
Influence of Exogenous Neuropeptides on the Astrocyte Response Under Conditions of Continuous and Cyclic Hypoxia and Red Blood Cell Lysate


 ,
,  , , , , and
, , , , and
Abstract
1. Introduction
2. Results
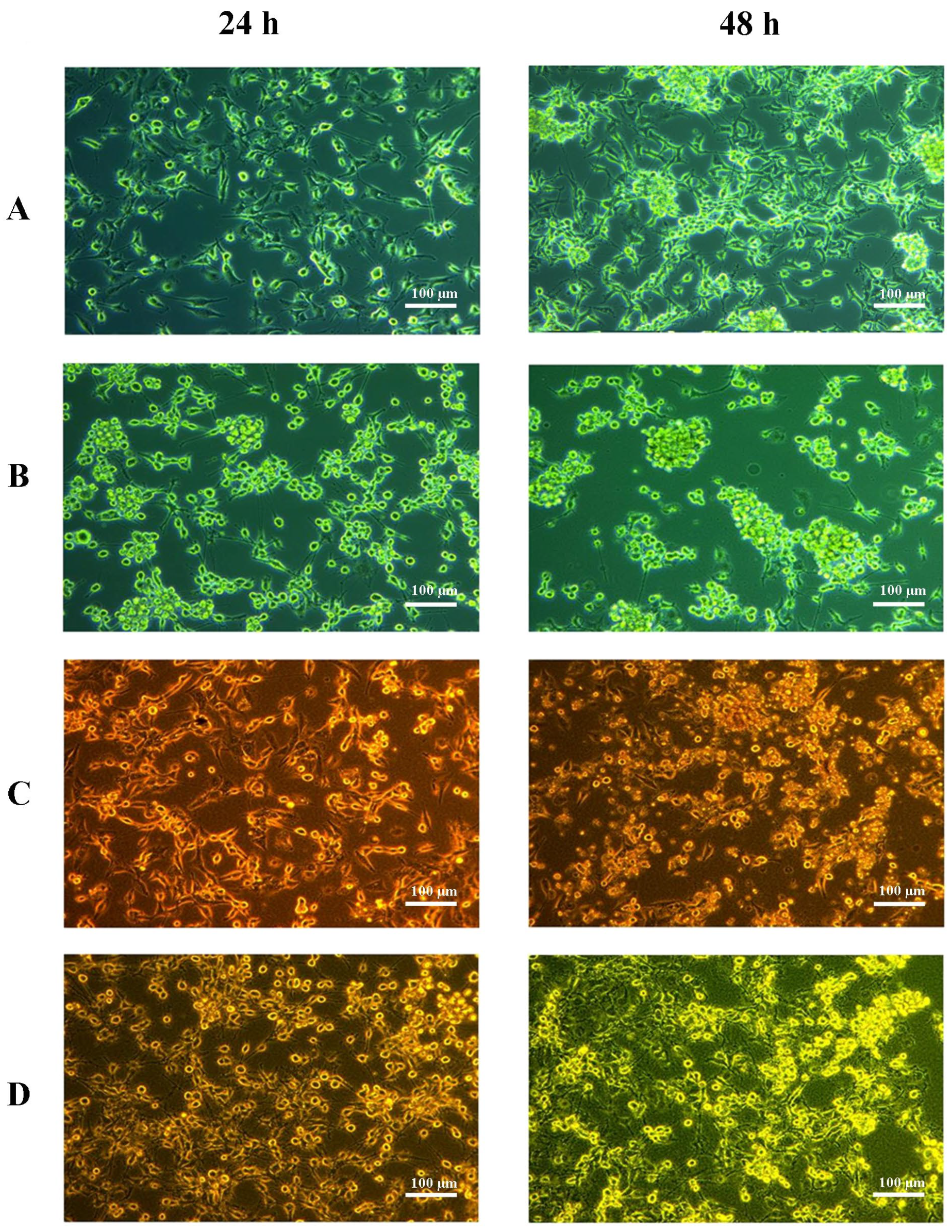
2.1. Cell Culture Visualization
2.2. The Effect of U87MG Cell Culture Exposure to CER, Red Blood Cells Lysate (RBC), and Red Blood Cells Lysate Together with (RBC + CER) on COX-1 and COX-2 Activity
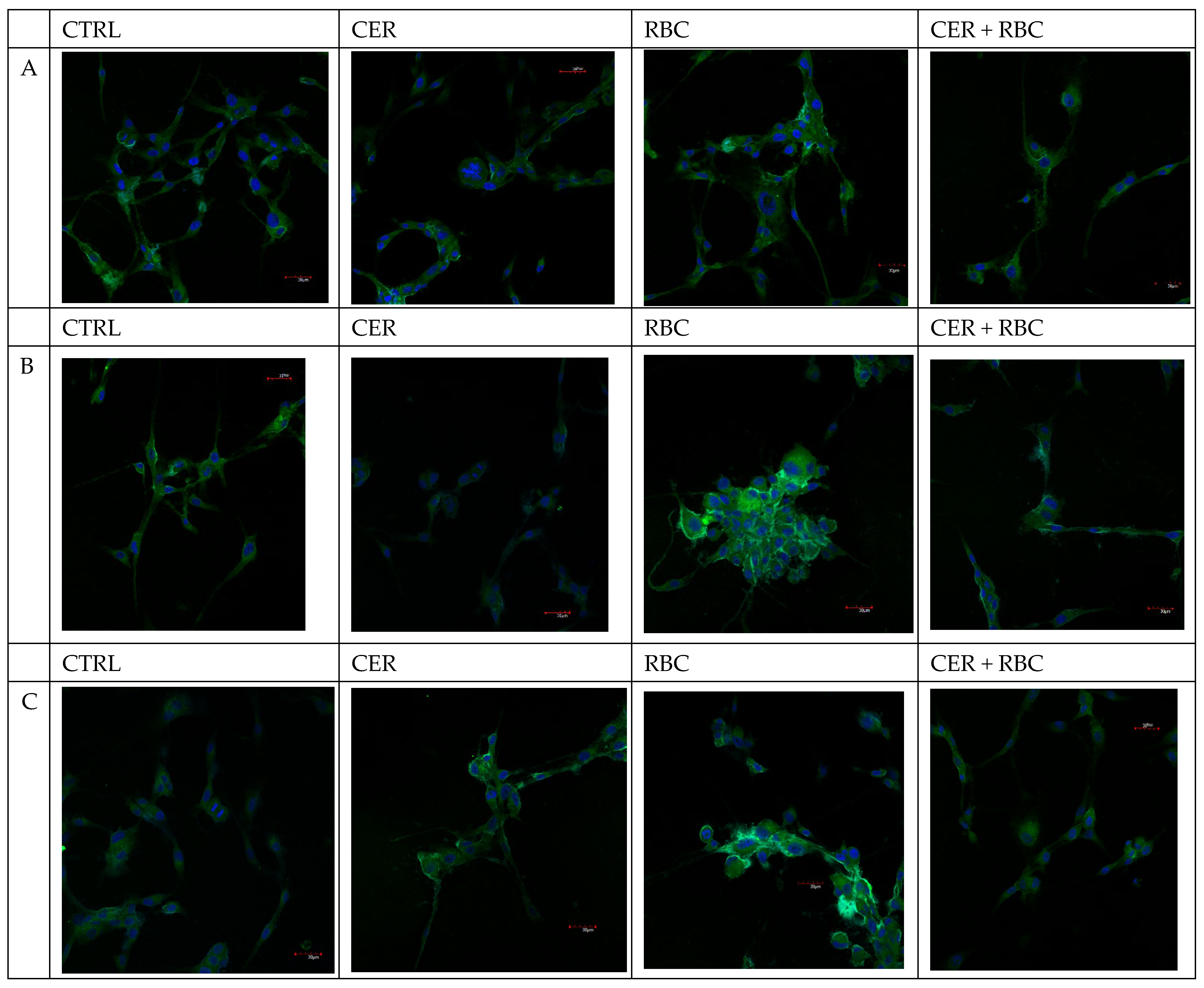
2.2.1. COX-1 and COX-2 Expression
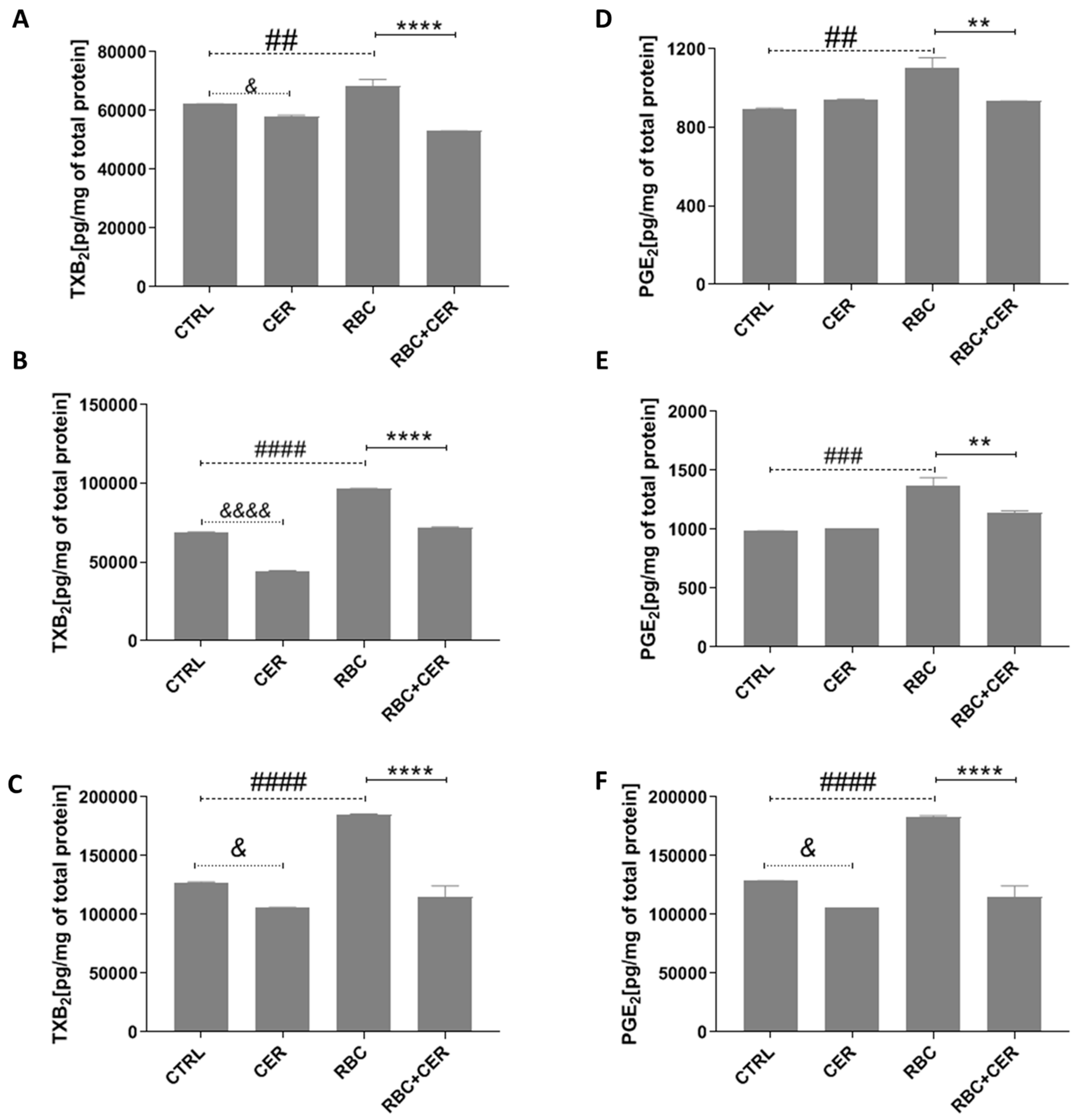
2.2.2. Effect of U87MG Cell Exposure to Cerebrolysin (CER), RBC Lysate (RBC), and RBC + CER on Prostaglandin E2 (PGE2) and Thromboxane B2 (TXB2) Levels
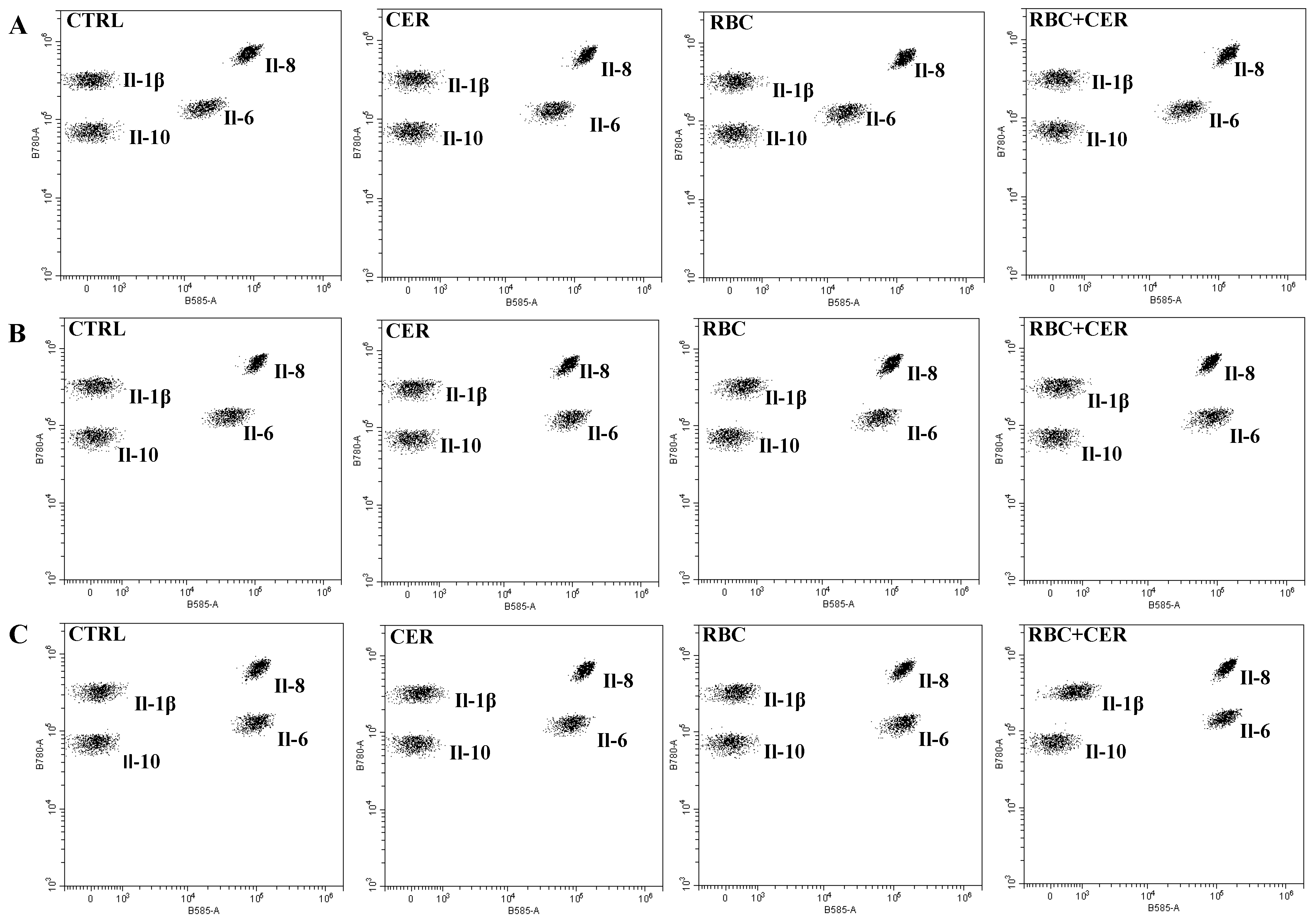
2.3. The Effect of U87MG Cell Culture Exposure to Cerebrolysine (CER), Red Blood Cells Lysate (RBC), and Red Blood Cells Lysate Together with Cerebrolysine (RBC + CER) on the Concentration of Cytokines: Il-1β, Il-6, Il-8, and Il-10
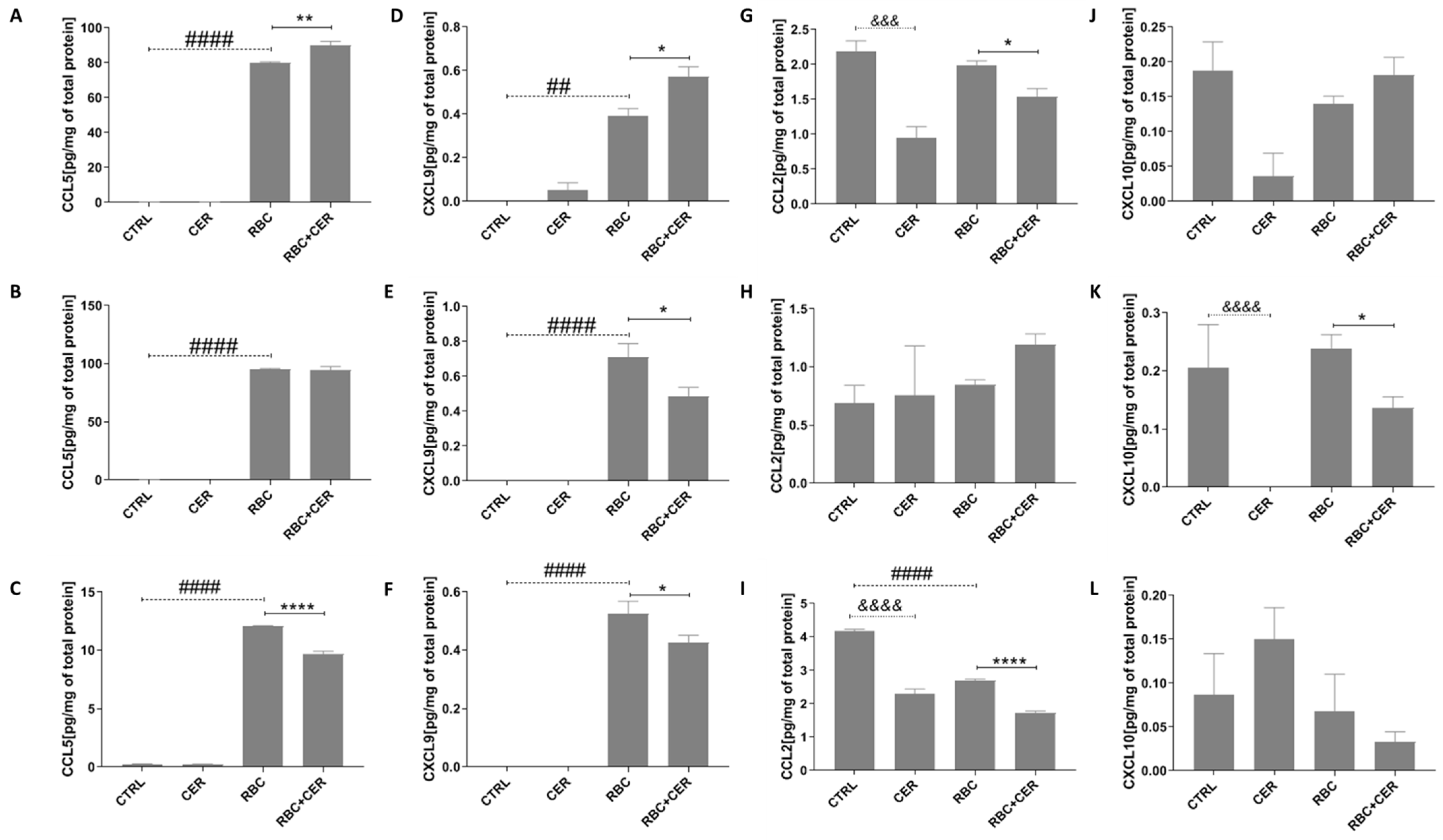
2.4. Effect of U87MG Cell Exposure to Cerebrolysin (CER), RBC Lysate (RBC), and RBC + CER on CCL5, CXCL9, CCL2, and CXCL10 Levels
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Determination of COX-1 and COX-2 Activity
4.3.1. Imaging of COX-1and COX-2 Expression
4.3.2. Determination of PGE2 and TXA2 Level
4.4. Determination of Cytokines and Chemokines Concentration
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Myer, D.J.; Gurkoff, G.G.; Lee, S.M.; Hovda, D.A.; Sofroniew, M.V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 2006, 129, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Brazinova, A.; Rehorcikova, V.; Taylor, M.S.; Buckova, V.; Majdan, M.; Psota, M.; Synnot, A. Epidemiology of traumatic brain injury in Europe: A living systematic review. J. Neurotrauma 2021, 38, 1411–1440. [Google Scholar] [CrossRef] [PubMed]
- Wallen, T.E.; Baucom, M.R.; England, L.G.; Schuster, R.M.; Pritts, T.A.; Goodman, M.D. Multimodal treatment approaches to combined traumatic brain injury and hemorrhagic shock alter postinjury inflammatory response. Shock 2022, 58, 565–572. [Google Scholar] [CrossRef]
- Xie, L.; He, M.; Ying, C.; Chu, H. Mechanisms of inflammation after ischemic stroke in brain-peripheral crosstalk. Front. Mol. Neurosci. 2024, 17, 1400808. [Google Scholar] [CrossRef] [PubMed]
- Bederson, J.B.; Levy, A.L.; Ding, W.H.; Kahn, R.; DiPerna, C.A.; Jenkins, A.L., III; Vallabhajosyula, P. Acute vasoconstriction after subarachnoid hemorrhage. Neurosurgery 1998, 42, 352–360. [Google Scholar] [CrossRef]
