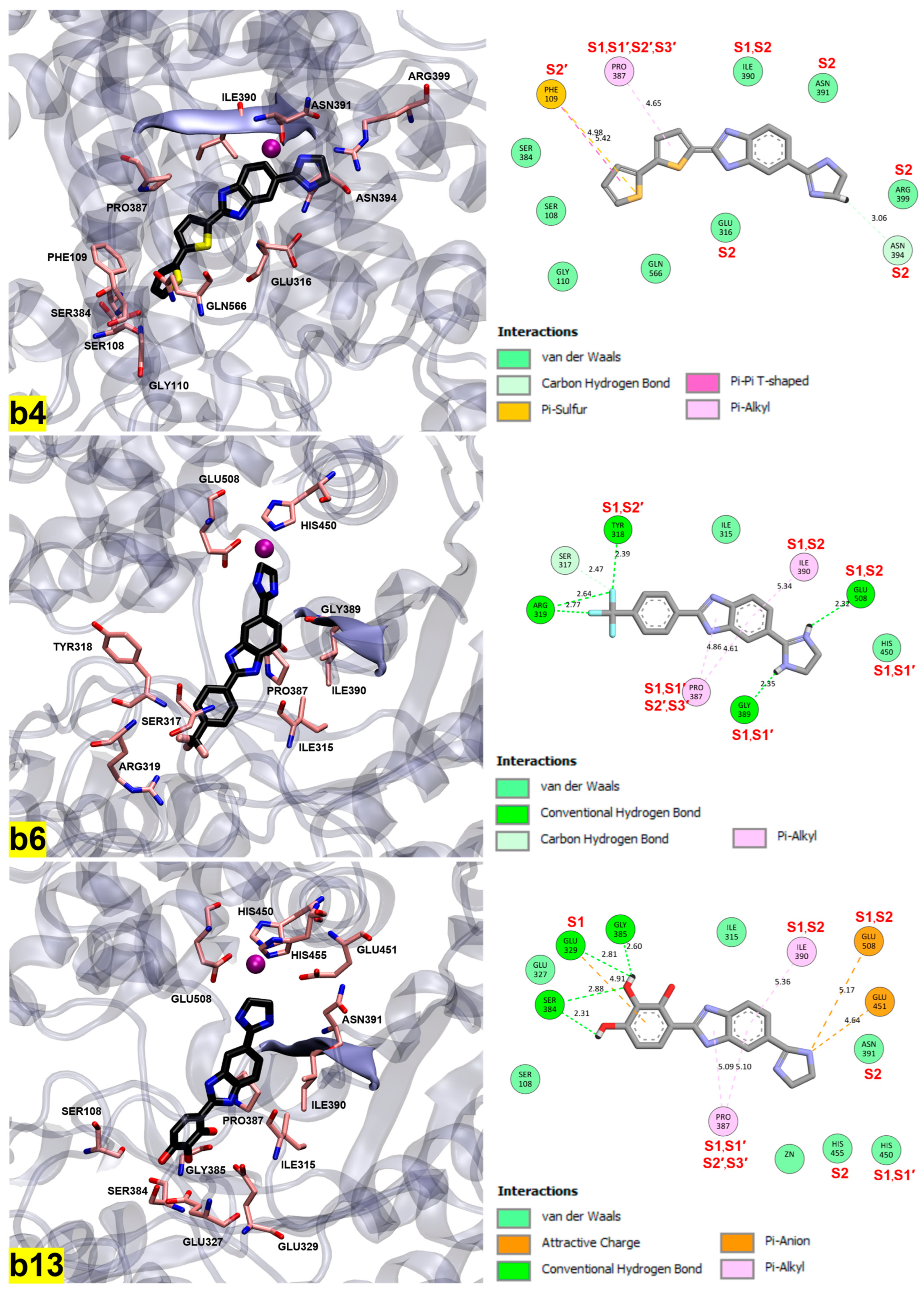
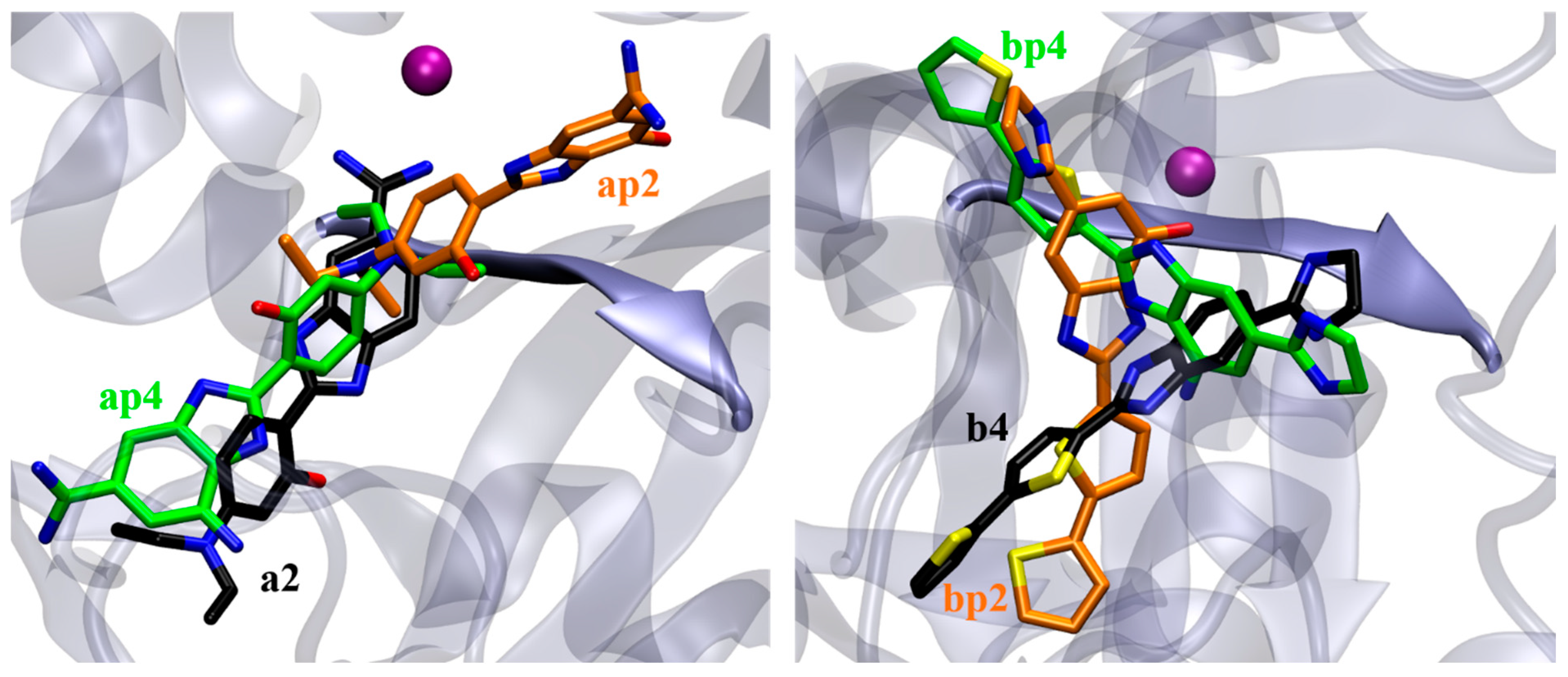
2.4. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. General Method for Preparation of Compounds a1, a2, b1, b2, c1, c2 and c3
3.2. Biochemistry
3.2.1. Heterologous Expression and Purification of Recombinant Human DPP III
3.2.2. Enzyme Activity Assay and IC50 Determination
3.3. Computational Study of Human DPP III Inhibitors
3.3.1. QSAR
3.3.2. Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrett, A.J.; Chen, J.-M. Dipeptidyl-peptidase III. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Salvesen, G.S., Eds.; Academic Press: London, UK, 2013; Volumes 1–2, pp. 1285–1289. [Google Scholar]
- Lee, C.M.; Snyder, S.H. Dipeptidyl-aminopeptidase III of Rat Brain. Selective Affinity for Enkephalin and Angiotensin. J. Biol. Chem. 1982, 257, 12043–12050. [Google Scholar] [CrossRef]
- Abramić, M.; Zubanović, M.; Vitale, L. Dipeptidyl Peptidase III from Human Erythrocytes. Biol. Chem. Hoppe-Seyler. 1988, 36, 29–38. [Google Scholar] [CrossRef]
- Baršun, M.; Jajčanin, N.; Vukelić, B.; Špoljarić, J.; Abramić, M. Human Dipeptidyl Peptidase III Acts as a Post-Proline-Cleaving Enzyme on Endomorphins. Biol. Chem. 2007, 388, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Abramić, M.; Špoljarić, J.; Šimaga, Š. Prokaryotic Homologs Help to Define Consensus Sequences in Peptidase Family M49. Period. Biol. 2004, 106, 161–168. [Google Scholar]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2017, 46, D624–D632. [Google Scholar] [CrossRef]
- Baral, P.K.; Jajčanin-Jozić, N.; Deller, S.; Macheroux, P.; Abramić, M.; Gruber, K. The First Structure of Dipeptidyl-Peptidase III Provides Insight into the Catalytic Mechanism and Mode of Substrate Binding. J. Biol. Chem. 2008, 283, 22316–22324. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, G.A.; Dobrovetsky, E.; Viertlmayr, R.; Dong, A.; Binter, A.; Abramić, M.; Macheroux, P.; Dhe-Paganon, S.; Gruber, K. Entropy-Driven Binding of Opioid Peptides Induces a Large Domain Motion in Human Dipeptidyl Peptidase III. Proc. Natl. Acad. Sci. USA 2012, 109, 6525–6530. [Google Scholar] [CrossRef]
- Tomić, A.; González, M.; Tomić, S. The Large Scale Conformational Change of the Human DPP III–Substrate Prefers the “Closed” Form. J. Chem. Inf. Model. 2012, 52, 1583–1594. [Google Scholar] [CrossRef]
- Jha, S.; Taschler, U.; Domenig, O.; Poglitsch, M.; Bourgeois, B.; Pollheimer, M.; Pusch, L.M.; Malovan, G.; Frank, S.; Madl, T.; et al. Dipeptidyl Peptidase 3 Modulates the Renin–Angiotensin System in Mice. J. Biol. Chem. 2020, 295, 13711–13723. [Google Scholar] [CrossRef]
