
Spatial Omics in Clinical Research: A Comprehensive Review of Technologies and Guidelines for Applications



Abstract
:
1. Introduction
2. Spatial Omics and Technologies
2.1. Spatial Transcriptomics
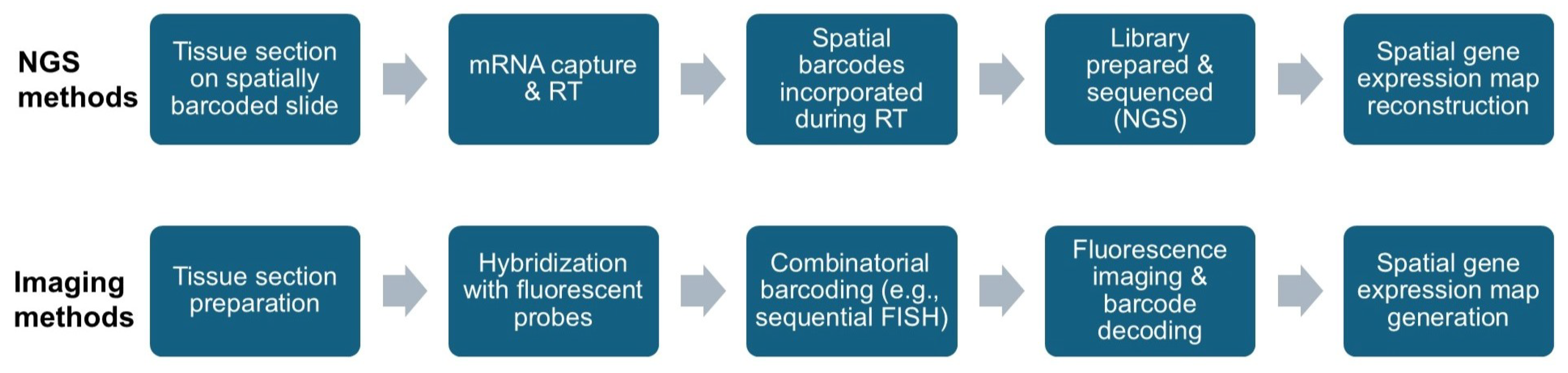
2.1.1. Next-Generation Sequencing-Based Methods
2.1.2. Imaging-Based Methods
2.1.3. Recent Advances in Imaging-Based Spatial Transcriptomics
2.2. Spatial Proteomics
2.2.1. Mass Spectrometry-Based Proteomics
2.2.2. Imaging-Based Proteomics
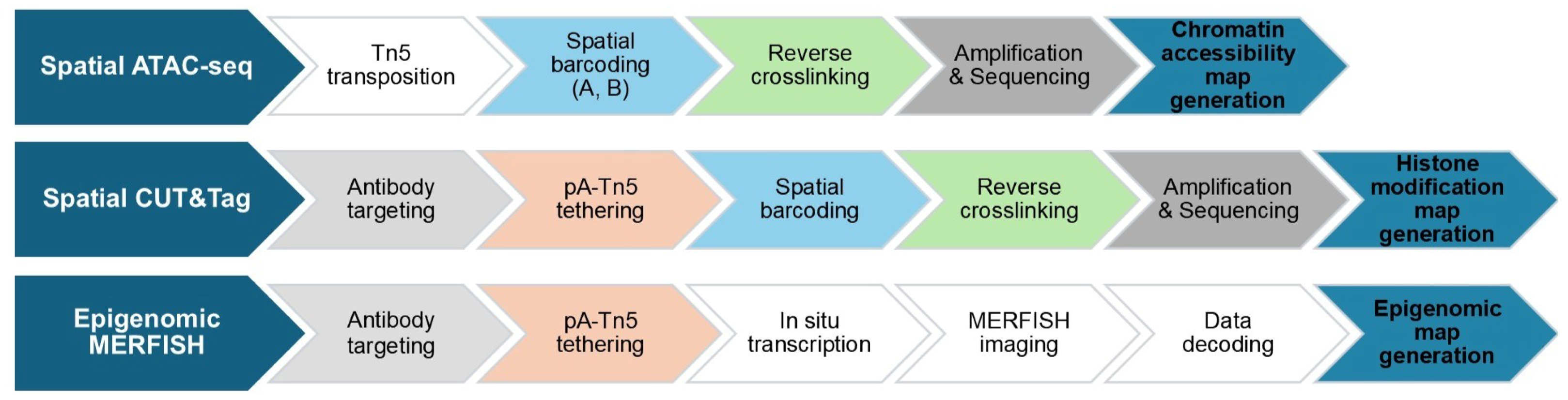
2.3. Spatial Epigenomics
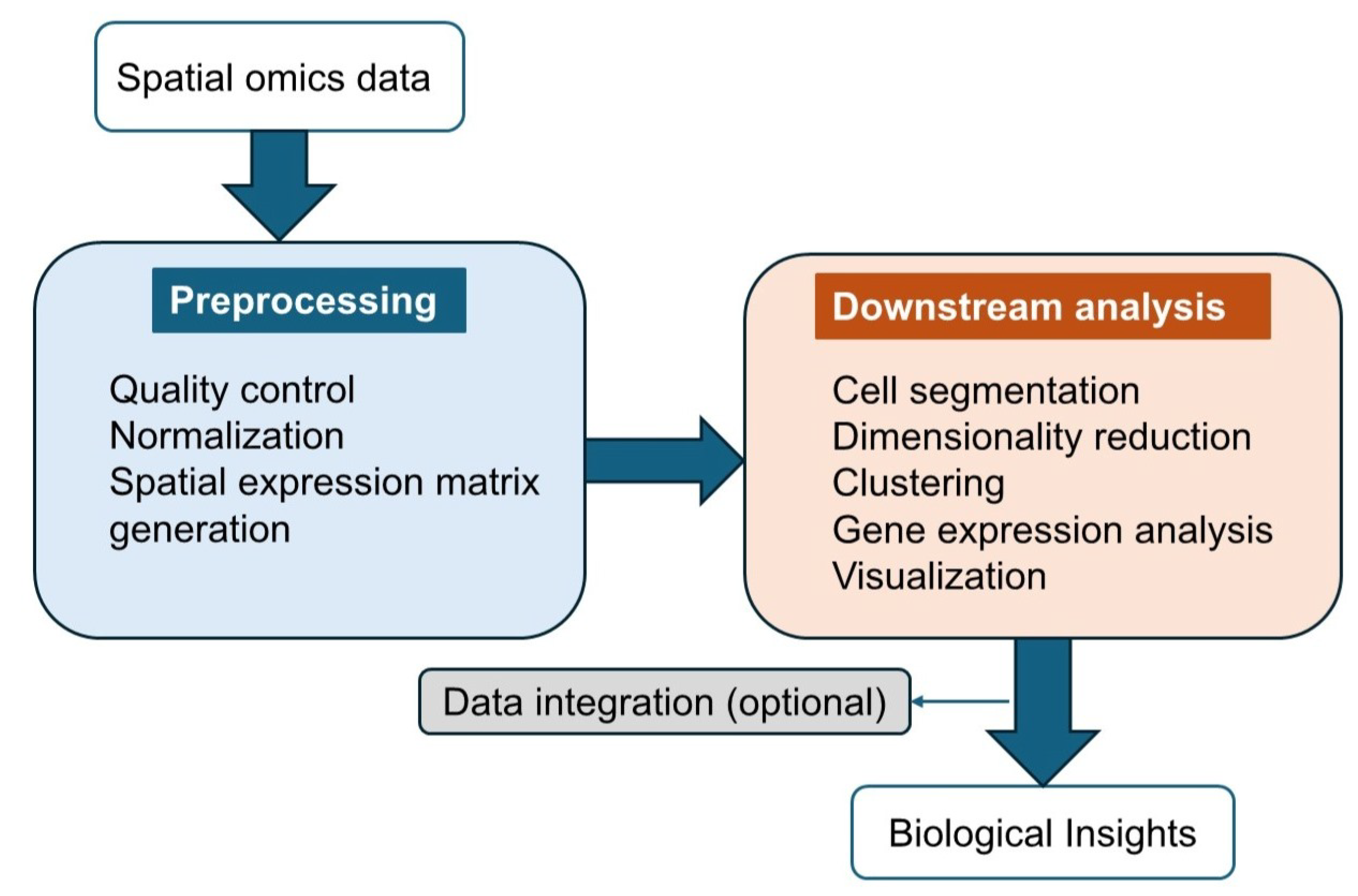
3. Spatial Omics Data Analysis Tools
4. Application in Clinical Research
4.1. Cancer Research
4.2. Neurological Diseases
4.3. Autoimmune Diseases
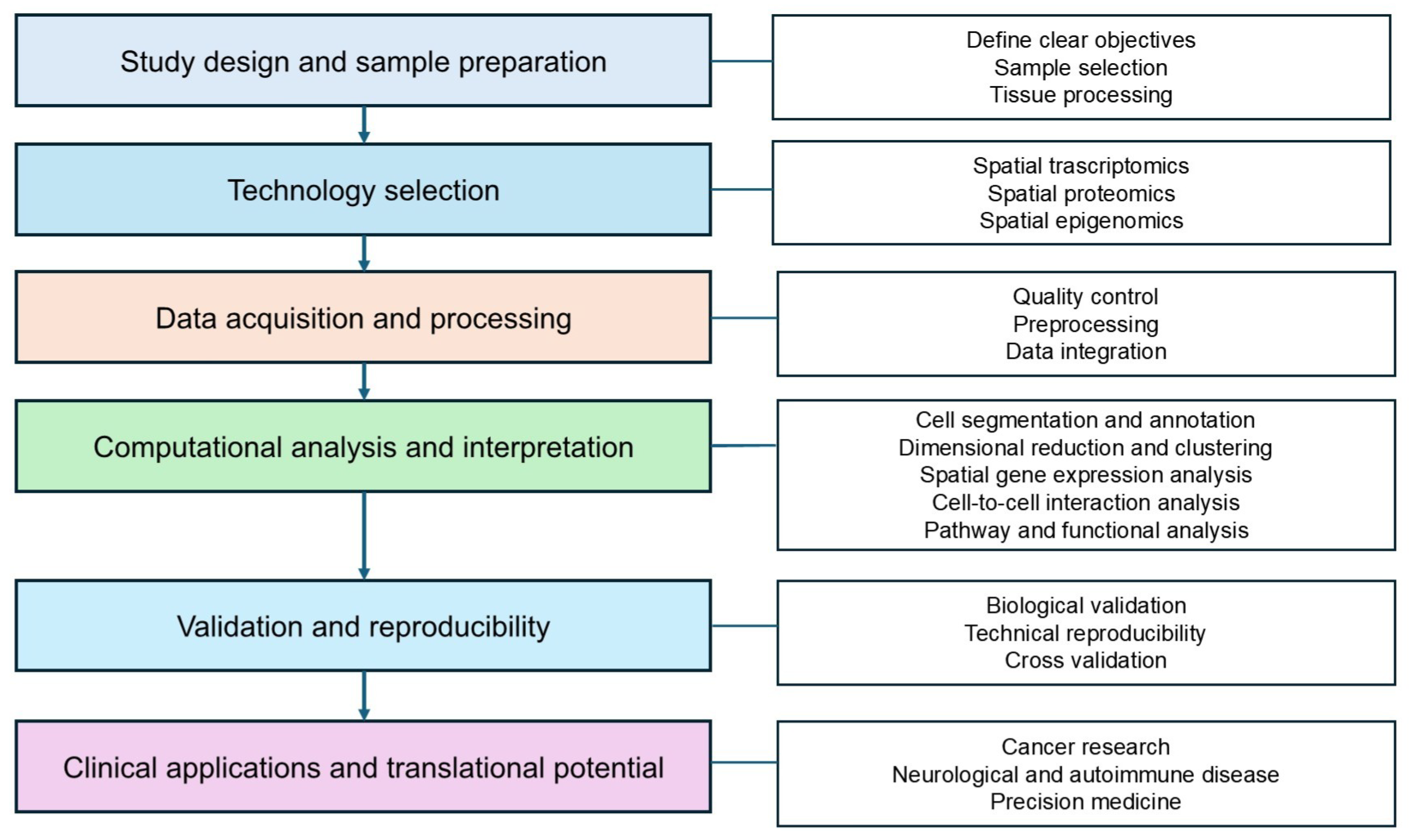
5. Guidelines to Design Spatial Omics in Clinical Research
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alberts, B.; Bray, D.; Hopkin, K.; Johnson, A.D.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Essential Cell Biology; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Andrews, T.S.; Hemberg, M. Identifying cell populations with scRNASeq. Mol. Asp. Med. 2018, 59, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Bressan, D.; Battistoni, G.; Hannon, G.J. The dawn of spatial omics. Science 2023, 381, eabq4964. [Google Scholar] [CrossRef]
- Liu, X.; Peng, T.; Xu, M.; Lin, S.; Hu, B.; Chu, T.; Liu, B.; Xu, Y.; Ding, W.; Li, L.; et al. Spatial multi-omics: Deciphering technological landscape of integration of multi-omics and its applications. J. Hematol. Oncol. 2024, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Barkley, D.; França, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Zhang, Y.; Wang, Y. Spatial transcriptomics in development and disease. Mol. Biomed. 2023, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; You, L.; Hardillo, J.A.U.; Chien, M.P. Spatial transcriptomic technologies. Cells 2023, 12, 2042. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Tian, L.; Chen, F.; Macosko, E.Z. The expanding vistas of spatial transcriptomics. Nat. Biotechnol. 2023, 41, 773–782. [Google Scholar] [CrossRef]
- Ren, J.; Luo, S.; Shi, H.; Wang, X. Spatial omics advances for in situ RNA biology. Mol. Cell 2024, 84, 3737–3757. [Google Scholar] [CrossRef]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Zhang, H.; Wang, M.; Ping, H. Visium spatial transcriptomics reveals intratumor heterogeneity and profiles of Gleason score progression in prostate cancer. iScience 2023, 26, 108429. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kong, H.; Yu, J.; Sun, M.; Lu, S.; Zhang, Y.; Hu, J.; Du, F.; Lian, Q.; Xin, H.; et al. Spatial transcriptomics reveals heterogeneity of histological subtypes between lepidic and acinar lung adenocarcinoma. Clin. Transl. Med. 2024, 14, e1573. [Google Scholar] [CrossRef]