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar] [CrossRef]
- Sehba, F.A.; Mostafa, G.; Knopman, J.; Friedrich, V.; Bederson, J.B. Acute alterations in microvascular basal lamina after subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 633–640. [Google Scholar] [CrossRef]
- Sehba, F.A.; Mostafa, G.; Friedrich, V.; Bederson, J.B. Acute microvascular platelet aggregation after subarachnoid hemorrhage. DidnJ. Neurosurg. 2005, 102, 1094–1100. [Google Scholar] [CrossRef]
- Prunell, G.F.; Svendgaard, N.A.; Alkass, K.; Mathiesen, T. Inflammation in the brain after experimental subarachnoid hemorrhage. Neurosurgery 2005, 56, 1082–1092. [Google Scholar]
- Ohashi, S.N.; DeLong, J.H.; Kozberg, M.G.; Mazur-Hart, D.J.; Van Veluw, S.J.; Alkayed, N.J.; Sansing, L.H. Role of inflammatory processes in hemorrhagic stroke. Stroke 2023, 54, 605–619. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef]
- Dumont, A.S.; Dumont, R.J.; Chow, M.M.; Lin, C.-L.; Calisaneller, T.; Ley, K.F.; Kassell, N.F.; Lee, K.S. Cerebral vasospasm after subarachnoid hemorrhage: Putative role of inflammation. Neurosurgery 2003, 53, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Yan, E.B.; Satgunaseelan, L.; Paul, E.; Bye, N.; Nguyen, P.; Agyapomaa, D.; Kossmann, T.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Post-traumatic hypoxia is associated with prolonged cerebral cytokine production, higher serum biomarker levels, and poor outcome in patients with severe traumatic brain injury. J. Neurotrauma 2014, 31, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 868. [Google Scholar] [CrossRef] [PubMed]
- Pappas, A.C.; Koide, M.; Wellman, G.C. Astrocyte Ca2+ signaling drives inversion of neurovascular coupling after subarachnoid hemorrhage. J. Neurosci. 2015, 35, 13375–13384. [Google Scholar] [CrossRef]
- Pekny, M.; Wilhelmsson, U.; Tatlisumak, T.; Pekna, M. Astrocyte activation and reactive gliosis—A new target in stroke? Neurosci. Lett. 2019, 689, 45–55. [Google Scholar] [CrossRef]
- Luo, C.; Yao, J.; Bi, H.; Li, Z.; Li, J.; Xue, G.; Li, K.; Zhang, S.; Zan, K.; Meng, W.; et al. Clinical value of inflammatory cytokines in patients with aneurysmal subarachnoid hemorrhage. Clin. Interv. Aging 2022, 615–626. [Google Scholar] [CrossRef]
- Wang, J.; Doré, S. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 27, 894–908. [Google Scholar] [CrossRef]
- Kurumbail, R.G.; Kiefer, J.R.; Marnett, L.J. Cyclooxygenase enzymes: Catalysis and inhibition. Curr. Opin. Struct. Biol. 2001, 11, 752–760. [Google Scholar] [CrossRef]