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.M.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.N.; Major, M.B. Proteomic Analysis of Ubiquitin Ligase KEAP1 Reveals Associated Proteins That Inhibit NRF2 Ubiquitination. Cancer Res. 2013, 73, 2199–2210. [Google Scholar] [CrossRef]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress–Induced DPP3–KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef]
- Tong, Y.; Huang, Y.; Zhang, Y.; Zeng, X.; Yan, M.; Xia, Z.; Lai, D. DPP3/CDK1 Contributes to the Progression of Colorectal Cancer through Regulating Cell Proliferation, Cell Apoptosis, and Cell Migration. Cell Death Dis. 2021, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; Abudula, A.; Yang, H.T.; Xu, L.X.; Nuerrula, Y.; Bai, G.; Tulahong, A.; Eli, M. DPP3 Expression Promotes Cell Proliferation and Migration in Vitro and Tumour Growth in Vivo, Which Is Associated with Poor Prognosis of Oesophageal Carcinoma. Oncol. Rep. 2022, 49, 9. [Google Scholar] [CrossRef]
- Alghamdi, R.A.; Al-Zahrani, M.H. Integrated Bioinformatics Analyses Identifying Key Transcriptomes Correlated with Prognosis and Immune Infiltrations in Lung Squamous Cell Carcinoma. Saudi J. Biol. Sci. 2023, 30, 103596. [Google Scholar] [CrossRef]
- Miettinen, J.J.; Kumari, R.; Traustadottir, G.A.; Huppunen, M.-E.; Sergeev, P.; Majumder, M.M.; Schepsky, A.; Gudjonsson, T.; Lievonen, J.; Bazou, D.; et al. Aminopeptidase Expression in Multiple Myeloma Associates with Disease Progression and Sensitivity to Melflufen. Cancers 2021, 13, 1527. [Google Scholar] [CrossRef] [PubMed]
- Šimaga, Š.; Babić, D.; Osmak, M.; Šprem, M.; Abramić, M. Tumor Cytosol Dipeptidyl Peptidase III Activity Is Increased with Histological Aggressiveness of Ovarian Primary Carcinomas. Gynecol. Oncol. 2003, 91, 194–200. [Google Scholar] [CrossRef]
- Takagi, K.; Blet, A.; Levy, B.; Deniau, B.; Azibani, F.; Feliot, E.; Bergmann, A.; Santos, K.; Hartmann, O.; Gayat, E.; et al. Circulating Dipeptidyl Peptidase 3 and Alteration in Haemodynamics in Cardiogenic Shock: Results from the OptimaCC Trial. Eur. J. Heart Fail. 2019, 22, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Deniau, B.; Blet, A.; Santos, K.; Ayar, P.V.; Genest, M.; Kästorf, M.; Sadoune, M.; de Sousa Jorge, A.; Samuel, J.L.; Vodovar, N.; et al. Inhibition of Circulating Dipeptidyl-Peptidase 3 Restores Cardiac Function in a Sepsis-Induced Model in Rats: A Proof of Concept Study. PLoS ONE 2020, 15, e0238039. [Google Scholar] [CrossRef]
