
New Insights and Implications of Cell–Cell Interactions in Developmental Biology

Abstract
:1. Introduction
2. Research Status of CCIs
2.1. Application of High-Throughput Sequencing Technology
2.2. Databases and Tools Related to CCIs
2.2.1. Database Related to CCIs
2.2.2. CCIs’ Tools and Analysis Process

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool Type | Tool Name | Note |
|---|---|---|
| Ligand–receptor co-expression and differential analysis | Cellphonedb [31], CellChat [89], iTALK [78], NATMI [94], CSOmap [95], NeuronChat [96], scLR [97], scConnect [98], SingleCellSignalR [37], ICELLNET [36], TraSig [99], TimiGP [100], NICHES [101], CCInx [102], Scriabin [103], ScSeqComm [104], Celltalker [105], dsCellNet [106], ScDiffCom [87], LRLoop [107], SPRUCE [108] | We will provide detailed key features and innovative points, as well as website information, for each tool in Supplementary Table S2, respectively. |
| Network analysis | NicheNet [34], Connectome [109], MDIC3 [110], CLARIFY [111], Giotto [26], CellAgentChat [112], cytotalk [113], exFINDER [114], SoptSC [115], SCENIC [116], ProximID [117], FlowSIg [118] | |
| Spatial distance and proximity analysis | SpatialCorr [119], SpaOTsc [120], BATCOM [121], COMMOT [122], SpaTalk [123], STcomm [124], SpaCET [125], CCPLS [126], CINS [127], RNA-Magnet [128], spaCI [129], SpatialDM [88], stLearn [130], MESSI [81], ScHOT [131], Squidpy [132], CellNeighborEX [133] | |
| Traditional machine learning and deep learning | DeepCCI [134], CytoCommunity [135], LR Hunting [136], NetPhosPan [137], GCNG [138], Neighbor-seq [139], HiVAE [140], DeepTalk [141], scMultiSim [142], RobustCCC [143], HoloNet [144], GraphComm [145], DeepLinc [146], CellEnBoost [147], ScTenifoldXct [148], SVCA [149], DIISCO [150], ISCHIA [151] | |
| Metabolic models and energy balance | scFBA [90], scFEA [152], COMPASS [153], MEBOCOST [154], MISTy [155] | |
| Tensor decomposition and matrix decomposition | ScTensor [156], Tensor-cell2cell [157], scITD [158], NCEM [159], DiSiR [160] | |
| TF and signaling pathway analysis | CellCall [46], CellComNet [161], CCCExplorer [83], Cell2cell [162], Commpath [163], ScMLnet [164], FunRes [165], TimeTalk [166], Domino [167], CellComm [168], DcjComm [169] | |
| Multiple combination methods | COMUNET [170], BulkSignalR [171], PyMINEr [172], CrossTalkeR [173], stMLnet [174] | |
| Comparative evaluation of cell communication tools | ESICCC/CCCbank [92], LIANA [91], LIANA+ [93] | |
| Analysis platform | InterCellar [175], TALKIEN [176] | |
| Analyze cell communication under various conditions | MOFAcell [177], DIALOGUE [178] |
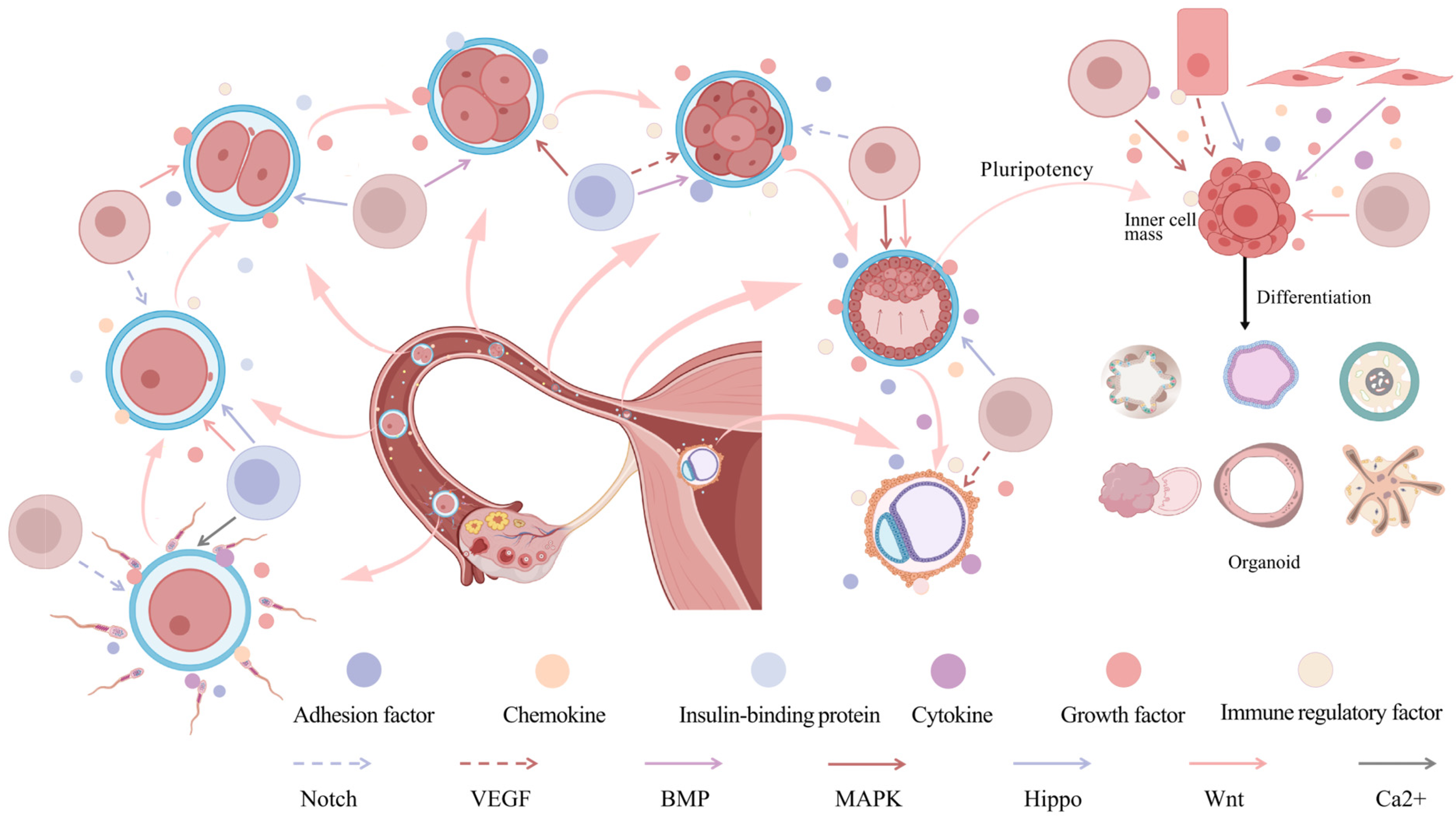
3. Effects of CCIs on Embryonic Development
3.1. Fertilization and Zygotic Genome Activation (ZGA)
3.2. Blastocyst Formation and Pluripotency Maintenance
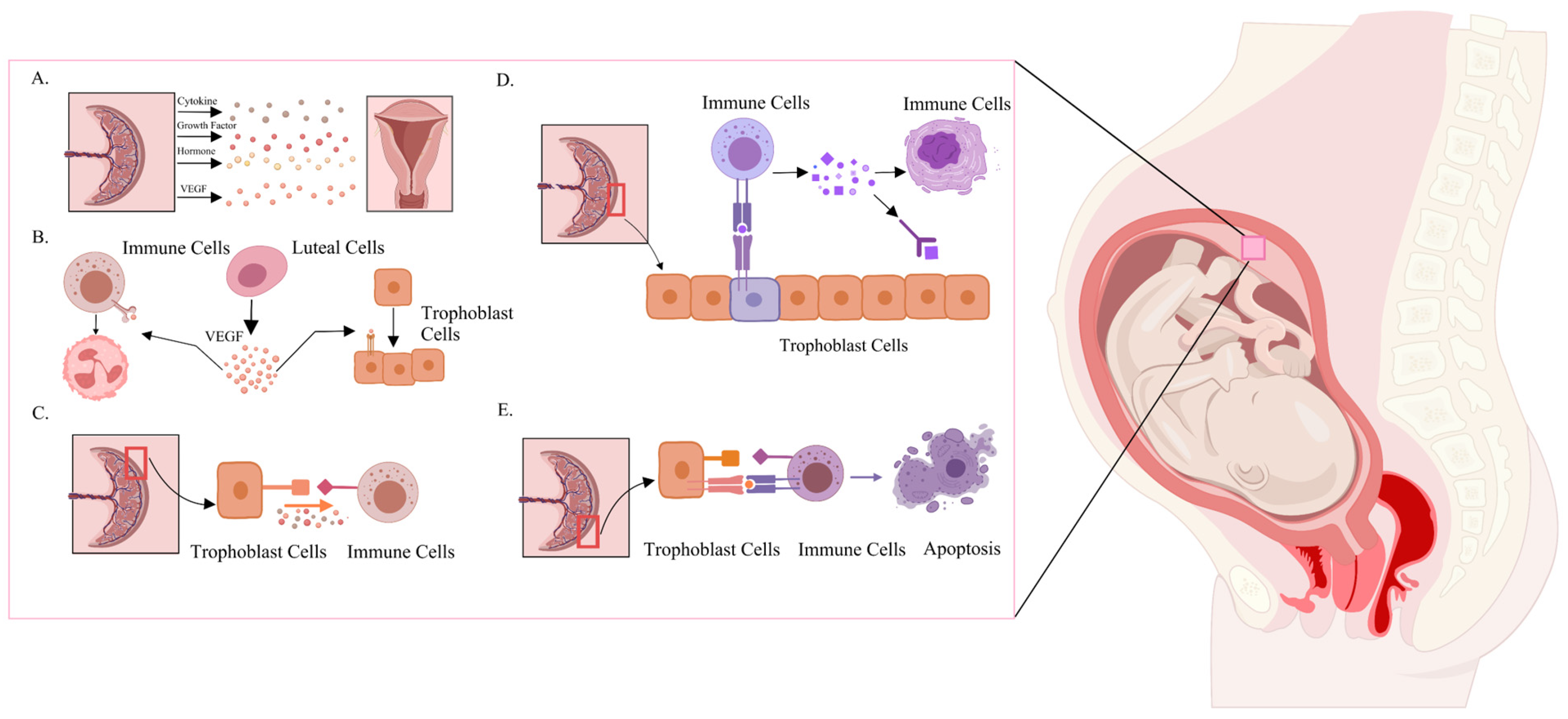
3.3. CCIs Between Implantation and Fetus
4. Challenges and Future Directions
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCIs | Cell–Cell Interactions |
| cAMP | Cyclic Adenosine Monophosphate |
| GFP | Green Fluorescent Protein |
| HGP | Human Genome Project |
| RNAi | RNA Interference |
| bulk RNA-seq | Bulk RNA sequencing |
| scRNA-seq | Single Cell RNA sequencing |
| ST | Spatial Transcriptomics |
| TF | Transcription Factor |
| MOSTA | Mouse Organogenesis Spatiotemporal Transcriptome Atlas |
| ODE | Ordinary Differential Equations |
| ORA | Over-Representation Analysis |
| FISH | Fluorescence In Situ Hybridization |
| ZGA | Zygotic Genome Activation |
| ICM | Inner Cell Mass |
| TE | Trophectoderm |
References
- Grosberg, R.K.; Strathmann, R.R. The evolution of multicellularity: A minor major transition? Annu. Rev. Ecol. Evol. Syst. 2007, 38, 621–654. [Google Scholar] [CrossRef]
- Pires-daSilva, A.; Sommer, R.J. The evolution of signalling pathways in animal development. Nat. Rev. Genet. 2003, 4, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.J. Intercellular communication and cell-cell adhesion. Science 1992, 255, 1671–1677. [Google Scholar] [CrossRef]
- Huang, L.; Sun, J.; Liu, B.H. Cell communication during cellular aging process. Chem. Life 2023, 43, 1169–1178. [Google Scholar] [CrossRef]
- Schwann, T.H. Microscopial researches into the accordance in the structure and growth of animals and plants. 1847. Obes. Res. 1993, 1, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI—Atheromatous affection of arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef]
- Piro, A.; Tagarelli, A.; Tagarelli, G.; Lagonia, P.; Quattrone, A. Paul Ehrlich: The Nobel Prize in physiology or medicine 1908. Int. Rev. Immunol. 2008, 27, 1–17. [Google Scholar] [CrossRef]
- Sutherland, E.W.; Rall, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091. [Google Scholar] [CrossRef]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef]
- Krebs, E.G.; Fischer, E.H. The phosphorylase b to a converting enzyme of rabbit skeletal muscle. Biochim. Biophys. Acta 1956, 20, 150–157. [Google Scholar] [CrossRef]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Shao, X.; Lu, X.; Liao, J.; Chen, H.; Fan, X. New avenues for systematically inferring cell-cell communication: Through single-cell transcriptomics data. Protein Cell 2020, 11, 866–880. [Google Scholar] [CrossRef] [PubMed]