- Khan, A.A.; Iadarola, M.; Yang, H.Y.; Dionne, R.A. Expression of COX-1 and COX-2 in a clinical model of acute inflammation. J. Pain 2007, 8, 349–354. [Google Scholar] [CrossRef]
- Lin, W.; Li, Z. Blueberries inhibit cyclooxygenase-1 and cyclooxygenase-2 activity in human epithelial ovarian cancer. Oncol. Lett. 2017, 13, 4897–4904. [Google Scholar] [CrossRef]
- Morita, I. Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002, 68–69, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Hong, K.; Jang, B.C. Transcriptional and translational regulation of COX-2 expression by cadmium in C6 glioma cells. Int. J. Mol. Med. 2012, 30, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chopp, M.; Zhang, Z.G.; Zhang, Y.; Zhang, L.; Lu, M.; Zhang, T.; Winter, S.; Brandstätter, H.; Mahmood, A.; et al. Prospective, randomized, blinded, and placebo-controlled study of Cerebrolysin dose-response effects on long-term functional outcomes in a rat model of mild traumatic brain injury. J. Neurosurg. 2018, 129, 1295–1304. [Google Scholar] [CrossRef]
- Avci, S.; Gunaydin, S.; Ari, N.S.; Sulukoglu, E.K.; Polat, O.E.; Gecili, I.; Yeni, Y.; Yilmaz, A.; Genc, S.; Hacimuftuoglu, A.; et al. Cerebrolysin alleviating effect on glutamate-mediated neuroinflammation via glutamate transporters and oxidative stress. J. Mol. Neurosci. 2022, 72, 2292–2302. [Google Scholar] [CrossRef]
- Lu, W.; Zhu, Z.; Shi, D.; Li, X.; Luo, J.; Liao, X. Cerebrolysin alleviates early brain injury after traumatic brain injury by inhibiting neuroinflammation and apoptosis via TLR signaling pathway. Acta Cir. Bras. 2022, 37, e370605. [Google Scholar] [CrossRef]
- Masliah, E.; Armasolo, F.; Veinbergs, I.; Mallory, M.; Samuel, W. Cerebrolysin ameliorates performance deficits, and neuronal damage in apolipoprotein E-deficient mice. Pharmacol. Biochem. Behav. 1999, 62, 239–245. [Google Scholar] [CrossRef]
- Chen, C.C.; Wei, S.T.; Tsaia, S.C.; Chen, X.X.; Cho, D.Y. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: Double-blind, placebo-controlled, randomized study. Br. J. Neurosurg. 2013, 27, 803–807. [Google Scholar] [CrossRef]
- Zhang, D.; Dong, Y.; Li, Y.; Chen, J.; Wang, J.; Hou, L. Efficacy and Safety of Cerebrolysin for Acute Ischemic Stroke: A Meta-Analysis of Randomized Controlled Trials. BioMed Res. Int. 2017, 2017, 4191670. [Google Scholar] [CrossRef]