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Catallini, J.L.; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 2021, 24, 425–436. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially resolved transcriptomes—next generation tools for tissue exploration. Bioessays 2020, 42, 1900221. [Google Scholar] [CrossRef]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ali, M.M.; Brook, M.A.; Li, Y. Rolling circle amplification: Applications in nanotechnology and biodetection with functional nucleic acids. Angew. Chem. Int. Ed. 2008, 47, 6330–6337. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B.; Zhao, G.; Lee, Y.; Buzdin, A.; Mu, X.; Zhao, J.; Chen, H.; Li, X. Spatial transcriptomics: Technologies, applications and experimental considerations. Genomics 2023, 115, 110671. [Google Scholar] [CrossRef]
- Wei, X.; Fu, S.; Li, H.; Liu, Y.; Wang, S.; Feng, W.; Yang, Y.; Liu, X.; Zeng, Y.Y.; Cheng, M.; et al. Single-cell Stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science 2022, 377, eabp9444. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 2020, 183, 1665–1681. [Google Scholar] [CrossRef] [PubMed]
- Enninful, A.; Foss, F.M.; Fan, R.; Xu, M. Spatial Multiomics Profiling of Angioimmunoblastic T-Cell Lymphoma. Blood 2024, 144, 1585. [Google Scholar] [CrossRef]
- Cheng, Y.; Dang, S.; Zhang, Y.; Chen, Y.; Yu, R.; Liu, M.; Jin, S.; Han, A.; Katz, S.; Wang, S. Sequencing-free whole genome spatial transcriptomics at molecular resolution in intact tissue. bioRxiv 2025. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Bambah-Mukku, D.; Eichhorn, S.W.; Vaughn, E.; Shekhar, K.; Perez, J.D.; Rubinstein, N.D.; Hao, J.; Regev, A.; Dulac, C.; et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 2018, 362, eaau5324. [Google Scholar] [CrossRef]
- Moses, L.; Pachter, L. Museum of spatial transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef]
- Henley, R.; Rapicavoli, N.; Janesick, A.; Shelansky, R.; Kim, A.; Hensel, J.; Meschi, F.; Farahani, N.; Kumar, V.; Qian, X.; et al. 95 Characterization of human breast cancer tissue with the Xenium In Situ platform reveals a novel marker for invasiveness. J. Immunother. Cancer 2022, 10, A104. [Google Scholar]
- Lu, T.; Gakhar, R.; Cheung, E.; Zhang, L.; Zhang, W.; Tran, T.; Ly, A.; Berger, B.; Verbeeck, N.; Patterson, H.; et al. 182 Xenium in situ analysis and reproducibility study of multiple carcinomas for clinical studies. J. Immunother. Cancer 2024, 12, A209. [Google Scholar]
- Moffet, J.J.; Fatunla, O.E.; Freytag, L.; Kriel, J.; Jones, J.J.; Roberts-Thomson, S.J.; Pavenko, A.; Scoville, D.K.; Zhang, L.; Liang, Y.; et al. Spatial architecture of high-grade glioma reveals tumor heterogeneity within distinct domains. Neuro-Oncol. Adv. 2023, 5, vdad142. [Google Scholar] [CrossRef]