- AlJanahi, A.A.; Danielsen, M.; Dunbar, C.E. An Introduction to the Analysis of Single-Cell RNA-Sequencing Data. Mol. Ther. Methods Clin. Dev. 2018, 10, 189–196. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef]
- Li, Z.; Lin, F.; Zhong, C.H.; Wang, S.; Xue, X.; Shao, Y. Single-Cell Sequencing to Unveil the Mystery of Embryonic Development. Adv. Biol. 2022, 6, e2101151. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Shahan, R.; Hsu, C.W.; Nolan, T.M.; Cole, B.J.; Taylor, I.W.; Greenstreet, L.; Zhang, S.; Afanassiev, A.; Vlot, A.H.C.; Schiebinger, G.; et al. A single-cell Arabidopsis root atlas reveals developmental trajectories in wild-type and cell identity mutants. Dev. Cell 2022, 57, 543–560.e9. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef]
- Du, J.; Yang, Y.C.; An, Z.J.; Zhang, M.H.; Fu, X.H.; Huang, Z.F.; Yuan, Y.; Hou, J. Advances in spatial transcriptomics and related data analysis strategies. J. Transl. Med. 2023, 21, 330. [Google Scholar] [CrossRef]
- Dries, R.; Zhu, Q.; Dong, R.; Eng, C.L.; Li, H.; Liu, K.; Fu, Y.; Zhao, T.; Sarkar, A.; Bao, F.; et al. Giotto: A toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021, 22, 78. [Google Scholar] [CrossRef]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef]
- Gao, J.; Mo, S.; Wang, J.; Zhang, M.; Shi, Y.; Zhu, C.; Shang, Y.; Tang, X.; Zhang, S.; Wu, X.; et al. MACC: A visual interactive knowledgebase of metabolite-associated cell communications. Nucleic Acids Res. 2024, 52, D633–D639. [Google Scholar] [CrossRef]
- Shao, X.; Liao, J.; Li, C.; Lu, X.; Cheng, J.; Fan, X. CellTalkDB: A manually curated database of ligand-receptor interactions in humans and mice. Brief Bioinform. 2021, 22, bbaa269. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Wang, J.; Zou, B.; Li, L.; Yao, L.; Chen, K.; Ning, L.; Wu, B.; Zhao, X.; et al. Cellinker: A platform of ligand-receptor interactions for intercellular communication analysis. Bioinformatics 2021, 37, 2025–2032. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Alterovitz, G.; Xiang, M.; Mohan, M.; Ramoni, M.F. GO PaD: The Gene Ontology Partition Database. Nucleic Acids Res. 2007, 35, D322–D327. [Google Scholar] [CrossRef] [PubMed]
- Noël, F.; Massenet-Regad, L.; Carmi-Levy, I.; Cappuccio, A.; Grandclaudon, M.; Trichot, C.; Kieffer, Y.; Mechta-Grigoriou, F.; Soumelis, V. Dissection of intercellular communication using the transcriptome-based framework ICELLNET. Nat. Commun. 2021, 12, 1089. [Google Scholar] [CrossRef]
- Cabello-Aguilar, S.; Alame, M.; Kon-Sun-Tack, F.; Fau, C.; Lacroix, M.; Colinge, J. SingleCellSignalR: Inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020, 48, e55. [Google Scholar] [CrossRef]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef]
- Graeber, T.G.; Eisenberg, D. Bioinformatic identification of potential autocrine signaling loops in cancers from gene expression profiles. Nat. Genet. 2001, 29, 295–300. [Google Scholar] [CrossRef]
- Noh, J.Y.; Lee, H.I.; Choi, J.H.; Cho, S.H.; Yi, Y.H.; Lim, J.H.; Myung, E.B.; Shin, Y.J.; Shin, H.J.; Woo, H.G. CCIDB: A manually curated cell-cell interaction database with cell context information. Database 2023, 2023, baad057. [Google Scholar] [CrossRef]
- Isserlin, R.; Voisin, V.; Ailles, L.; Bader, G.D. Cell-Cell Interaction Database. Zenodo 2020. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Hodge, M.R.; Horton, W.; Brown, T.; Herrick, R.; Olsen, T.; Hileman, M.E.; McKay, M.; Archie, K.A.; Cler, E.; Harms, M.P.; et al. ConnectomeDB--Sharing human brain connectivity data. Neuroimage 2016, 124, 1102–1107. [Google Scholar] [CrossRef]
- Sharman, J.L.; Mpamhanga, C.P.; Spedding, M.; Germain, P.; Staels, B.; Dacquet, C.; Laudet, V.; Harmar, A.J. IUPHAR-DB: New receptors and tools for easy searching and visualization of pharmacological data. Nucleic Acids Res. 2011, 39, D534–D538. [Google Scholar] [CrossRef]
- Shan, N.; Lu, Y.; Guo, H.; Li, D.; Jiang, J.; Yan, L.; Gao, J.; Ren, Y.; Zhao, X.; Hou, L. CITEdb: A manually curated database of cell-cell interactions in human. Bioinformatics 2022, 38, 5144–5148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Hu, X.; Wang, M.; Wang, J.; Zou, B.; Tan, P.; Cui, T.; Dou, Y.; Ning, L.; et al. CellCall: Integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res. 2021, 49, 8520–8534. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Li, Q.; Zheng, X.; Pan, J. CellCommuNet: An atlas of cell-cell communication networks from single-cell RNA sequencing of human and mouse tissues in normal and disease states. Nucleic Acids Res. 2024, 52, D597–D606. [Google Scholar] [CrossRef]
- Pawson, A.J.; Sharman, J.L.; Benson, H.E.; Faccenda, E.; Alexander, S.P.; Buneman, O.P.; Davenport, A.P.; McGrath, J.C.; Peters, J.A.; Southan, C.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY: An expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014, 42, D1098–D1106. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015, 6, 7866. [Google Scholar] [CrossRef]