- Staszewski, J.; Stȩpień, A.; Piusińska-Macoch, R.; Dȩbiec, A.; Gniadek-Olejniczak, K.; Frankowska, E.; Maliborski, A.; Chadaide, Z.; Balo, D.; Król, B.; et al. Efficacy of cerebrolysin treatment as an add-on therapy to mechanical thrombectomy in patients with acute ischemic stroke due to large vessel occlusion: Study protocol for a prospective, open label, single-center study with 12 Months of follow-up. Front. Neurol. 2022, 13, 910697. [Google Scholar] [CrossRef]
- Zhang, Y.; Chopp, M.; Zhang, Z.G.; Zhang, Y.; Zhang, L.; Lu, M.; Zhang, T.; Winter, S.; Doppler, E.; Brandstäetter, H.; et al. Cerebrolysin reduces astrogliosis and axonal injury and enhances neurogenesis in rats after closed head injury. Neurorehabilit. Neural Repair 2019, 33, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Wu, D.; Windisch, M. In vivo upregulation of the blood–brain barrier GLUT1 glucose transporter by brain-derived peptides. Neurosci. Res. 1999, 34, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Onose, G.; Mureşanu, D.F.; Ciurea, A.V.; Chendreanu, C.D.; Mihaescu, A.S.; Mardare, D.C.; Spircu, T. Neuroprotective and consequent neurorehabilitative clinical outcomes, in patients treated with the pleiotropic drug cerebrolysin. J. Med. Life 2009, 2, 350. [Google Scholar] [PubMed]
- Guan, X.; Wang, Y.; Kai, G.; Zhao, S.; Huang, T.; Li, Y.; Xu, Y.; Zhang, L.; Pang, T. Cerebrolysin ameliorates focal cerebral ischemia injury through neuroinflammatory inhibition via CREB/PGC-1α pathway. Front. Pharmacol. 2019, 10, 1245. [Google Scholar] [CrossRef]
- Heiss, W.D.; Brainin, M.; Bornstein, N.M.; Tuomilehto, J.; Hong, Z. Cerebrolysin in patients with acute ischemic stroke in Asia: Results of a double-blind, placebo-controlled randomized trial. Stroke 2012, 43, 630–636. [Google Scholar] [CrossRef]
- Amiri-Nikpour, M.R.; Nazarbaghi, S.; Ahmadi-Salmasi, B.; Mokari, T.; Tahamtan, U.; Rezaei, Y. Cerebrolysin effects on neurological outcomes and cerebral blood flow in acute ischemic stroke. Neuropsychiatr. Dis. Treat. 2014, 10, 2299–2306. [Google Scholar]
- Hartbauer, M.; Hutter-Paier, B.; Skofitsch, G.; Windisch, M. Antiapoptotic effects of the peptidergic drug cerebrolysin on primary cultures of embryonic chick cortical neurons. J. Neural Transm. 2001, 108, 459–473. [Google Scholar] [CrossRef]
- Gutmann, B.; Hutter-Paier, B.; Skofitsch, G.; Windisch, M.; Gmeinbauer, R. In vitro models of brain ischemia: The peptidergic drug cerebrolysin protects cultured chick cortical neurons from cell death. Neurotox. Res. 2002, 4, 59–65. [Google Scholar] [CrossRef]