- Innelli, P.; Lopizzo, T.; Paternò, G.; Bruno, N.; Radice, R.P.; Bertini, P.; Marabotti, A.; Luzi, G.; Stabile, E.; Di Fazio, A.; et al. Dipeptidyl Amino-Peptidase 3 (DPP3) as an Early Marker of Severity in a Patient Population with Cardiogenic Shock. Diagnostics 2023, 13, 1350. [Google Scholar] [CrossRef]
- Magliocca, A.; Omland, T.; Latini, R. Dipeptidyl Peptidase 3, a Biomarker in Cardiogenic Shock and Hopefully Much More. Eur. J. Heart Fail. 2020, 22, 300–302. [Google Scholar] [CrossRef]
- Abramić, M.; Agić, D. Survey of Dipeptidyl Peptidase III Inhibitors: From Small Molecules of Microbial or Synthetic Origin to Aprotinin. Molecules 2022, 27, 3006. [Google Scholar] [CrossRef] [PubMed]
- Agić, D.; Hranjec, M.; Jajčanin, N.; Starčević, K.; Karminski-Zamola, G.; Abramić, M. Novel Amidino-Substituted Benzimidazoles: Synthesis of Compounds and Inhibition of Dipeptidyl Peptidase III. Bioorg. Chem. 2007, 35, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Shahar Yar, M. Structure-Activity Relationship (SAR) Study and Design Strategies of Nitrogen-Containing Heterocyclic Moieties for Their Anticancer Activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef]
- Nardi, M.; Cano, N.C.H.; Simeonov, S.; Bence, R.; Kurutos, A.; Scarpelli, R.; Wunderlin, D.; Procopio, A. A Review on the Green Synthesis of Benzimidazole Derivatives and Their Pharmacological Activities. Catalysts 2023, 13, 392. [Google Scholar] [CrossRef]
- Perin, N.; Hok, L.; Beč, A.; Persoons, L.; Vanstreels, E.; Daelemans, D.; Vianello, R.; Hranjec, M. N-Substituted Benzimidazole Acrylonitriles as in Vitro Tubulin Polymerization Inhibitors: Synthesis, Biological Activity and Computational Analysis. Eur. J. Med. Chem. 2021, 211, 113003. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Mukherjee, S.; Nath, P.; Mukherjee, A.; Mukherjee, S.; Ashok Kumar, S.K.; De, S.; Banerjee, S. A Critical Review of Benzimidazole: Sky-High Objectives towards the Lead Molecule to Predict the Future in Medicinal Chemistry. Results Chem. 2023, 6, 101013. [Google Scholar] [CrossRef]
- Bai, S.; Zhang, M.; Tang, S.; Li, M.; Wu, R.; Wan, S.; Chen, L.; Wei, X.; Li, F. Research Progress on Benzimidazole Fungicides: A Review. Molecules 2024, 29, 1218. [Google Scholar] [CrossRef]
- Hranjec, M.; Kralj, M.; Piantanida, I.; Sedić, M.; Šuman, L.; Pavelić, K.; Karminski-Zamola, G. Novel Cyano- and Amidino-Substituted Derivatives of Styryl-2-Benzimidazoles and Benzimidazo[1,2-a]quinolines. Synthesis, Photochemical Synthesis, DNA Binding, and Antitumor Evaluation, Part 3. J. Med. Chem. 2007, 50, 5696–5711. [Google Scholar] [CrossRef] [PubMed]