- Ma, X.; Chen, P.; Wei, J.; Zhang, J.; Chen, C.; Zhao, H.; Ferguson, D.; McGee, A.W.; Dai, Z.; Qiu, S. Protocol for Xenium spatial transcriptomics studies using fixed frozen mouse brain sections. STAR Protoc. 2024, 5, 103420. [Google Scholar] [CrossRef]
- Janesick, A.; Shelansky, R.; Gottscho, A.D.; Wagner, F.; Williams, S.R.; Rouault, M.; Beliakoff, G.; Morrison, C.A.; Oliveira, M.F.; Sicherman, J.T.; et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 2023, 14, 8353. [Google Scholar] [CrossRef]
- National Cancer Institute Center for Cancer Research. Xenium In Situ. 2025. Available online: https://ostr.ccr.cancer.gov/emerging-technologies/spatial-biology/xenium/ (accessed on 6 April 2025).
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Owusu-Boaitey, K.E.; Valdes, G.M.; Reddien, P.W. Fate specification is spatially intermingled across planarian stem cells. Nat. Commun. 2023, 14, 7422. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Kong, X.; Johnston, K.G.; Mortazavi, A.; Holmes, T.C.; Tan, Z.; Yokomori, K.; Xu, X. Single-cell spatial transcriptomics reveals a dystrophic trajectory following a developmental bifurcation of myoblast cell fates in facioscapulohumeral muscular dystrophy. Genome Res. 2024, 34, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.E.; Blosser, T.R.; Sullivan, Z.A.; Dulac, C.; Zhuang, X. Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 2023, 186, 194–208. [Google Scholar] [CrossRef]
- Yao, Z.; van Velthoven, C.T.; Kunst, M.; Zhang, M.; McMillen, D.; Lee, C.; Jung, W.; Goldy, J.; Abdelhak, A.; Aitken, M.; et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 2023, 624, 317–332. [Google Scholar] [CrossRef]
- Tejwani, L.; Ravindra, N.G.; Lee, C.; Cheng, Y.; Nguyen, B.; Luttik, K.; Ni, L.; Zhang, S.; Morrison, L.M.; Gionco, J.; et al. Longitudinal single-cell transcriptional dynamics throughout neurodegeneration in SCA1. Neuron 2024, 112, 362–383. [Google Scholar] [CrossRef]
- Klughammer, J.; Abravanel, D.L.; Segerstolpe, Å.; Blosser, T.R.; Goltsev, Y.; Cui, Y.; Goodwin, D.R.; Sinha, A.; Ashenberg, O.; Slyper, M.; et al. A multi-modal single-cell and spatial expression map of metastatic breast cancer biopsies across clinicopathological features. Nat. Med. 2024, 30, 3236–3249. [Google Scholar] [CrossRef]
- Eng, C.H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Polonsky, M.; Gerhardt, L.M.; Yun, J.; Koppitch, K.; Colón, K.L.; Amrhein, H.; Wold, B.; Zheng, S.; Yuan, G.C.; Thomson, M.; et al. Spatial transcriptomics defines injury specific microenvironments and cellular interactions in kidney regeneration and disease. Nat. Commun. 2024, 15, 7010. [Google Scholar] [CrossRef]
- Cai Lab. seqFISH+ Official Website. 2019. Available online: https://spatial.caltech.edu/seqfish/ (accessed on 21 November 2024).