- Xu, C.; Ma, D.; Ding, Q.; Zhou, Y.; Zheng, H.L. PlantPhoneDB: A manually curated pan-plant database of ligand-receptor pairs infers cell-cell communication. Plant Biotechnol. J. 2022, 20, 2123–2134. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.S.S.; Rodiger, J.; Comjean, A.; Attrill, H.; Antonazzo, G.; Brown, N.H.; Hu, Y.; Perrimon, N. FlyPhoneDB: An integrated web-based resource for cell-cell communication prediction in Drosophila. Genetics 2022, 220, iyab235. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Zhang, X.; Dai, X.; Huang, J.; Hu, X.; Zhang, J.; Shi, L. InterCellDB: A User-Defined Database for Inferring Intercellular Networks. Adv. Sci. 2022, 9, e2200045. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, I.; Yu Hsu, S.; Rauch, R.; Kowalski, H.W.; Hsueh, A.J. Signaling receptome: A genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci. STKE 2003, 2003, Re9. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulos, G.N.; Klapa, M.I.; Moschonas, N.K. PICKLE 3.0: Enriching the human meta-database with the mouse protein interactome extended via mouse-human orthology. Bioinformatics 2021, 37, 145–146. [Google Scholar] [CrossRef]
- Alonso-López, D.; Campos-Laborie, F.J.; Gutiérrez, M.A.; Lambourne, L.; Calderwood, M.A.; Vidal, M.; De Las Rivas, J. APID database: Redefining protein-protein interaction experimental evidences and binary interactomes. Database 2019, 2019, baz005. [Google Scholar] [CrossRef]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010, 38, D525–D531. [Google Scholar] [CrossRef]
- Rodchenkov, I.; Babur, O.; Luna, A.; Aksoy, B.A.; Wong, J.V.; Fong, D.; Franz, M.; Siper, M.C.; Cheung, M.; Wrana, M.; et al. Pathway Commons 2019 Update: Integration, analysis and exploration of pathway data. Nucleic Acids Res. 2020, 48, D489–D497. [Google Scholar] [CrossRef]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef]
- UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Chatr-Aryamontri, A.; Boucher, L.; Oughtred, R.; Livstone, M.S.; Nixon, J.; Van Auken, K.; Wang, X.; Shi, X.; et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011, 39, D698–D704. [Google Scholar] [CrossRef]
- Almén, M.S.; Nordström, K.J.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef]
- Fahey, M.E.; Bennett, M.J.; Mahon, C.; Jäger, S.; Pache, L.; Kumar, D.; Shapiro, A.; Rao, K.; Chanda, S.K.; Craik, C.S.; et al. GPS-Prot: A web-based visualization platform for integrating host-pathogen interaction data. BMC Bioinform. 2011, 12, 298. [Google Scholar] [CrossRef] [PubMed]
- Orii, N.; Ganapathiraju, M.K. Wiki-pi: A web-server of annotated human protein-protein interactions to aid in discovery of protein function. PLoS ONE 2012, 7, e49029. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.M.; Hoffmann, R.; Valencia, A. iHOP web services. Nucleic Acids Res. 2007, 35, W21–W26. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Kleshchevnikov, V.; Shmatko, A.; Dann, E.; Aivazidis, A.; King, H.W.; Li, T.; Elmentaite, R.; Lomakin, A.; Kedlian, V.; Gayoso, A.; et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 2022, 40, 661–671. [Google Scholar] [CrossRef]
- Shao, X.; Liao, J.; Lu, X.; Xue, R.; Ai, N.; Fan, X. scCATCH: Automatic Annotation on Cell Types of Clusters from Single-Cell RNA Sequencing Data. iScience 2020, 23, 100882. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. scmap: Projection of single-cell RNA-seq data across data sets. Nat. Methods 2018, 15, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef] [PubMed]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The Human Cell Atlas. Elife 2017, 6, e27041. [Google Scholar] [CrossRef]
- Quan, F.; Liang, X.; Cheng, M.; Yang, H.; Liu, K.; He, S.; Sun, S.; Deng, M.; He, Y.; Liu, W.; et al. Annotation of cell types (ACT): A convenient web server for cell type annotation. Genome Med. 2023, 15, 91. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Zheng, L.; Liang, P.; Long, C.; Li, H.; Li, H.; Liang, Y.; He, X.; Xi, Q.; Xing, Y.; Zuo, Y. EmAtlas: A comprehensive atlas for exploring spatiotemporal activation in mammalian embryogenesis. Nucleic Acids Res. 2023, 51, D924–D932. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Zhang, S.; Song, S.; Jiang, C.; Han, G.; Wang, M.; Ajani, J.; Futreal, A.; Wang, L. iTALK: An R Package to Characterize and Illustrate Intercellular Communication. bioRxiv 2019. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Navarro, J.F.; Sjöstrand, J.; Salmén, F.; Lundeberg, J.; Ståhl, P.L. ST Pipeline: An automated pipeline for spatial mapping of unique transcripts. Bioinformatics 2017, 33, 2591–2593. [Google Scholar] [CrossRef]
- Li, D.; Ding, J.; Bar-Joseph, Z. Identifying signaling genes in spatial single-cell expression data. Bioinformatics 2021, 37, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell-cell interactions and communication from gene expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef]
- Choi, H.; Sheng, J.; Gao, D.; Li, F.; Durrans, A.; Ryu, S.; Lee, S.B.; Narula, N.; Rafii, S.; Elemento, O.; et al. Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. Cell Rep. 2015, 10, 1187–1201. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.; Nelson, C.E.; Nabavi, S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinform. 2019, 20, 40. [Google Scholar] [CrossRef]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468.e4. [Google Scholar] [CrossRef] [PubMed]