- Racané, L.; Zlatar, I.; Perin, N.; Cindrić, M.; Radovanović, V.; Banjanac, M.; Shanmugam, S.; Stojković, M.R.; Brajša, K.; Hranjec, M. Biological Activity of Newly Synthesized Benzimidazole and Benzothizole 2,5-Disubstituted Furane Derivatives. Molecules 2021, 26, 4935. [Google Scholar] [CrossRef]
- Greenhill, J.V.; Lue, P. Amidines and Guanidines in Medicinal Chemistry. Prog. Med. Chem. 1993, 30, 203–326. [Google Scholar]
- Racané, L.; Rep, V.; Pavelić, S.K.; Grbčić, P.; Zonjić, I.; Radić Stojković, M.; Taylor, M.C.; Kelly, J.M.; Raić-Malić, S. Synthesis, Antiproliferative and Antitrypanosomal Activities, and DNA Binding of Novel 6-Amidino-2-Arylbenzothiazoles. J. Enzyme Inhib. Med. Chem. 2021, 36, 1952–1967. [Google Scholar] [CrossRef]
- Racané, L.; Pavelić, S.K.; Nhili, R.; Depauw, S.; Paul-Constant, C.; Ratkaj, I.; David-Cordonnier, M.-H.; Pavelić, K.; Tralić-Kulenović, V.; Karminski-Zamola, G. New Anticancer Active and Selective Phenylene-Bisbenzothiazoles: Synthesis, Antiproliferative Evaluation and DNA Binding. Eur. J. Med. Chem. 2013, 63, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Farahat, A.A.; Paul, A.; Boykin, D.W.; Wilson, W.D. Engineered Modular Heterocyclic-Diamidines for Sequence-Specific Recognition of Mixed AT/GC Base Pairs at the DNA Minor Groove. Chem. Sci. 2021, 12, 15849–15861. [Google Scholar] [CrossRef] [PubMed]
- Racané, L.; Cindrić, M.; Zlatar, I.; Kezele, T.; Milić, A.; Brajša, K.; Hranjec, M. Preclinical in Vitro Screening of Newly Synthesised Amidino Substituted Benzimidazoles and Benzothiazoles. J. Enzyme Inhib. Med. Chem. 2021, 36, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Racané, L.; Zlatić, K.; Cindrić, M.; Mehić, E.; Karminski-Zamola, G.; Taylor, M.C.; Kelly, J.M.; Malić, S.R.; Stojković, M.R.; Kralj, M.; et al. Synthesis and Biological Activity of 2-Benzo[B]Thienyl and 2-Bithienyl Amidino-Substituted Benzothiazole and Benzimidazole Derivatives. ChemMedChem 2023, 18, e202300261. [Google Scholar] [CrossRef]
- Perin, N.; Starčević, K.; Perić, M.; Čipčić Paljetak, H.; Matijašić, M.; Stepanić, V.; Verbanac, D.; Karminski-Zamola, G.; Hranjec, M. Synthesis and SAR Study of Novel Amidino 2-Substituted Benzimidazoles as Potential Antibacterial Agents. Croat. Chem. Acta 2017, 90, 145–154. [Google Scholar] [CrossRef]
- Racané, L.; Cindrić, M.; Perin, N.; Roškarić, P.; Starčević, K.; Mašek, T.; Maurić, M.; Dogan, J.; Karminski-Zamola, G. Synthesis and Antioxidative Potency of Novel Amidino Substituted Benzimidazole and Benzothiazole Derivatives. Croat. Chem. Acta 2017, 90, 187–195. [Google Scholar] [CrossRef]