- Riley, C.; Hutter-Paier, B.; Windisch, M.; Doppler, E.; Moessler, H.; Wronski, R. A peptide preparation protects cells in organotypic brain slices against cell death after glutamate intoxication. J. Neural Transm. 2006, 113, 103–110. [Google Scholar] [CrossRef]
- Schauer, E.; Wronski, R.; Patockova, J.; Moessler, H.; Doppler, E.; Hutter-Paier, B.; Windisch, M. Neuroprotection of cerebrolysin in tissue culture models of brain ischemia: Post lesion application indicates a wide therapeutic window. J. Neural Transm. 2006, 113, 855–868. [Google Scholar] [CrossRef]
- Ren, J.; Sietsma, D.; Qiu, S.; Moessler, H.; Finklestein, S.P. Cerebrolysin enhances functional recovery following focal cerebral infarction in rats. Restor. Neurol. Neurosci. 2007, 25, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Liu, X.F.; Ross, T.M.; Doppler, E.; Zimmermann-Meinzingen, S.; Moessler, H.; Frey, I.I. Cerebrolysin reduces infarct volume in a rat model of focal cerebral ischemic damage. Am. J. Neuroprot. Neuroregener. 2009, 1, 60–66. [Google Scholar] [CrossRef]
- Woo, P.Y.M.; Ho, J.W.K.; Ko, N.M.W.; Li, R.P.T.; Jian, L.; Chu, A.C.H.; Kwan, M.C.L.; Chan, Y.; Wong, A.K.S.; Wong, H.-T.; et al. Randomized, placebo-controlled, double-blind, pilot trial to investigate safety and efficacy of Cerebrolysin in patients with aneurysmal subarachnoid hemorrhage. BMC Neurol. 2020, 20, 401. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.M.; Stillings, S.A.; Leclerc, J.L.; Phillips, H.; Edwards, N.J.; Robicsek, S.A.; Hoh, B.L.; Blackburn, S.; Doré, S. Role of interleukin-10 in acute brain injuries. Front. Neurol. 2017, 8, 244. [Google Scholar] [CrossRef]
- Yang, X.L.; Kim, C.K.; Kim, T.J.; Sun, J.; Rim, D.; Kim, Y.J.; Ko, S.B.; Jang, H.; Yoon, B.W. Anti-inflammatory effects of fimasartan via Akt, ERK, and NFkappaB pathways on astrocytes stimulated by hemolysate. Inflamm. Res. 2016, 65, 115–123. [Google Scholar] [CrossRef]
- Bajenaru, O.; Tiu, C.; Moessler, H.; Antochi, F.; Muresanu, D.; Popescu, B.O.; Novak, P. Efficacy and safety of Cerebrolysin in patients with hemorrhagic stroke. J. Med. Life 2010, 3, 137. [Google Scholar]
- Jacquens, A.; Needham, E.J.; Zanier, E.R.; Degos, V.; Gressens, P.; Menon, D. Neuro-inflammation modulation and post-traumatic brain injury lesions: From bench to bed-side. Int. J. Mol. Sci. 2022, 23, 11193. [Google Scholar] [CrossRef]
- Cieri, M.B.; Ramos, A.J. Astrocytes, reactive astrogliosis, and glial scar formation in traumatic brain injury. Neural Regen. Res. 2025, 20, 973–989. [Google Scholar] [CrossRef]