- Lagger, C.; Ursu, E.; Equey, A.; Avelar, R.A.; Pisco, A.O.; Tacutu, R.; de Magalhães, J.P. scDiffCom: A tool for differential analysis of cell-cell interactions provides a mouse atlas of aging changes in intercellular communication. Nat. Aging 2023, 3, 1446–1461. [Google Scholar] [CrossRef]
- Li, Z.; Wang, T.; Liu, P.; Huang, Y. SpatialDM for rapid identification of spatially co-expressed ligand-receptor and revealing cell-cell communication patterns. Nat. Commun. 2023, 14, 3995. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell-cell communication from single-cell transcriptomics. Nat. Protoc. 2024, 20, 180–219. [Google Scholar] [CrossRef]
- Damiani, C.; Maspero, D.; Di Filippo, M.; Colombo, R.; Pescini, D.; Graudenzi, A.; Westerhoff, H.V.; Alberghina, L.; Vanoni, M.; Mauri, G. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019, 15, e1006733. [Google Scholar] [CrossRef]
- Dimitrov, D.; Türei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Ramirez Flores, R.O.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Deng, M.; Zhang, X.; Sun, X. ESICCC as a systematic computational framework for evaluation, selection, and integration of cell-cell communication inference methods. Genome Res. 2023, 33, 1788–1805. [Google Scholar] [CrossRef]
- Dimitrov, D.; Schäfer, P.S.L.; Farr, E.; Rodriguez-Mier, P.; Lobentanzer, S.; Badia, I.M.P.; Dugourd, A.; Tanevski, J.; Ramirez Flores, R.O.; Saez-Rodriguez, J. LIANA+ provides an all-in-one framework for cell-cell communication inference. Nat. Cell Biol. 2024, 26, 1613–1622. [Google Scholar] [CrossRef]
- Hou, R.; Denisenko, E.; Ong, H.T.; Ramilowski, J.A.; Forrest, A.R.R. Predicting cell-to-cell communication networks using NATMI. Nat. Commun. 2020, 11, 5011. [Google Scholar] [CrossRef]
- Ren, X.; Zhong, G.; Zhang, Q.; Zhang, L.; Sun, Y.; Zhang, Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 2020, 30, 763–778. [Google Scholar] [CrossRef]
- Zhao, W.; Johnston, K.G.; Ren, H.; Xu, X.; Nie, Q. Inferring neuron-neuron communications from single-cell transcriptomics through NeuronChat. Nat. Commun. 2023, 14, 1128. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hsu, C.Y.; Li, J.; Shyr, Y. Dysregulated ligand-receptor interactions from single-cell transcriptomics. Bioinformatics 2022, 38, 3216–3221. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, J.E.T.; Spjuth, O.; Lagerström, M.C. scConnect: A method for exploratory analysis of cell-cell communication based on single-cell RNA-sequencing data. Bioinformatics 2021, 37, 3501–3508. [Google Scholar] [CrossRef]
- Li, D.; Velazquez, J.J.; Ding, J.; Hislop, J.; Ebrahimkhani, M.R.; Bar-Joseph, Z. TraSig: Inferring cell-cell interactions from pseudotime ordering of scRNA-Seq data. Genome Biol. 2022, 23, 73. [Google Scholar] [CrossRef]
- Li, C.; Zhang, B.; Schaafsma, E.; Reuben, A.; Wang, L.; Turk, M.J.; Zhang, J.; Cheng, C. TimiGP: Inferring cell-cell interactions and prognostic associations in the tumor immune microenvironment through gene pairs. Cell Rep. Med. 2023, 4, 101121. [Google Scholar] [CrossRef]
- Raredon, M.S.B.; Yang, J.; Kothapalli, N.; Lewis, W.; Kaminski, N.; Niklason, L.E.; Kluger, Y. Comprehensive visualization of cell-cell interactions in single-cell and spatial transcriptomics with NICHES. Bioinformatics 2023, 39, btac775. [Google Scholar] [CrossRef] [PubMed]
- Ximerakis, M.; Lipnick, S.L.; Innes, B.T.; Simmons, S.K.; Adiconis, X.; Dionne, D.; Mayweather, B.A.; Nguyen, L.; Niziolek, Z.; Ozek, C.; et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019, 22, 1696–1708. [Google Scholar] [CrossRef]
- Wilk, A.J.; Shalek, A.K.; Holmes, S.; Blish, C.A. Comparative analysis of cell-cell communication at single-cell resolution. Nat. Biotechnol. 2024, 42, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Baruzzo, G.; Cesaro, G.; Di Camillo, B. Identify, quantify and characterize cellular communication from single-cell RNA sequencing data with scSeqComm. Bioinformatics 2022, 38, 1920–1929. [Google Scholar] [CrossRef]
- Vahid, M.R.; Brown, E.L.; Steen, C.B.; Zhang, W.; Jeon, H.S.; Kang, M.; Gentles, A.J.; Newman, A.M. High-resolution alignment of single-cell and spatial transcriptomes with CytoSPACE. Nat. Biotechnol. 2023, 41, 1543–1548. [Google Scholar] [CrossRef]
- Song, Z.; Wang, T.; Wu, Y.; Fan, M.; Wu, H. dsCellNet: A new computational tool to infer cell-cell communication networks in the developing and aging brain. Comput. Struct. Biotechnol. J. 2022, 20, 4072–4081. [Google Scholar] [CrossRef]
- Xin, Y.; Lyu, P.; Jiang, J.; Zhou, F.; Wang, J.; Blackshaw, S.; Qian, J. LRLoop: A method to predict feedback loops in cell-cell communication. Bioinformatics 2022, 38, 4117–4126. [Google Scholar] [CrossRef] [PubMed]
- Subedi, S.; Park, Y.P. Single-cell pair-wise relationships untangled by composite embedding model. iScience 2023, 26, 106025. [Google Scholar] [CrossRef]
- Raredon, M.S.B.; Yang, J.; Garritano, J.; Wang, M.; Kushnir, D.; Schupp, J.C.; Adams, T.S.; Greaney, A.M.; Leiby, K.L.; Kaminski, N.; et al. Computation and visualization of cell-cell signaling topologies in single-cell systems data using Connectome. Sci. Rep. 2022, 12, 4187. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Chang, X.; Liu, X. MDIC3: Matrix decomposition to infer cell-cell communication. Patterns 2024, 5, 100911. [Google Scholar] [CrossRef]