- Racané, L.; Butković, K.; Martin-Kleiner, I.; Kralj, M.; Karminski-Zamola, G.; Hranjec, M. Synthesis and Antiproliferative Activity in Vitro of Amidino Substituted 2 Phenylbenzazoles. Croat. Chem. Acta 2019, 92, 181–189. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V.; Maiocchi, A. Theory Development and Its Application in Chemometrics. Chemom. Intell. Lab. Syst. 1999, 46, 13–29. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR Models Validation: Internal and External. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The Importance of Being Earnest: Validation Is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Kiralj, R.; Ferreira, M.M.C. Basic Validation Procedures for Regression Models in QSAR and QSPR Studies: Theory and Application. J. Braz. Chem. Soc. 2009, 20, 770–787. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Trinajstić, N.; Nikolić, S.; Basak, S.C.; Lukovits, I. Distance Indices and Their Hyper-Counterparts: Intercorrelation and Use in the Structure-Property Modeling. SAR QSAR Environ. Res. 2001, 12, 31–54. [Google Scholar] [CrossRef]
- Sharma, B.K.; Singh, P.; Pilania, P.; Shekhawat, M.; Prabhakar, Y.S. QSAR of 2-(4-Methylsulphonylphenyl) Pyrimidine Derivatives as Cyclooxygenase-2 Inhibitors: Simple Structural Fragments as Potential Modulators of Activity. J. Enzyme Inhib. Med. Chem. 2011, 27, 249–260. [Google Scholar] [CrossRef]
- Stanton, D.T. Evaluation and Use of BCUT Descriptors in QSAR and QSPR Studies. J. Chem. Inf. Comput. Sci. 1998, 39, 11–20. [Google Scholar] [CrossRef]
- Cao, D.-S.; Xu, Q.-S.; Liang, Y.-Z.; Chen, X.; Li, H.-D. Prediction of Aqueous Solubility of Druglike Organic Compounds Using Partial Least Squares, Back-Propagation Network and Support Vector Machine. J. Chemom. 2010, 24, 584–595. [Google Scholar] [CrossRef]
- Agić, D.; Karnaš, M.; Tomić, S.; Komar, M.; Karačić, Z.; Rastija, V.; Bešlo, D.; Šubarić, D.; Molnar, M. Experimental and Computational Evaluation of Dipeptidyl Peptidase III Inhibitors Based on Quinazolinone-Schiff’s Bases. J. Biomol. Struct. Dyn. 2022, 41, 7567–7581. [Google Scholar] [CrossRef]
- Rastija, V.; Agić, D.; Tomić, S.; Nikolić, S.; Hranjec, M.; Karminski-Zamola, G.; Abramić, M. Synthesis, QSAR, and Molecular Dynamics Simulation of Amidino-substituted Benzimidazoles as Dipeptidyl Peptidase III Inhibitors. Acta Chim. Slov. 2015, 62, 867–878. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Li, X.; Yu, Z.; Song, C.; Du, Y. Nitrile-Containing Pharmaceuticals: Target, Mechanism of Action, and Their SAR Studies. RSC Med. Chem. 2021, 12, 1650–1671. [Google Scholar] [CrossRef]