- Cai, L.; Eng, C.H. RNA seqFISH+ Supplementary Protocol. 2019. Available online: https://scispace.com/pdf/rna-seqfish-supplementary-protocol-3iwcvg3wmm.pdf (accessed on 21 November 2024).
- He, S.; Bhatt, R.; Brown, C.; Brown, E.A.; Buhr, D.L.; Chantranuvatana, K.; Danaher, P.; Dunaway, D.; Garrison, R.G.; Geiss, G.; et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 2022, 40, 1794–1806. [Google Scholar] [CrossRef]
- Williams, C.; Reeves, J.W.; Danaher, P.; He, S.; Kim, S.; Patrick, M.; Preciado, J.; Gregory, M.; Reitz, Z.; Jenkins, J.; et al. Spatial insights into tumor immune evasion illuminated with 1000-plex RNA profiling with CosMx Spatial Molecular Imager. Cancer Res. 2023, 83, 10–1158. [Google Scholar] [CrossRef]
- Denisenko, E.; de Kock, L.; Tan, A.; Beasley, A.B.; Beilin, M.; Jones, M.E.; Hou, R.; Muirí, D.Ó.; Bilic, S.; Mohan, G.R.K.; et al. Spatial transcriptomics reveals discrete tumour microenvironments and autocrine loops within ovarian cancer subclones. Nat. Commun. 2024, 15, 2860. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pai, R.; Gupta, S.; Currenti, J.; Guo, W.; Di Bartolomeo, A.; Feng, H.; Zhang, Z.; Li, Z.; Liu, L.; et al. Presence of onco-fetal neighborhoods in hepatocellular carcinoma is associated with relapse and response to immunotherapy. Nat. Cancer 2024, 5, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Goralski, T.M.; Meyerdirk, L.; Breton, L.; Brasseur, L.; Kurgat, K.; DeWeerd, D.; Turner, L.; Becker, K.; Adams, M.; Newhouse, D.J.; et al. Spatial transcriptomics reveals molecular dysfunction associated with cortical Lewy pathology. Nat. Commun. 2024, 15, 2642. [Google Scholar] [CrossRef]
- Tan, C.W.; Chen, J.; Liu, N.; Bhuva, D.D.; Blick, T.; Monkman, J.; Cooper, C.; Kharbanda, M.; Feher, K.; Phipson, B.; et al. In situ single-cell profiling sheds light on IFI27 localisation during SARS-CoV-2 infection. eBioMedicine 2024, 101, 105016. [Google Scholar] [CrossRef] [PubMed]
- Mennillo, E.; Kim, Y.J.; Lee, G.; Rusu, I.; Patel, R.K.; Dorman, L.C.; Flynn, E.; Li, S.; Bain, J.L.; Andersen, C.; et al. Single-cell and spatial multi-omics highlight effects of anti-integrin therapy across cellular compartments in ulcerative colitis. Nat. Commun. 2024, 15, 1493. [Google Scholar] [CrossRef]
- Zhuang, X. Spatially resolved single-cell genomics and transcriptomics by imaging. Nat. Methods 2021, 18, 18–22. [Google Scholar] [CrossRef]
- Chen, J.; Ke, R. Spatial analysis toolkits for RNA in situ sequencing. Wiley Interdiscip. Rev. RNA 2024, 15, e1842. [Google Scholar] [CrossRef]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef]
- Zeng, H.; Huang, J.; Zhou, H.; Meilandt, W.J.; Dejanovic, B.; Zhou, Y.; Bohlen, C.J.; Lee, S.H.; Ren, J.; Liu, A.; et al. Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 430–446. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.C.; Church, G.M.; Lee, J.H.; Zador, A.M. Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res. 2018, 46, e22. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, Y.C.; Zhan, H.; Kebschull, J.M.; Fischer, S.; Matho, K.; Huang, Z.J.; Gillis, J.; Zador, A.M. High-throughput mapping of long-range neuronal projection using in situ sequencing. Cell 2019, 179, 772–786. [Google Scholar] [CrossRef]
- Wyss Institute for Biologically Inspired Engineering at Harvard University. Fluorescent In Situ Sequencing (FISSEQ). Available online: https://wyss.harvard.edu/technology/fluorescent-in-situ-sequencing-fisseq/ (accessed on 11 December 2024).