- Wang, X.; Fu, S.; Wang, Y.; Yu, P.; Hu, J.; Gu, W.; Xu, X.-M.; Lu, P. Interleukin-1β mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol. Cell. Neurosci. 2007, 36, 343–354. [Google Scholar] [CrossRef]
- Bosetti, F. Arachidonic acid metabolism in brain physiology and pathology: Lessons from genetically altered mouse models. J. Neurochem. 2007, 102, 577–586. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Chung, J.; Burger, M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1094–L1103. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, T.; Stahel, P.F.; Lenzlinger, P.M.; Redl, H.; Dubs, R.W.; Trentz, O.; Morganti-Kossmann, M.C. Interleukin-8 released into the cerebrospinal fluid after brain injury is associated with blood–brain barrier dysfunction and nerve growth factor production. J. Cereb. Blood Flow Metab. 1997, 17, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.C.B.; Regner, A.; Miotto, K.D.L.; Moura, S.D.; Ikuta, N.; Vargas, A.E.; Simon, D. Increased levels of interleukin-6,-8 and-10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014, 28, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Gopcevic, A.; Mazul-Sunko, B.; Marout, J.; Sekulic, A.; Antoljak, N.; Siranovic, M.; Zarkovic, N. Plasma interleukin-8 as a potential predictor of mortality in adult patients with severe traumatic brain injury. Tohoku J. Exp. Med. 2007, 211, 387–393. [Google Scholar] [CrossRef]
- Rodney, T.; Osier, N.; Gill, J. Pro-and anti-inflammatory biomarkers and traumatic brain injury outcomes: A review. Cytokine 2018, 110, 248–256. [Google Scholar] [CrossRef]
- Rasmussen, R.; Bache, S.; Stavngaard, T.; Møller, K. Plasma levels of IL-6, IL-8, IL-10, ICAM-1, VCAM-1, IFNγ, and TNFα are not associated with delayed cerebral ischemia, cerebral vasospasm, or clinical outcome in patients with subarachnoid hemorrhage. World Neurosurg. 2019, 128, e1131–e1136. [Google Scholar] [CrossRef]
- Hendryk, S.; Jarzab, B.; Josko, J. Increase of the IL-1beta and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAH. Neuroendocrinol. Lett. 2004, 25, 141–147. [Google Scholar]
- Samatra, D.P.G.P.; Pratiwi, N.M.D.; Widyadharma, I.P.E. High Il-1β Serum as a Predictor of Decreased Cognitive Function in Mild Traumatic Brain Injury Patients. Open Access Maced. J. Med. Sci. 2018, 6, 1674. [Google Scholar] [CrossRef]
- Sozen, T.; Tsuchiyama, R.; Hasegawa, Y.; Suzuki, H.; Jadhav, V.; Nishizawa, S.; Zhang, J.H. Role of interleukin-1β in early brain injury after subarachnoid hemorrhage in mice. Stroke 2009, 40, 2519–2525. [Google Scholar] [CrossRef]