- Agić, D.; Karnaš, M.; Šubarić, D.; Lončarić, M.; Tomić, S.; Karačić, Z.; Bešlo, D.; Rastija, V.; Molnar, M.; Popović, B.M.; et al. Coumarin Derivatives Act as Novel Inhibitors of Human Dipeptidyl Peptidase III: Combined in Vitro and in Silico Study. Pharmaceuticals 2021, 14, 540. [Google Scholar] [CrossRef]
- Kumar, P.; Reithofer, V.; Reisinger, M.; Wallner, S.; Pavkov-Keller, T.; Macheroux, P.; Gruber, K. Substrate Complexes of Human Dipeptidyl Peptidase III Reveal the Mechanism of Enzyme Inhibition. Sci. Rep. 2016, 6, 23787. [Google Scholar] [CrossRef] [PubMed]
- Tomić, A.; Kovačević, B.; Tomić, S. Concerted Nitrogen Inversion and Hydrogen Bonding to Glu451 Are Responsible for Protein-Controlled Suppression of the Reverse Reaction in Human DPP III. Phys. Chem. Chem. Phys. 2016, 18, 27245–27256. [Google Scholar] [CrossRef] [PubMed]
- Salopek-Sondi, B.; Vukelić, B.; Špoljarić, J.; Šimaga, Š.; Vujaklija, D.; Makarević, J.; Jajčanin, N.; Abramić, M. Functional tyrosine residue in the active center of human dipeptidyl peptidase III. Biol. Chem. 2008, 389, 163–167. [Google Scholar] [CrossRef]
- Tomić, A.; Abramić, M.; Špoljarić, J.; Agić, D.; Smith, D.M.; Tomić, S. Human dipeptidyl peptidase III: Insights into ligand binding from a combined experimental and computational approach. J. Mol. Recognit. 2011, 24, 804–814. [Google Scholar] [CrossRef]
- Nonlinear Regression Analysis was Performed Using GraphPad Prism, Version 10.4.1 for Windows; GraphPad Software; GraphPad Prism: Boston, MA, USA, 2024; Available online: www.graphpad.com (accessed on 12 January 2025).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Hocquet, A.; Langgård, M. An Evaluation of the MM+ Force Field. J. Mol. Model. 1998, 4, 94–112. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods, I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual Computational Chemistry Laboratory—Design and Description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A New Software for the Development, Analysis, and Validation of QSAR MLR Models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-D.; Lee, K.-H.; Tropsha, A. Rational Selection of Training and Test Sets for the Development of Validated QSAR Models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR Modeling. Int. J. Quant. Struct—Prop. Relatsh. 