- Bassiouni, R.; Gibbs, L.D.; Craig, D.W.; Carpten, J.D.; McEachron, T.A. Applicability of spatial transcriptional profiling to cancer research. Mol. Cell 2021, 81, 1631–1639. [Google Scholar] [CrossRef]
- Marblestone, A.H.; Daugharthy, E.R.; Kalhor, R.; Peikon, I.D.; Kebschull, J.M.; Shipman, S.L.; Mishchenko, Y.; Lee, J.H.; Kording, K.P.; Boyden, E.S.; et al. Rosetta brains: A strategy for molecularly-annotated connectomics. arXiv 2014, arXiv:1404.5103. [Google Scholar]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Ferrante, T.C.; Terry, R.; Turczyk, B.M.; Yang, J.L.; Lee, H.S.; Aach, J.; et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 2015, 10, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Khafizov, R.; Piazza, E.; Cui, Y.; Patrick, M.; Metzger, E.; McGuire, D.; Dunaway, D.; Danaher, P.; Hoang, M.L.; Grootsky, A.; et al. Sub-cellular Imaging of the Entire Protein-Coding Human Transcriptome (18933-plex) on FFPE Tissue Using Spatial Molecular Imaging. bioRxiv 2024. [Google Scholar] [CrossRef]
- Unterauer, E.M.; Boushehri, S.S.; Jevdokimenko, K.; Masullo, L.A.; Ganji, M.; Sograte-Idrissi, S.; Kowalewski, R.; Strauss, S.; Reinhardt, S.C.; Perovic, A.; et al. Spatial proteomics in neurons at single-protein resolution. Cell 2024, 187, 1785–1800. [Google Scholar] [CrossRef]
- Mund, A.; Brunner, A.D.; Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 2022, 82, 2335–2349. [Google Scholar] [CrossRef]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-omics: Novel approaches to probe cell heterogeneity and extracellular matrix biology. Matrix Biol. 2020, 91, 152–166. [Google Scholar] [CrossRef]
- Lundberg, E.; Borner, G.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Kuett, L.; Catena, R.; Özcan, A.; Plüss, A.; Schraml, P.; Moch, H.; de Souza, N.; Bodenmiller, B. Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment. Nat. Cancer 2022, 3, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.; Lazcano, R.; Serrano, A.; Powell, S.; Kostousov, L.; Mehta, J.; Khan, K.; Lu, W.; Solis, L.M. Challenges and opportunities for immunoprofiling using a spatial high-plex technology: The NanoString GeoMx® digital spatial profiler. Front. Oncol. 2022, 12, 890410. [Google Scholar] [CrossRef]
- Liu, C.C.; Bosse, M.; Kong, A.; Kagel, A.; Kinders, R.; Hewitt, S.M.; Varma, S.; van de Rijn, M.; Nowak, S.H.; Bendall, S.C.; et al. Reproducible, high-dimensional imaging in archival human tissue by multiplexed ion beam imaging by time-of-flight (MIBI-TOF). Lab. Investig. 2022, 102, 762–770. [Google Scholar] [CrossRef]
- Keren, L.; Bosse, M.; Thompson, S.; Risom, T.; Vijayaragavan, K.; McCaffrey, E.; Marquez, D.; Angoshtari, R.; Greenwald, N.F.; Fienberg, H.; et al. MIBI-TOF: A multiplexed imaging platform relates cellular phenotypes and tissue structure. Sci. Adv. 2019, 5, eaax5851. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, T.; Becker, M.; Guntinas-Lichius, O.; Ernst, G.; von Eggeling, F. MALDI-imaging segmentation is a powerful tool for spatial functional proteomic analysis of human larynx carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 85–95. [Google Scholar] [CrossRef]
- Gessel, M.M.; Norris, J.L.; Caprioli, R.M. MALDI imaging mass spectrometry: Spatial molecular analysis to enable a new age of discovery. J. Proteom. 2014, 107, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef]
- Gustafsson, J.O.; Oehler, M.K.; Ruszkiewicz, A.; McColl, S.R.; Hoffmann, P. MALDI imaging mass spectrometry (MALDI-IMS)―Application of spatial proteomics for ovarian cancer classification and diagnosis. Int. J. Mol. Sci. 2011, 12, 773–794. [Google Scholar] [CrossRef]
- Cornett, D.S.; Reyzer, M.L.; Chaurand, P.; Caprioli, R.M. MALDI imaging mass spectrometry: Molecular snapshots of biochemical systems. Nat. Methods 2007, 4, 828–833. [Google Scholar] [CrossRef]
- Black, S.; Phillips, D.; Hickey, J.W.; Kennedy-Darling, J.; Venkataraaman, V.G.; Samusik, N.; Goltsev, Y.; Schürch, C.M.; Nolan, G.P. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 2021, 16, 3802–3835. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, Y.; Wang, X.; Fan, J.; Gao, Q. Spatial omics: Navigating to the golden era of cancer research. Clin. Transl. Med. 2022, 12, e696. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.K.; Patterson, N.H.; Rivera, E.S.; Allen, J.L.; Brewer, M.; deCaestecker, M.P.; Caprioli, R.M.; Fogo, A.B.; Spraggins, J.M. Highly multiplexed immunofluorescence of the human kidney using co-detection by indexing. Kidney Int. 2022, 101, 137–143. [Google Scholar] [CrossRef]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 2018, 174, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Schürch, C.M.; Bhate, S.S.; Barlow, G.L.; Phillips, D.J.; Noti, L.; Zlobec, I.; Chu, P.; Black, S.; Demeter, J.; McIlwain, D.R.; et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 2020, 182, 1341–1359. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.T.; Holman, D.R.; Sood, A.; Tandon, U.; Bhate, S.S.; Bodapati, S.; Barlow, G.L.; Chang, J.; Black, S.; Crenshaw, E.C.; et al. A tissue atlas of ulcerative colitis revealing evidence of sex-dependent differences in disease-driving inflammatory cell types and resistance to TNF inhibitor therapy. Sci. Adv. 2023, 9, eadd1166. [Google Scholar] [CrossRef]
- Ferreira, R.M.; Sabo, A.R.; Winfree, S.; Collins, K.S.; Janosevic, D.; Gulbronson, C.J.; Cheng, Y.H.; Casbon, L.; Barwinska, D.; Ferkowicz, M.J.; et al. Integration of spatial and single-cell transcriptomics localizes epithelial cell–immune cross-talk in kidney injury. JCI Insight 2021, 6, e147703. [Google Scholar] [CrossRef]
- Jungmann, R.; Avendaño, M.S.; Woehrstein, J.B.; Dai, M.; Shih, W.M.; Yin, P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 2014, 11, 313–318. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Lin, J.R.; Fallahi-Sichani, M.; Sorger, P.K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 2015, 6, 8390. [Google Scholar] [CrossRef]
- Lin, J.R.; Fallahi-Sichani, M.; Chen, J.Y.; Sorger, P.K. Cyclic immunofluorescence (CycIF), a highly multiplexed method for single-cell imaging. Curr. Protoc. Chem. Biol. 2016, 8, 251–264. [Google Scholar] [CrossRef]