- Bafna, M.; Li, H.; Zhang, X. CLARIFY: Cell-cell interaction and gene regulatory network refinement from spatially resolved transcriptomics. Bioinformatics 2023, 39, i484–i493. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, V.; Li, Y.; Ding, J. Harnessing Agent-Based Modeling in CellAgentChat to Unravel Cell-Cell Interactions from Single-Cell Data. bioRxiv 2024. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, T.; Gao, L.; Tan, K. CytoTalk: De novo construction of signal transduction networks using single-cell transcriptomic data. Sci. Adv. 2021, 7, eabf1356. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhou, P.; Nie, Q. exFINDER: Identify external communication signals using single-cell transcriptomics data. Nucleic Acids Res. 2023, 51, e58. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Karikomi, M.; MacLean, A.L.; Nie, Q. Cell lineage and communication network inference via optimization for single-cell transcriptomics. Nucleic Acids Res. 2019, 47, e66. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Boisset, J.C.; Vivié, J.; Grün, D.; Muraro, M.J.; Lyubimova, A.; van Oudenaarden, A. Mapping the physical network of cellular interactions. Nat. Methods 2018, 15, 547–553. [Google Scholar] [CrossRef]
- Almet, A.A.; Tsai, Y.C.; Watanabe, M.; Nie, Q. Inferring pattern-driving intercellular flows from single-cell and spatial transcriptomics. Nat. Methods 2024, 21, 1806–1817. [Google Scholar] [CrossRef]
- Bernstein, M.N.; Ni, Z.; Prasad, A.; Brown, J.; Mohanty, C.; Stewart, R.; Newton, M.A.; Kendziorski, C. SpatialCorr identifies gene sets with spatially varying correlation structure. Cell Rep. Methods 2022, 2, 100369. [Google Scholar] [CrossRef]
- Cang, Z.; Nie, Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 2020, 11, 2084. [Google Scholar] [CrossRef]
- Wu, D.; Gaskins, J.T.; Sekula, M.; Datta, S. Inferring Cell-Cell Communications from Spatially Resolved Transcriptomics Data Using a Bayesian Tweedie Model. Genes 2023, 14, 1368. [Google Scholar] [CrossRef] [PubMed]
- Cang, Z.; Zhao, Y.; Almet, A.A.; Stabell, A.; Ramos, R.; Plikus, M.V.; Atwood, S.X.; Nie, Q. Screening cell-cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 2023, 20, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Li, C.; Yang, H.; Lu, X.; Liao, J.; Qian, J.; Wang, K.; Cheng, J.; Yang, P.; Chen, H.; et al. Knowledge-graph-based cell-cell communication inference for spatially resolved transcriptomic data with SpaTalk. Nat. Commun. 2022, 13, 4429. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Li, W.; Xu, J.; Zhang, R.; Ke, J.; Ren, X.; Meng, X.; Qin, L.; Zhang, J.; Lu, F.; et al. Three-dimensional molecular architecture of mouse organogenesis. Nat. Commun. 2023, 14, 4599. [Google Scholar] [CrossRef]
- Ru, B.; Huang, J.; Zhang, Y.; Aldape, K.; Jiang, P. Estimation of cell lineages in tumors from spatial transcriptomics data. Nat. Commun. 2023, 14, 568. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Hori, H.; Ozaki, H. CCPLS reveals cell-type-specific spatial dependence of transcriptomes in single cells. Bioinformatics 2022, 38, 4868–4877. [Google Scholar] [CrossRef]
- Yuan, Y.; Cosme, C., Jr.; Adams, T.S.; Schupp, J.; Sakamoto, K.; Xylourgidis, N.; Ruffalo, M.; Li, J.; Kaminski, N.; Bar-Joseph, Z. CINS: Cell Interaction Network inference from Single cell expression data. PLoS Comput. Biol. 2022, 18, e1010468. [Google Scholar] [CrossRef]
- Baccin, C.; Al-Sabah, J.; Velten, L.; Helbling, P.M.; Grünschläger, F.; Hernández-Malmierca, P.; Nombela-Arrieta, C.; Steinmetz, L.M.; Trumpp, A.; Haas, S. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 2020, 22, 38–48. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, T.; Yang, B.; Su, J.; Song, Q. spaCI: Deciphering spatial cellular communications through adaptive graph model. Brief Bioinform. 2023, 24, bbac563. [Google Scholar] [CrossRef]
- Pham, D.; Tan, X.; Balderson, B.; Xu, J.; Grice, L.F.; Yoon, S.; Willis, E.F.; Tran, M.; Lam, P.Y.; Raghubar, A.; et al. Robust mapping of spatiotemporal trajectories and cell-cell interactions in healthy and diseased tissues. Nat. Commun. 2023, 14, 7739. [Google Scholar] [CrossRef]
- Ghazanfar, S.; Lin, Y.; Su, X.; Lin, D.M.; Patrick, E.; Han, Z.G.; Marioni, J.C.; Yang, J.Y.H. Investigating higher-order interactions in single-cell data with scHOT. Nat. Methods 2020, 17, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Palla, G.; Spitzer, H.; Klein, M.; Fischer, D.; Schaar, A.C.; Kuemmerle, L.B.; Rybakov, S.; Ibarra, I.L.; Holmberg, O.; Virshup, I.; et al. Squidpy: A scalable framework for spatial omics analysis. Nat. Methods 2022, 19, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kumar, A.; Lövkvist, C.; Palma, A.M.; Martin, P.; Kim, J.; Bhoopathi, P.; Trevino, J.; Fisher, P.; Madan, E.; et al. CellNeighborEX: Deciphering neighbor-dependent gene expression from spatial transcriptomics data. Mol. Syst. Biol. 2023, 19, e11670. [Google Scholar] [CrossRef]