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M.; Gramatica, P. Methods for Reliability and Uncertainty Assessment and for Applicability Evaluations of Classification- and Regression-Based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Agić, D.; Brkić, H.; Tomić, S.; Karačić, Z.; Špoljarević, M.; Lisjak, M.; Bešlo, D.; Abramić, M. Validation of Flavonoids as Potential Dipeptidyl Peptidase III Inhibitors: Experimental and Computational Approach. Chem. Biol. Drug Des. 2016, 89, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Tomić, A.; Berynskyy, M.; Wade, R.C.; Tomić, S. Molecular Simulations Reveal That the Long Range Fluctuations of Human DPP III Change upon Ligand Binding. Mol. Biosyst. 2015, 11, 3068–3080. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer, Release 2019; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Cpd | Am | Ar | % inh. exp. * | log % inh. exp. | log % inh. calc. ** |
| a1 | 4-(N,N-diethylamino) phenyl | 62.9 ± 0.8 | 1.80 | 2.07 | |
| a2 | 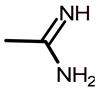 | 4-(N,N-diethylamino)-2-hydroxyphenyl | 100.0 ± 0.0 | 2.00 | 2.17 |
| a3 | benzo[b]thiophene-2-yl | 59.4 ± 1.4 | 1.77 | 1.60 | |
| a4 | 2,2′-bithiophenyl | 99.6 ± 0.8 | 2.00 | 1.95 | |
| a5 | 4-cyanophenyl | 67.8 ± 1.2 | 1.83 | 1.59 | |
| a6 | 4-trifluoromethylphenyl | 98.7 ± 0.0 | 1.99 | 2.01 | |
| a7 | 3H-benzimidazolyl | 21.6 ± 0.2 | 1.33 | 1.38 | |
| a8 | naphthalen-1-yl | 38.7 ± 0.8 | 1.59 | 1.51 | |
| a9 | quinolin-2-yl | 82.9 ± 0.2 | 1.92 | 1.73 | |
| a10 | 2-hydroxyphenyl | 24.3 ± 0.2 | 1.39 | 1.32 | |
| a11 | 2,4-dihydroxyphenyl | 14.6 ± 3.1 | 1.16 | 1.45 | |
| a12 | 2-hydroxy-4-methoxyphenyl | 51.5 ± 1.3 | 1.71 | 1.65 | |
| b1 | 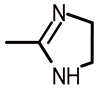 | 4-(N,N-dimethylamino)phenyl | 52.9 ± 1.7 | 1.72 | 1.95 |
| b2 | phenanthren-9-yl | 100.0 ± 0.0 | 2.00 | 1.97 | |
| b3 | benzo[b]thiophene-2-yl | 60.6 ± 2.1 | 1.77 | 1.81 | |
| b4 | 2,2′-bithiophenyl | 100.0 ± 0.0 | 2.00 | 2.06 | |
| b5 | 4-cyanophenyl | 34.4 ± 0.2 | 1.54 | 1.57 | |
| b6 | 4-trifluoromethylphenyl | 99.4 ± 1.0 | 2.00 | 1.95 | |
| b7 | 3H-benzimidazolyl | 51.1 ± 1.8 | 1.71 | 1.59 | |
| b8 | naphthalen-1-yl | 29.5 ± 1.2 | 1.47 | 1.71 | |
| b9 | quinolin-2-yl | 73.4 ± 2.6 | 1.87 | 1.95 | |
| b10 | 2-hydroxyphenyl | 42.8 ± 3.0 | 1.63 | 1.28 | |
| b11 | 2,4-dihydroxyphenyl | 41.1 ± 0.8 | 1.61 | 1.40 | |
| b12 | 2-hydroxy-4-methoxyphenyl | 47.1 ± 0.9 | 1.67 | 1.59 | |
| b13 | 2,3,4-trihydroxyphenyl | 100.0 ± 0.0 | 2.00 | 1.86 | |
| c1 | 4-(N,N-dimethylamino)phenyl | 9.9 ± 1.3 | 1.00 | 0.98 | |
| c2 | 4-(N,N-diethylamino) phenyl | 100.0 ± 0.0 | 2.00 | 1.71 | |