- Li, X. Harnessing the potential of spatial multiomics: A timely opportunity. Signal Transduct. Target. Ther. 2023, 8, 234. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Ang, C.E.; Zhuang, X. Spatially resolved epigenomic profiling of single cells in complex tissues. Cell 2022, 185, 4448–4464. [Google Scholar] [CrossRef]
- Farzad, N.; Enninful, A.; Bao, S.; Zhang, D.; Deng, Y.; Fan, R. Spatially resolved epigenome sequencing via Tn5 transposition and deterministic DNA barcoding in tissue. Nat. Protoc. 2024, 19, 3389–3425. [Google Scholar] [CrossRef]
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 2022, 609, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Bartosovic, M.; Kukanja, P.; Zhang, D.; Liu, Y.; Su, G.; Enninful, A.; Bai, Z.; Castelo-Branco, G.; Fan, R. Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 2022, 375, 681–686. [Google Scholar]
- Schueder, F.; Bewersdorf, J. Omics goes spatial epigenomics. Cell 2022, 185, 4253–4255. [Google Scholar] [CrossRef]
- Koch, L. Spatial epigenomics in single cells. Nat. Rev. Genet. 2023, 24, 1. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef]
- Liu, M.; Yang, B.; Hu, M.; Radda, J.S.; Chen, Y.; Jin, S.; Cheng, Y.; Wang, S. Chromatin tracing and multiplexed imaging of nucleome architectures (MINA) and RNAs in single mammalian cells and tissue. Nat. Protoc. 2021, 16, 2667–2697. [Google Scholar] [CrossRef]
- Hu, M.; Wang, S. Chromatin tracing: Imaging 3D genome and nucleome. Trends Cell Biol. 2021, 31, 5–8. [Google Scholar] [CrossRef]
- Gizzi, A.M.C.; Cattoni, D.I.; Fiche, J.B.; Espinola, S.M.; Gurgo, J.; Messina, O.; Houbron, C.; Ogiyama, Y.; Papadopoulos, G.L.; Cavalli, G.; et al. Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell 2019, 74, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef]
- Lewis, S.M.; Asselin-Labat, M.L.; Nguyen, Q.; Berthelet, J.; Tan, X.; Wimmer, V.C.; Merino, D.; Rogers, K.L.; Naik, S.H. Spatial omics and multiplexed imaging to explore cancer biology. Nat. Methods 2021, 18, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yang, Y.C.; An, Z.J.; Zhang, M.H.; Fu, X.H.; Huang, Z.F.; Yuan, Y.; Hou, J. Advances in spatial transcriptomics and related data analysis strategies. J. Transl. Med. 2023, 21, 330. [Google Scholar] [CrossRef]
- Cho, C.S.; Xi, J.; Si, Y.; Park, S.R.; Hsu, J.E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021, 184, 3559–3572. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Sun, L.; Chen, J.Y.; Dong, R.; Lin, Y.; Palmiter, R.D.; Lin, S.; Gu, L. Continuous polony gels for tissue mapping with high resolution and RNA capture efficiency. BioRxiv 2021. [Google Scholar] [CrossRef]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A generalist algorithm for cellular segmentation. Nat. Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Marco Salas, S.; Kuemmerle, L.B.; Mattsson-Langseth, C.; Tismeyer, S.; Avenel, C.; Hu, T.; Rehman, H.; Grillo, M.; Czarnewski, P.; Helgadottir, S.; et al. Optimizing Xenium In Situ data utility by quality assessment and best-practice analysis workflows. Nat. Methods 2025, 22, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, J.; Lewy, T.; Rice, C.M.; Elemento, O.; Rendeiro, A.F.; Mason, C.E. Spatial omics technologies at multimodal and single cell/subcellular level. Genome Biol. 2022, 23, 256. [Google Scholar] [CrossRef]
- Xu, Z.; Li, W.; Dong, X.; Chen, Y.; Zhang, D.; Wang, J.; Zhou, L.; He, G. Precision medicine in colorectal cancer: Leveraging multi-omics, spatial omics, and artificial intelligence. Clin. Chim. Acta 2024, 559, 119686. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-r.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Argelaguet, R.; Arnol, D.; Bredikhin, D.; Deloro, Y.; Velten, B.; Marioni, J.C.; Stegle, O. MOFA+: A statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 2020, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Singhal, V.; Chou, N.; Lee, J.; Yue, Y.; Liu, J.; Chock, W.K.; Lin, L.; Chang, Y.C.; Teo, E.M.L.; Aow, J.; et al. BANKSY unifies cell typing and tissue domain segmentation for scalable spatial omics data analysis. Nat. Genet. 2024, 56, 431–441. [Google Scholar] [CrossRef]
- Birk, S.; Bonafonte-Pardàs, I.; Feriz, A.M.; Boxall, A.; Agirre, E.; Memi, F.; Maguza, A.; Yadav, A.; Armingol, E.; Fan, R.; et al. Quantitative characterization of cell niches in spatially resolved omics data. Nat. Genet. 2025, 57, 897–909. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Satija Lab. Seurat—Guided Analysis of Single-Cell RNA Sequencing Data. 2024. Available online: https://satijalab.org/seurat/ (accessed on 27 December 2024).
- Wolf, F.A.; Angerer, P.; Theis, F.J. Scanpy: Single-Cell Analysis in Python—Documentation. 2024. Available online: https://scanpy.readthedocs.io/en/stable/ (accessed on 27 December 2024).
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Palla, G.; Spitzer, H.; Theis, F.J. Squidpy: Scalable Analysis of Spatial Omics Data—Documentation. 2024. Available online: https://squidpy.readthedocs.io/ (accessed on 27 December 2024).
- Palla, G.; Spitzer, H.; Klein, M.; Fischer, D.; Schaar, A.C.; Kuemmerle, L.B.; Rybakov, S.; Ibarra, I.L.; Holmberg, O.; Virshup, I.; et al. Squidpy: A scalable framework for spatial omics analysis. Nat. Methods 2022, 19, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Giotto. Giotto: Documentation for Spatial Data Analysis and Visualization. 2024. Available online: https://giotto.ai (accessed on 27 December 2024).
- Dries, R.; Zhu, Q.; Dong, R.; Eng, C.H.L.; Li, H.; Liu, K.; Fu, Y.; Zhao, T.; Sarkar, A.; Bao, F.; et al. Giotto: A toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021, 22, 78. [Google Scholar] [CrossRef]
- Hu, J.; Li, X.; Coleman, K.; Schroeder, A.; Ma, N.; Irwin, D.J.; Lee, E.B.; Shinohara, R.T.; Li, M. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 2021, 18, 1342–1351. [Google Scholar] [CrossRef]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]
- Gayoso, A.; Lopez, R.; Xing, G.; Boyeau, P.; Valiollah Pour Amiri, V.; Hong, J.; Wu, K.; Jayasuriya, M.; Mehlman, E.; Langevin, M.; et al. A Python library for probabilistic analysis of single-cell omics data. Nat. Biotechnol. 2022, 40, 163–166. [Google Scholar] [CrossRef]
- nf-core Community. nf-core/Spatialvi: Reproducible Pipeline for Spatial Transcriptomics with SpatialVI. 2025. Available online: https://nf-co.re/spatialvi/dev/ (accessed on 12 April 2025).