- Jedrzejowska-Szypulka, H.; Straszak, G.; Larysz-Brysz, M.; Karpe, J.; Marcol, W.; Olakowska, E.; Woszczycka-Korczynska, I.; Lewin-Kowalik, J. Interleukin-1β plays a role in the activation of peripheral leukocytes after blood-brain barrier rupture in the course of subarachnoid hemorrhage. Curr. Neurovascular Res. 2010, 7, 39–48. [Google Scholar] [CrossRef]
- Lv, S.Y.; Wu, Q.; Liu, J.P.; Shao, J.; Wen, L.L.; Xue, J.; Zhang, X.S.; Zhang, Q.R.; Zhang, X. Levels of interleukin-1β, interleukin-18, and tumor necrosis factor-α in cerebrospinal fluid of aneurysmal subarachnoid hemorrhage patients may be predictors of early brain injury and clinical prognosis. World Neurosurg. 2018, 111, e362–e373. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hu, S.; Li, Y.; Sun, Y.; Xiong, X.; Hu, X.; Chen, J.; Qiu, S. Interleukins and ischemic stroke. Front. Immunol. 2022, 13, 828447. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, N.M.; Guekht, A.; Vester, J.; Heiss, W.D.; Gusev, E.; Hoemberg, V.; Muresanu, D. Safety and efficacy of Cerebrolysin in early post-stroke recovery: A meta-analysis of nine randomized clinical trials. Neurol. Sci. 2018, 39, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Muresanu, D.F.; Florian, S.; Hömberg, V.; Matula, C.; von Steinbüchel, N.; Vos, P.E.; Vester, J. Efficacy and safety of cerebrolysin in neurorecovery after moderate-severe traumatic brain injury: Results from the CAPTAIN II trial. Neurol. Sci. 2020, 41, 1171–1181. [Google Scholar] [CrossRef]
- Jarosz, K.; Kojder, K.; Andrzejewska, A.; Solek-Pastuszka, J.; Jurczak, A. Cerebrolysin in patients with TBI: Systematic review and meta-analysis. Brain Sci. 2023, 13, 507. [Google Scholar] [CrossRef]
- Park, Y.K.; Yi, H.J.; Choi, K.S.; Lee, Y.J.; Kim, D.W.; Kwon, S.M. Cerebrolysin for the treatment of aneurysmal subarachnoid hemorrhage in adults: A retrospective chart review. Adv. Ther. 2018, 35, 2224–2235. [Google Scholar] [CrossRef]
- Deroide, N.; Li, X.; Lerouet, D.; Van Vré, E.; Baker, L.; Harrison, J.; Poittevin, M.; Masters, L.; Nih, L.; Margaill, I.; et al. MFGE8 inhibits inflammasome-induced IL-1β production and limits postischemic cerebral injury. J. Clin. Investig. 2013, 123, 1176–1181. [Google Scholar] [CrossRef]
- Wagner, K.R.; Sharp, F.R.; Ardizzone, T.D.; Lu, A.; Clark, J.F. Heme and iron metabolism: Role in cerebral hemorrhage. J. Cereb. Blood Flow Metab. 2003, 23, 629–652. [Google Scholar] [CrossRef]