- Yang, W.; Wang, P.; Luo, M.; Cai, Y.; Xu, C.; Xue, G.; Jin, X.; Cheng, R.; Que, J.; Pang, F.; et al. DeepCCI: A deep learning framework for identifying cell-cell interactions from single-cell RNA sequencing data. Bioinformatics 2023, 39, btad596. [Google Scholar] [CrossRef]
- Hu, Y.; Rong, J.; Xu, Y.; Xie, R.; Peng, J.; Gao, L.; Tan, K. Unsupervised and supervised discovery of tissue cellular neighborhoods from cell phenotypes. Nat. Methods 2024, 21, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sha, Y.; Silva, T.C.; Colaprico, A.; Sun, X.; Ban, Y.; Wang, L.; Lehmann, B.D.; Chen, X.S. LR Hunting: A Random Forest Based Cell-Cell Interaction Discovery Method for Single-Cell Gene Expression Data. Front. Genet. 2021, 12, 708835. [Google Scholar] [CrossRef]
- Fenoy, E.; Izarzugaza, J.M.G.; Jurtz, V.; Brunak, S.; Nielsen, M. A generic deep convolutional neural network framework for prediction of receptor-ligand interactions-NetPhosPan: Application to kinase phosphorylation prediction. Bioinformatics 2019, 35, 1098–1107. [Google Scholar] [CrossRef]
- Yuan, Y.; Bar-Joseph, Z. GCNG: Graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 2020, 21, 300. [Google Scholar] [CrossRef]
- Ghaddar, B.; De, S. Reconstructing physical cell interaction networks from single-cell data using Neighbor-seq. Nucleic Acids Res. 2022, 50, e82. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Peng, J.; Shang, X. An improved hierarchical variational autoencoder for cell-cell communication estimation using single-cell RNA-seq data. Brief Funct. Genom. 2024, 23, 118–127. [Google Scholar] [CrossRef]
- Yang, W.; Wang, P.; Xu, S.; Wang, T.; Luo, M.; Cai, Y.; Xu, C.; Xue, G.; Que, J.; Ding, Q.; et al. Deciphering cell-cell communication at single-cell resolution for spatial transcriptomics with subgraph-based graph attention network. Nat. Commun. 2024, 15, 7101. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.; Squires, M.; Chen, X.; Zhang, X. scMultiSim: Simulation of multi-modality single cell data guided by cell-cell interactions and gene regulatory networks. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, L.; Hu, Y.; Huang, Z. RobustCCC: A robustness evaluation tool for cell-cell communication methods. Front. Genet. 2023, 14, 1236956. [Google Scholar] [CrossRef]
- Li, H.; Ma, T.; Hao, M.; Guo, W.; Gu, J.; Zhang, X.; Wei, L. Decoding functional cell-cell communication events by multi-view graph learning on spatial transcriptomics. Brief Bioinform. 2023, 24, bbad359. [Google Scholar] [CrossRef]
- So, E.; Hayat, S.; Nair, S.K.; Wang, B.; Haibe-Kains, B. GraphComm: A Graph-based Deep Learning Method to Predict Cell-Cell Communication in single-cell RNAseq data. bioRxiv 2024. [Google Scholar] [CrossRef]
- Li, R.; Yang, X. De novo reconstruction of cell interaction landscapes from single-cell spatial transcriptome data with DeepLinc. Genome Biol. 2022, 23, 124. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Yuan, R.; Han, C.; Han, G.; Tan, J.; Wang, Z.; Chen, M.; Chen, X. CellEnBoost: A Boosting-Based Ligand-Receptor Interaction Identification Model for Cell-to-Cell Communication Inference. IEEE Trans. Nanobiosci. 2023, 22, 705–715. [Google Scholar] [CrossRef]
- Yang, Y.; Li, G.; Zhong, Y.; Xu, Q.; Lin, Y.T.; Roman-Vicharra, C.; Chapkin, R.S.; Cai, J.J. scTenifoldXct: A semi-supervised method for predicting cell-cell interactions and mapping cellular communication graphs. Cell Syst. 2023, 14, 302–311.e4. [Google Scholar] [CrossRef]
- Arnol, D.; Schapiro, D.; Bodenmiller, B.; Saez-Rodriguez, J.; Stegle, O. Modeling Cell-Cell Interactions from Spatial Molecular Data with Spatial Variance Component Analysis. Cell Rep. 2019, 29, 202–211.e6. [Google Scholar] [CrossRef]
- Park, C.; Mani, S.; Beltran-Velez, N.; Maurer, K.; Huang, T.; Li, S.; Gohil, S.; Livak, K.J.; Knowles, D.A.; Wu, C.J.; et al. A Bayesian framework for inferring dynamic intercellular interactions from time-series single-cell data. Genome Res. 2024, 34, 1384–1396. [Google Scholar] [CrossRef]
- Lafzi, A.; Borrelli, C.; Baghai Sain, S.; Bach, K.; Kretz, J.A.; Handler, K.; Regan-Komito, D.; Ficht, X.; Frei, A.; Moor, A. Identifying Spatial Co-occurrence in Healthy and InflAmed tissues (ISCHIA). Mol. Syst. Biol. 2024, 20, 98–119. [Google Scholar] [CrossRef]
- Alghamdi, N.; Chang, W.; Dang, P.; Lu, X.; Wan, C.; Gampala, S.; Huang, Z.; Wang, J.; Ma, Q.; Zang, Y.; et al. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome Res. 2021, 31, 1867–1884. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Wang, C.; Fessler, J.; DeTomaso, D.; Avila-Pacheco, J.; Kaminski, J.; Zaghouani, S.; Christian, E.; Thakore, P.; Schellhaass, B.; et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 2021, 184, 4168–4185.e21. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Gao, X.; Wang, B.; Wu, H.; Arulsamy, K.; Dong, Y.; Xiao, Y.; Jiang, X.; Malovichko, M.V.; Li, K.; et al. Epsin Nanotherapy Regulates Cholesterol Transport to Fortify Atheroma Regression. Circ. Res. 2023, 132, e22–e42. [Google Scholar] [CrossRef]
- Tanevski, J.; Flores, R.O.R.; Gabor, A.; Schapiro, D.; Saez-Rodriguez, J. Explainable multiview framework for dissecting spatial relationships from highly multiplexed data. Genome Biol. 2022, 23, 97. [Google Scholar] [CrossRef] [PubMed]