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Marconato, L.; Palla, G.; Yamauchi, K.A.; Virshup, I.; Heidari, E.; Treis, T.; Vierdag, W.M.; Toth, M.; Stockhaus, S.; Shrestha, R.B.; et al. SpatialData: An open and universal data framework for spatial omics. Nat. Methods 2025, 22, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Ederveen, T.H.; Niehues, A.; de Visser, C.; Huang, J.; Badmus, F.; Doornbos, C.; Orlova, Y.; Kulkarni, P.; van der Velde, K.J.; et al. FAIR Data Cube, a FAIR data infrastructure for integrated multi-omics data analysis. J. Biomed. Semant. 2024, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Valdeolivas, A.; Amberg, B.; Giroud, N.; Richardson, M.; Gálvez, E.J.; Badillo, S.; Julien-Laferrière, A.; Túrós, D.; Voith von Voithenberg, L.; Wells, I.; et al. Profiling the heterogeneity of colorectal cancer consensus molecular subtypes using spatial transcriptomics. NPJ Precis. Oncol. 2024, 8, 10. [Google Scholar] [CrossRef]
- Wang, X.; Venet, D.; Lifrange, F.; Larsimont, D.; Rediti, M.; Stenbeck, L.; Dupont, F.; Rouas, G.; Garcia, A.J.; Craciun, L.; et al. Spatial transcriptomics reveals substantial heterogeneity in triple-negative breast cancer with potential clinical implications. Nat. Commun. 2024, 15, 10232. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Hsieh, W.C.; Budiarto, B.R.; Wang, Y.F.; Lin, C.Y.; Gwo, M.C.; So, D.K.; Tzeng, Y.S.; Chen, S.Y. Spatial multi-omics analyses of the tumor immune microenvironment. J. Biomed. Sci. 2022, 29, 96. [Google Scholar] [CrossRef]
- Lu, X.; Luo, Y.; Nie, X.; Zhang, B.; Wang, X.; Li, R.; Liu, G.; Zhou, Q.; Liu, Z.; Fan, L.; et al. Single-cell multi-omics analysis of human testicular germ cell tumor reveals its molecular features and microenvironment. Nat. Commun. 2023, 14, 8462. [Google Scholar] [CrossRef] [PubMed]
- Stur, E.; Corvigno, S.; Xu, M.; Chen, K.; Tan, Y.; Lee, S.; Liu, J.; Ricco, E.; Kraushaar, D.; Castro, P.; et al. Spatially resolved transcriptomics of high-grade serous ovarian carcinoma. iScience 2022, 25, 103923. [Google Scholar] [CrossRef]
- Hirz, T.; Mei, S.; Sarkar, H.; Kfoury, Y.; Wu, S.; Verhoeven, B.M.; Subtelny, A.O.; Zlatev, D.V.; Wszolek, M.W.; Salari, K.; et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat. Commun. 2023, 14, 663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, R.Y.; Tan, C.W.; Guo, X.; Yim, W.W.; Lim, J.C.; Wee, F.Y.; Yang, W.; Kharbanda, M.; Lee, J.Y.J.; et al. Spatial omics techniques and data analysis for cancer immunotherapy applications. Curr. Opin. Biotechnol. 2024, 87, 103111. [Google Scholar] [CrossRef]
- Monkman, J.; Moradi, A.; Yunis, J.; Ivison, G.; Mayer, A.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Spatial insights into immunotherapy response in non-small cell lung cancer (NSCLC) by multiplexed tissue imaging. J. Transl. Med. 2024, 22, 239. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougoüin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Erazo, L.; Rhodes, J.L.; Marion, V.C.; Kemp, R.A. Tertiary lymphoid structures in cancer–considerations for patient prognosis. Cell. Mol. Immunol. 2020, 17, 570–575. [Google Scholar] [CrossRef]
- Sadeghirad, H.; Monkman, J.; Tan, C.W.; Liu, N.; Yunis, J.; Donovan, M.L.; Moradi, A.; Jhaveri, N.; Perry, C.; Adams, M.N.; et al. Spatial dynamics of tertiary lymphoid aggregates in head and neck cancer: Insights into immunotherapy response. J. Transl. Med. 2024, 22, 677. [Google Scholar] [CrossRef]
- Jung, N.; Kim, T.K. Spatial transcriptomics in neuroscience. Exp. Mol. Med. 2023, 55, 2105–2115. [Google Scholar] [CrossRef]
- Zhang, L.; Xiong, Z.; Xiao, M. A Review of the Application of Spatial Transcriptomics in Neuroscience. Interdiscip. Sci. Comput. Life Sci. 2024, 16, 243–260. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Frigerio, C.S.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 2020, 182, 976–991. [Google Scholar] [CrossRef]
- Johnston, K.G.; Berackey, B.T.; Tran, K.M.; Gelber, A.; Yu, Z.; MacGregor, G.R.; Mukamel, E.A.; Tan, Z.; Green, K.N.; Xu, X. Single-cell spatial transcriptomics reveals distinct patterns of dysregulation in non-neuronal and neuronal cells induced by the Trem2R47H Alzheimer’s risk gene mutation. Mol. Psychiatry 2025, 30, 461–477. [Google Scholar] [CrossRef]
- Yu, M.; Risacher, S.L.; Nho, K.T.; Wen, Q.; Oblak, A.L.; Unverzagt, F.W.; Apostolova, L.G.; Farlow, M.R.; Brosch, J.R.; Clark, D.G.; et al. Spatial transcriptomic patterns underlying amyloid-β and tau pathology are associated with cognitive dysfunction in Alzheimer’s disease. Cell Rep. 2024, 43, 113691. [Google Scholar] [CrossRef]
- Hirota, T.; King, B.H. Autism spectrum disorder: A review. JAMA 2023, 329, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Wang, M.; Huang, L.; Chen, Y.; Ge, Y.; Zhang, J.; Shi, Y.; Dong, H.; Zhou, X.; Wang, B.; et al. Single-cell epigenomics and spatiotemporal transcriptomics reveal human cerebellar development. Nat. Commun. 2023, 14, 7613. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z. Biological hypotheses, risk factors, and biomarkers of schizophrenia. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2023, 120, 110626. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Tyler, T.; Dragicevic, K.; Mei, S.; Rydbirk, R.; Petukhov, V.; Deviatiiarov, R.; Sedmak, D.; Frank, E.; Feher, V.; et al. Upper cortical layer–driven network impairment in schizophrenia. Sci. Adv. 2022, 8, eabn8367. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J.M. Common mechanisms of autoimmune diseases (the autoimmune tautology). Autoimmun. Rev. 2012, 11, 781–784. [Google Scholar] [CrossRef]
- Pisetsky, D.S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 2023, 19, 509–524. [Google Scholar] [CrossRef]
- Zheng, F.; Tang, D.; Li, S.; Luo, Z.; Song, Y.; Huang, Y.; Gan, Q.; Liu, H.; Zhang, X.; Liu, D.; et al. Spatial proteomics landscape and immune signature analysis of renal sample of lupus nephritis based on laser-captured microsection. Inflamm. Res. 2023, 72, 1603–1620. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, Y.; Li, X.; Xu, R.; Ji, Y.; Liu, J.; Liu, J.; Zhuang, Q.; Zhang, H. Immune landscape and the key role of APOE+ monocytes of lupus nephritis under the single-cell and spatial transcriptional vista. Clin. Transl. Med. 2023, 13, e1237. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, K.; Korotkova, M.; Larsson, L.; Catrina, A.I.; Ståhl, P.L.; Malmström, V. Exploring inflammatory signatures in arthritic joint biopsies with spatial transcriptomics. Sci. Rep. 2019, 9, 18975. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Sánchez de la Blanca, N.; Sacristán-Gómez, P.; Serrano-Somavilla, A.; Muñoz De Nova, J.L.; Sánchez Cabo, F.; Heyn, H.; Sampedro-Núñez, M.; Marazuela, M. Unraveling the molecular architecture of autoimmune thyroid diseases at spatial resolution. Nat. Commun. 2024, 15, 5895. [Google Scholar] [CrossRef] [PubMed]