- Xi, G.; Hua, Y.; Bhasin, R.R.; Ennis, S.R.; Keep, R.F.; Hoff, J.T. Mechanisms of edema formation after intracerebral hemorrhage: Effects of extravasated red blood cells on blood flow and blood-brain barrier integrity. Stroke 2001, 32, 2932–2938. [Google Scholar] [CrossRef]
- Aoki, T.; Takenaka, K.; Suzuki, S.; Kassell, N.F.; Sagher, O.; Lee, K.S. The role of hemolysate in the facilitation of oxyhemoglobin-induced contraction in rabbit basilar arteries. J. Neurosurg. 1994, 81, 261–266. [Google Scholar] [CrossRef]
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ctrl | Cer | RBC | Cer + RBC | |
|---|---|---|---|---|
| A | + | + | ++ | + |
| B | + | + | ++ | + |
| C | ± | + | ++ | + |
| Ctrl | Cer | RBC | Cer + RBC | |
|---|---|---|---|---|
| A | + | + | + | + |
| B | + | + | +++ | + |
| C | + | + | +++ | ± |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kojder, K.; Gąssowska-Dobrowolska, M.; Żwierełło, W.; Kłos, P.; Piotrowska, K.; Wszołek, A.; Maruszewska, A.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Influence of Exogenous Neuropeptides on the Astrocyte Response Under Conditions of Continuous and Cyclic Hypoxia and Red Blood Cell Lysate. Int. J. Mol. Sci. 2025, 26, 3953. https://doi.org/10.3390/ijms26093953
Kojder K, Gąssowska-Dobrowolska M, Żwierełło W, Kłos P, Piotrowska K, Wszołek A, Maruszewska A, Gutowska I, Chlubek D, Baranowska-Bosiacka I. Influence of Exogenous Neuropeptides on the Astrocyte Response Under Conditions of Continuous and Cyclic Hypoxia and Red Blood Cell Lysate. International Journal of Molecular Sciences. 2025; 26(9):3953. https://doi.org/10.3390/ijms26093953
Chicago/Turabian StyleKojder, Klaudyna, Magdalena Gąssowska-Dobrowolska, Wojciech Żwierełło, Patrycja Kłos, Katarzyna Piotrowska, Agata Wszołek, Agnieszka Maruszewska, Izabela Gutowska, Dariusz Chlubek, and Irena Baranowska-Bosiacka. 2025. "Influence of Exogenous Neuropeptides on the Astrocyte Response Under Conditions of Continuous and Cyclic Hypoxia and Red Blood Cell Lysate" International Journal of Molecular Sciences 26, no. 9: 3953. https://doi.org/10.3390/ijms26093953
APA StyleKojder, K., Gąssowska-Dobrowolska, M., Żwierełło, W., Kłos, P., Piotrowska, K., Wszołek, A., Maruszewska, A., Gutowska, I., Chlubek, D., & Baranowska-Bosiacka, I. (2025). Influence of Exogenous Neuropeptides on the Astrocyte Response Under Conditions of Continuous and Cyclic Hypoxia and Red Blood Cell Lysate. International Journal of Molecular Sciences, 26(9), 3953. https://doi.org/10.3390/ijms26093953