- Tsuyuzaki, K.; Ishii, M.; Nikaido, I. Sctensor detects many-to-many cell-cell interactions from single cell RNA-sequencing data. BMC Bioinform. 2023, 24, 420. [Google Scholar] [CrossRef]
- Armingol, E.; Baghdassarian, H.M.; Martino, C.; Perez-Lopez, A.; Aamodt, C.; Knight, R.; Lewis, N.E. Context-aware deconvolution of cell-cell communication with Tensor-cell2cell. Nat. Commun. 2022, 13, 3665. [Google Scholar] [CrossRef]
- Mitchel, J.; Gordon, M.G.; Perez, R.K.; Biederstedt, E.; Bueno, R.; Ye, C.J.; Kharchenko, P.V. Coordinated, multicellular patterns of transcriptional variation that stratify patient cohorts are revealed by tensor decomposition. Nat. Biotechnol. 2024, 39313646. [Google Scholar] [CrossRef]
- Fischer, D.S.; Schaar, A.C.; Theis, F.J. Modeling intercellular communication in tissues using spatial graphs of cells. Nat. Biotechnol. 2023, 41, 332–336. [Google Scholar] [CrossRef]
- Lenczner, G.; Le Saux, B.; Luminari, N.; Chan-Hon-Tong, A.; Le Besnerais, G. DISIR: Deep image segmentation with interactive refinement. arXiv 2020. [Google Scholar] [CrossRef]
- Peng, L.; Tan, J.; Xiong, W.; Zhang, L.; Wang, Z.; Yuan, R.; Li, Z.; Chen, X. Deciphering ligand-receptor-mediated intercellular communication based on ensemble deep learning and the joint scoring strategy from single-cell transcriptomic data. Comput. Biol. Med. 2023, 163, 107137. [Google Scholar] [CrossRef] [PubMed]
- Armingol, E.; Ghaddar, A.; Joshi, C.J.; Baghdassarian, H.; Shamie, I.; Chan, J.; Her, H.L.; Berhanu, S.; Dar, A.; Rodriguez-Armstrong, F.; et al. Inferring a spatial code of cell-cell interactions across a whole animal body. PLoS Comput. Biol. 2022, 18, e1010715. [Google Scholar] [CrossRef]
- Lu, H.; Ping, J.; Zhou, G.; Zhao, Z.; Gao, W.; Jiang, Y.; Quan, C.; Lu, Y.; Zhou, G. CommPath: An R package for inference and analysis of pathway-mediated cell-cell communication chain from single-cell transcriptomics. Comput. Struct. Biotechnol. J. 2022, 20, 5978–5983. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, J.; Wu, Z.; Sun, X. Inferring microenvironmental regulation of gene expression from single-cell RNA sequencing data using scMLnet with an application to COVID-19. Brief Bioinform. 2021, 22, 988–1005. [Google Scholar] [CrossRef]
- Jung, S.; Singh, K.; Del Sol, A. FunRes: Resolving tissue-specific functional cell states based on a cell-cell communication network model. Brief Bioinform. 2021, 22, bbaa283. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, Y.; Sun, Y.; Mao, S.; Li, H.; Bo, X.; Li, C.; Chen, H. TimeTalk uses single-cell RNA-seq datasets to decipher cell-cell communication during early embryo development. Commun. Biol. 2023, 6, 901. [Google Scholar] [CrossRef] [PubMed]
- Cherry, C.; Maestas, D.R.; Han, J.; Andorko, J.I.; Cahan, P.; Fertig, E.J.; Garmire, L.X.; Elisseeff, J.H. Computational reconstruction of the signalling networks surrounding implanted biomaterials from single-cell transcriptomics. Nat. Biomed. Eng. 2021, 5, 1228–1238. [Google Scholar] [CrossRef]
- Lummertz da Rocha, E.; Kubaczka, C.; Sugden, W.W.; Najia, M.A.; Jing, R.; Markel, A.; LeBlanc, Z.C.; Dos Santos Peixoto, R.; Falchetti, M.; Collins, J.J.; et al. CellComm infers cellular crosstalk that drives haematopoietic stem and progenitor cell development. Nat. Cell Biol. 2022, 24, 579–589. [Google Scholar] [CrossRef]
- Ding, Q.; Yang, W.; Xue, G.; Liu, H.; Cai, Y.; Que, J.; Jin, X.; Luo, M.; Pang, F.; Yang, Y.; et al. Dimension reduction, cell clustering, and cell-cell communication inference for single-cell transcriptomics with DcjComm. Genome Biol. 2024, 25, 241. [Google Scholar] [CrossRef]
- Solovey, M.; Scialdone, A. COMUNET: A tool to explore and visualize intercellular communication. Bioinformatics 2020, 36, 4296–4300. [Google Scholar] [CrossRef]
- Villemin, J.P.; Bassaganyas, L.; Pourquier, D.; Boissière, F.; Cabello-Aguilar, S.; Crapez, E.; Tanos, R.; Cornillot, E.; Turtoi, A.; Colinge, J. Inferring ligand-receptor cellular networks from bulk and spatial transcriptomic datasets with BulkSignalR. Nucleic Acids Res. 2023, 51, 4726–4744. [Google Scholar] [CrossRef] [PubMed]
- Tyler, S.R.; Rotti, P.G.; Sun, X.; Yi, Y.; Xie, W.; Winter, M.C.; Flamme-Wiese, M.J.; Tucker, B.A.; Mullins, R.F.; Norris, A.W.; et al. PyMINEr Finds Gene and Autocrine-Paracrine Networks from Human Islet scRNA-Seq. Cell Rep. 2019, 26, 1951–1964.e8. [Google Scholar] [CrossRef]
- Nagai, J.S.; Leimkühler, N.B.; Schaub, M.T.; Schneider, R.K.; Costa, I.G. CrossTalkeR: Analysis and visualization of ligand-receptorne tworks. Bioinformatics 2021, 37, 4263–4265. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Yan, L.; Nie, Q.; Sun, X. Modeling and inference of spatial intercellular communications and multilayer signaling regulations using stMLnet. bioRxiv 2023. [Google Scholar] [CrossRef]
- Interlandi, M.; Kerl, K.; Dugas, M. InterCellar enables interactive analysis and exploration of cell-cell communication in single-cell transcriptomic data. Commun. Biol. 2022, 5, 21. [Google Scholar] [CrossRef]
- Moratalla-Navarro, F.; Moreno, V.; Sanz-Pamplona, R. TALKIEN: crossTALK IntEraction Network. A web-based tool for deciphering molecular communication through ligand-receptor interactions. Mol. Omics 2023, 19, 688–696. [Google Scholar] [CrossRef]