- Gniadecki, R.; Osman, M.; Hennesey, D.; O’Keefe, S.; Thomsen, S.F.; Iyer, A. Architecture of skin inflammation in psoriasis revealed by spatial transcriptomics. Clin. Immunol. 2023, 256, 109771. [Google Scholar] [CrossRef]
- Gaffney, E.; Riegman, P.; Grizzle, W.; Watson, P. Factors that drive the increasing use of FFPE tissue in basic and translational cancer research. Biotech. Histochem. 2018, 93, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Dezem, F.S.; Morosini, N.S.; Arjumand, W.; DuBose, H.; Plummer, J. Spatially resolved single-cell omics: Methods, challenges, and future perspectives. Annu. Rev. Biomed. Data Sci. 2024, 7, 131–153. [Google Scholar] [CrossRef]
- Walsh, L.A.; Quail, D.F. Decoding the tumor microenvironment with spatial technologies. Nat. Immunol. 2023, 24, 1982–1993. [Google Scholar] [CrossRef]
- Huan, C.; Li, J.; Li, Y.; Zhao, S.; Yang, Q.; Zhang, Z.; Li, C.; Li, S.; Guo, Z.; Yao, J.; et al. Spatially Resolved Multiomics: Data Analysis from Monoomics to Multiomics. BME Front. 2025, 6, 0084. [Google Scholar] [CrossRef]
- Gurkar, A.U.; Gerencser, A.A.; Mora, A.L.; Nelson, A.C.; Zhang, A.R.; Lagnado, A.B.; Enninful, A.; Benz, C.; Furman, D.; Beaulieu, D.; et al. Spatial mapping of cellular senescence: Emerging challenges and opportunities. Nat. Aging 2023, 3, 776–790. [Google Scholar] [CrossRef]
- Lee, S.; Kim, G.; Lee, J.; Lee, A.C.; Kwon, S. Mapping cancer biology in space: Applications and perspectives on spatial omics for oncology. Mol. Cancer 2024, 23, 26. [Google Scholar] [CrossRef]
- Vickovic, S.; Lötstedt, B.; Klughammer, J.; Mages, S.; Segerstolpe, Å.; Rozenblatt-Rosen, O.; Regev, A. SM-Omics is an automated platform for high-throughput spatial multi-omics. Nat. Commun. 2022, 13, 795. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, H.; Rudin, C.; Shaposhnik, Y. Understanding how dimension reduction tools work: An empirical approach to deciphering t-SNE, UMAP, TriMAP, and PaCMAP for data visualization. J. Mach. Learn. Res. 2021, 22, 1–73. [Google Scholar]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef] [PubMed]
- Bollhagen, A.; Bodenmiller, B. Highly Multiplexed Tissue Imaging in Precision Oncology and Translational Cancer Research. Cancer Discov. 2024, 14, 2071–2088. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Lundberg, E.; Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet. 2022, 23, 741–759. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Resolution | Tissue Compatibility | Features |
|---|---|---|---|
| Visium | 55 m | FFPE, FF | Barcoded spots |
| Slide-seq | 10 m | FF | Barcoded beads |
| Stereo-seq | 220 nm | FF | DNA nanoball patterned array chips |
| DBiT-seq | 10 m | FFPE, FF | Microfluidic spatial barcoding |
| Method | Detection Principle | Spatial Resolution | Genomic Resolution | Tissue Compatibility | Key Applications |
|---|---|---|---|---|---|
| Xenium | FISH with RCA | Subcellular | Gene-level (1000+) | FFPE, FF | Cancer research, Brain mapping, TME analysis |
| MERFISH | Combinatorial barcoding + sequential FISH | Single-molecule | Gene-level (1000+) | FF, Cultured cells | Neuroscience, Developmental biology, Cancer research |
| SeqFISH+ | Sequential FISH with temporal barcoding | Subcellular | Gene-level (10,000+) | FF, Cultured cells | Cell-type identification, Tissue architecture, Cell–cell interactions |
| CosMx SMI | Combinatorial barcoding + sequential FISH | Subcellular | Gene-level (1000+) | FFPE, FF | Cancer research, Neuroscience, Immunology |
| STARmap | Padlock probe-based RCA | Subcellular | Targeted genes | FF, Hydrogel-embedded tissues | Brain studies, Molecularly defined cell typing, Gene expression in intact tissue |
| BaristaSeq | Gap padlock probe-based sequencing | Subcellular | Targeted barcode sequencing | FF, Cultured cells | Lineage tracing, Neuronal projection mapping |
| FISSEQ | Reverse transcription + RCA + SBL | Subcellular | Genome-wide (untargeted) | FFPE, FF, Cultured cells | Cancer research, Neuronal mapping, RNA splicing analysis |
| Method | Focus | Target | Barcoding Method | Resolution |
|---|---|---|---|---|
| Spatial ATAC-seq | Chromatin accessibility | Open chromatin, TF binding sites | Microfluidic dual-barcode grid | 20 μm |
| Spatial-CUT&Tag | Histone modifications | H3K27me3, H3K4me3, H3K27ac | Microfluidic dual-barcode grid | 20 μm |
| Epigenomic MERFISH | Epigenetic modifications | H3K27me3, H3K4me3, H3K27ac, Enhancer regions | Sequential fluorescent barcoding | Sub-nuclear (1 kb) |
| Chromatin tracing | 3D genome organization | Locus-specific chromatin structure | Multiplexed FISH | Sub-nuclear (30 kb) |
| Hi-F | 3D genome organization | Chromatin contacts (TADs, loops) | Multiplexed FISH | Sub-nuclear (2 kb) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.; Lee, M.; Shin, Y.; Kim, K.; Kim, T. Spatial Omics in Clinical Research: A Comprehensive Review of Technologies and Guidelines for Applications. Int. J. Mol. Sci. 2025, 26, 3949. https://doi.org/10.3390/ijms26093949
Lee Y, Lee M, Shin Y, Kim K, Kim T. Spatial Omics in Clinical Research: A Comprehensive Review of Technologies and Guidelines for Applications. International Journal of Molecular Sciences. 2025; 26(9):3949. https://doi.org/10.3390/ijms26093949
Chicago/Turabian StyleLee, Yoonji, Mingyu Lee, Yoojin Shin, Kyuri Kim, and Taejung Kim. 2025. "Spatial Omics in Clinical Research: A Comprehensive Review of Technologies and Guidelines for Applications" International Journal of Molecular Sciences 26, no. 9: 3949. https://doi.org/10.3390/ijms26093949
APA StyleLee, Y., Lee, M., Shin, Y., Kim, K., & Kim, T. (2025). Spatial Omics in Clinical Research: A Comprehensive Review of Technologies and Guidelines for Applications. International Journal of Molecular Sciences, 26(9), 3949. https://doi.org/10.3390/ijms26093949