| c3 |  | 4-(N,N-diethylamino)-2-hydroxyphenyl | 92.6 ± 0.5 | 1.97 | 1.83 |
| c4 | 2,2′-bithiophenyl | 99.1 ± 0.8 | 2.00 | 1.70 | |
| c5 | 4-cyanophenyl | 0.1 ± 0.2 | −1.00 (0.00 ***) | 0.60 | |
| c6 | 4-trifluoromethylphenyl | 15.5 ± 0.9 | 1.19 | 1.02 | |
| c7 | 3H-benzimidazolyl | 29.8 ± 3.8 | 1.47 | 1.24 | |
| c8 | phenanthren-9-yl | 51.7 ± 0.2 | 1.71 | 1.73 | |
| c9 | phenyl | 2.1 ± 0.7 | 0.32 | 0.20 | |
| c10 | naphthalene-2-yl | 8.0 ± 0.5 | 0.90 | 1.22 | |
| c11 | benzo[b]thiophene-2-yl | 28.9 ± 3.1 | 1.46 | 1.47 | |
| Statistical Parameters | QSAR Model Values |
|---|---|
| Ntraining set | 29 |
| Ntest set | 7 (a3, a9, b3, b5, b12, c1, c4) |
| R2 | 0.82 |
| R2adj | 0.79 |
| s | 0.23 |
| F | 26.93 |
| Kxx | 0.26 |
| ΔK | 0.10 |
| RMSE | 0.21 |
| MAE | 0.16 |
| CCC | 0.90 |
| Q2LOO | 0.72 |
| Q2LMO | 0.69 |
| RMSEcv | 0.26 |
| MAEcv | 0.19 |
| CCCcv | 0.85 |
| R2Yscr | 0.15 |
| Q2Yscr | −0.26 |
| R2ext | 0.87 |
| CCCext | 0.86 |
| RMSEext | 0.15 |
| MAEext | 0.12 |
| Q2F1 | 0.77 |
| Q2F2 | 0.75 |
| Q2F3 | 0.90 |
| r2maver | 0.75 |
| r2mdiff | 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agić, D.; Karnaš Babić, M.; Hranjec, M.; Šubarić, D.; Karačić, Z.; Abramić, M. New Amidino-Substituted Benzimidazole Derivatives as Human Dipeptidyl Peptidase III Inhibitors: Synthesis, In Vitro Evaluation, QSAR, and Molecular Docking Studies. Int. J. Mol. Sci. 2025, 26, 3899. https://doi.org/10.3390/ijms26083899
Agić D, Karnaš Babić M, Hranjec M, Šubarić D, Karačić Z, Abramić M. New Amidino-Substituted Benzimidazole Derivatives as Human Dipeptidyl Peptidase III Inhibitors: Synthesis, In Vitro Evaluation, QSAR, and Molecular Docking Studies. International Journal of Molecular Sciences. 2025; 26(8):3899. https://doi.org/10.3390/ijms26083899
Chicago/Turabian StyleAgić, Dejan, Maja Karnaš Babić, Marijana Hranjec, Domagoj Šubarić, Zrinka Karačić, and Marija Abramić. 2025. "New Amidino-Substituted Benzimidazole Derivatives as Human Dipeptidyl Peptidase III Inhibitors: Synthesis, In Vitro Evaluation, QSAR, and Molecular Docking Studies" International Journal of Molecular Sciences 26, no. 8: 3899. https://doi.org/10.3390/ijms26083899
APA StyleAgić, D., Karnaš Babić, M., Hranjec, M., Šubarić, D., Karačić, Z., & Abramić, M. (2025). New Amidino-Substituted Benzimidazole Derivatives as Human Dipeptidyl Peptidase III Inhibitors: Synthesis, In Vitro Evaluation, QSAR, and Molecular Docking Studies. International Journal of Molecular Sciences, 26(8), 3899. https://doi.org/10.3390/ijms26083899