- Ramirez Flores, R.O.; Lanzer, J.D.; Dimitrov, D.; Velten, B.; Saez-Rodriguez, J. Multicellular factor analysis of single-cell data for a tissue-centric understanding of disease. Elife 2023, 12, e93161. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Regev, A. DIALOGUE maps multicellular programs in tissue from single-cell or spatial transcriptomics data. Nat. Biotechnol. 2022, 40, 1467–1477. [Google Scholar] [CrossRef]
- Deneke, V.E.; Blaha, A.; Lu, Y.; Suwita, J.P.; Draper, J.M.; Phan, C.S.; Panser, K.; Schleiffer, A.; Jacob, L.; Humer, T.; et al. A conserved fertilization complex bridges sperm and egg in vertebrates. Cell 2024, 187, 7066–7078.e22. [Google Scholar] [CrossRef]
- Liu, B.; Xu, Q.; Wang, Q.; Feng, S.; Lai, F.; Wang, P.; Zheng, F.; Xiang, Y.; Wu, J.; Nie, J.; et al. The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 2020, 587, 139–144. [Google Scholar] [CrossRef]
- Li, H.; Long, C.; Xiang, J.; Liang, P.; Li, X.; Zuo, Y. Dppa2/4 as a trigger of signaling pathways to promote zygote genome activation by binding to CG-rich region. Brief Bioinform. 2021, 22, bbaa342. [Google Scholar] [CrossRef] [PubMed]
- Vastenhouw, N.L.; Cao, W.X.; Lipshitz, H.D. The maternal-to-zygotic transition revisited. Development 2019, 146, dev161471. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, B.; Haider, S.; Meinhardt, G.; Pollheimer, J.; Knöfler, M. WNT and NOTCH signaling in human trophoblast development and differentiation. Cell Mol. Life Sci. 2022, 79, 292. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guan, K.L. Hippo Signaling in Embryogenesis and Development. Trends Biochem. Sci. 2021, 46, 51–63. [Google Scholar] [CrossRef]
- Li, L.; Lai, F.; Hu, X.; Liu, B.; Lu, X.; Lin, Z.; Liu, L.; Xiang, Y.; Frum, T.; Halbisen, M.A.; et al. Multifaceted SOX2-chromatin interaction underpins pluripotency progression in early embryos. Science 2023, 382, eadi5516. [Google Scholar] [CrossRef]
- Lai, F.; Li, L.; Hu, X.; Liu, B.; Zhu, Z.; Liu, L.; Fan, Q.; Tian, H.; Xu, K.; Lu, X.; et al. NR5A2 connects zygotic genome activation to the first lineage segregation in totipotent embryos. Cell Res. 2023, 33, 952–966. [Google Scholar] [CrossRef]
- Suryawanshi, H.; Morozov, P.; Straus, A.; Sahasrabudhe, N.; Max, K.E.A.; Garzia, A.; Kustagi, M.; Tuschl, T.; Williams, Z. A single-cell survey of the human first-trimester placenta and decidua. Sci. Adv. 2018, 4, eaau4788. [Google Scholar] [CrossRef]
- Ruane, P.T.; Garner, T.; Parsons, L.; Babbington, P.A.; Wangsaputra, I.; Kimber, S.J.; Stevens, A.; Westwood, M.; Brison, D.R.; Aplin, J.D. Trophectoderm differentiation to invasive syncytiotrophoblast is promoted by endometrial epithelial cells during human embryo implantation. Hum. Reprod. 2022, 37, 777–792. [Google Scholar] [CrossRef]
- Barrientos, G.; Freitag, N.; Tirado-González, I.; Unverdorben, L.; Jeschke, U.; Thijssen, V.L.; Blois, S.M. Involvement of galectin-1 in reproduction: Past, present and future. Hum. Reprod. Update 2014, 20, 175–193. [Google Scholar] [CrossRef]
- Nowak, D.G.; Amin, E.M.; Rennel, E.S.; Hoareau-Aveilla, C.; Gammons, M.; Damodoran, G.; Hagiwara, M.; Harper, S.J.; Woolard, J.; Ladomery, M.R.; et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J. Biol. Chem. 2010, 285, 5532–5540. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, Q.; Shao, X.; Zhang, T.; Xue, C.; Shi, S.; Zhao, D.; Lin, Y. IGF-1 promotes angiogenesis in endothelial cells/adipose-derived stem cells co-culture system with activation of PI3K/Akt signal pathway. Cell Prolif. 2017, 50, e12390. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zhang, X.D.; Miao, W.Y.; Sun, Y.J.; Xiong, G.; Wu, Q.; Li, G.; Yang, P.; Yu, H.; Li, H.; et al. PDGFRβ Cells Rapidly Relay Inflammatory Signal from the Circulatory System to Neurons via Chemokine CCL2. Neuron 2018, 100, 183–200.e8. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ong, J.; Meng, F.; Zhang, F.; Shen, H.; Kitt, K.; Liu, T.; Tao, W.; Du, P. Spatiotemporal insight into early pregnancy governed by immune-featured stromal cells. Cell 2023, 186, 4271–4288.e24. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Zhang, D.; Wang, W.; Ding, Y.; Wang, Y.; Gu, S.; Shang, Y.; Gan, J.; Jiang, L.; Meng, F.; et al. Cross-Species Insights into Trophoblast Invasion During Placentation Governed by Immune-Featured Trophoblast Cells. Adv. Sci. 2024, 11, e2407221. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Bondareva, O.; Guhathakurta, S.; Tsang, T.H.; Sikora, K.; Aizarani, N.; Sagar; Holz, H.; Grün, D.; Hein, L.; et al. Systematic Identification of Cell-Cell Communication Networks in the Developing Brain. iScience 2019, 21, 273–287. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Armingol, E.; Baghdassarian, H.M.; Lewis, N.E. The diversification of methods for studying cell-cell interactions and communication. Nat. Rev. Genet. 2024, 25, 381–400. [Google Scholar] [CrossRef]

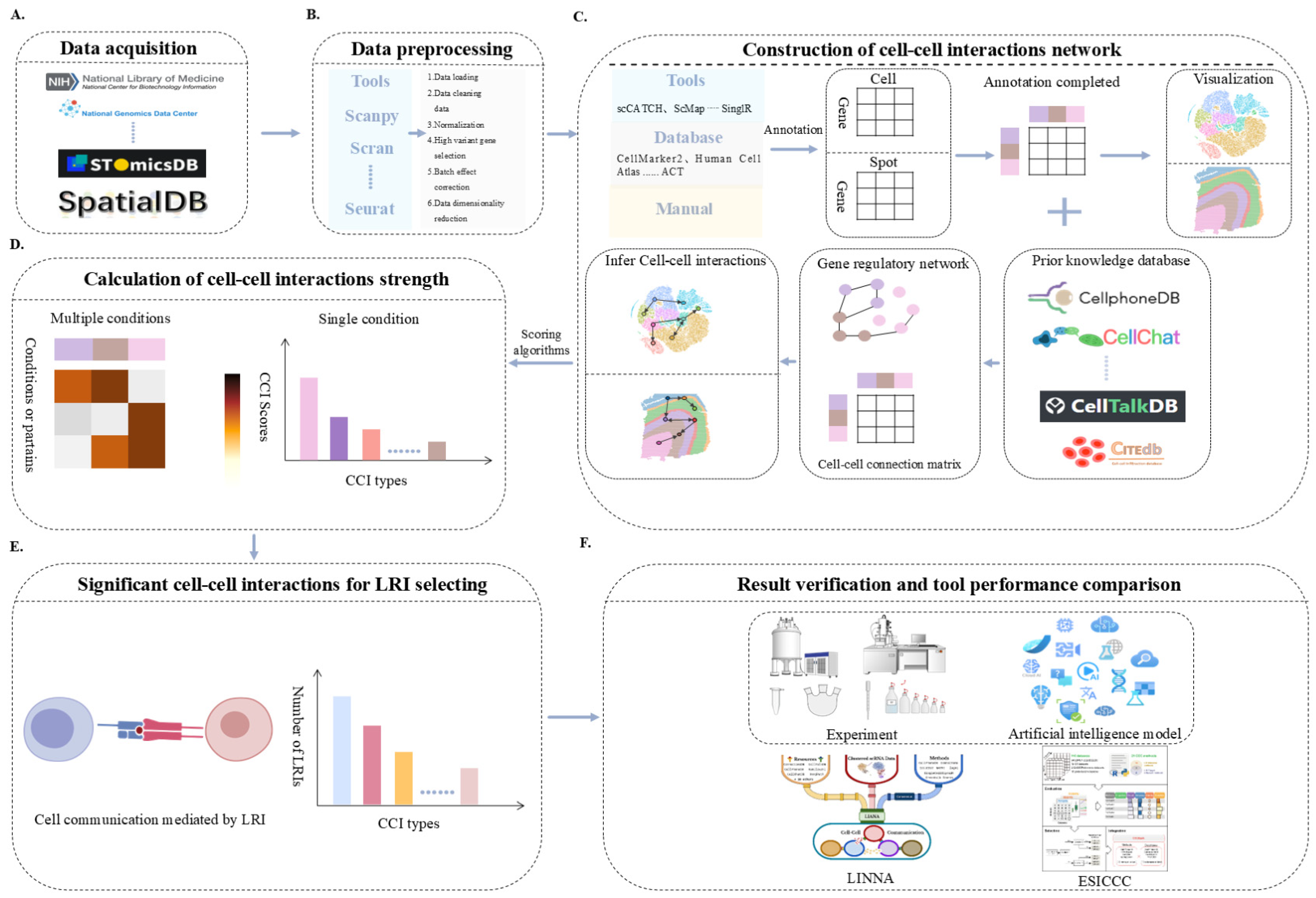
| Data Type | Database Name | Note |
|---|---|---|
| Ligand–receptor pair database | celltalkDB [29], cellphoneDB [31], KEGG [32], cellchatDB [30], Cellinker [33], NicheNet [34], Gene Ontology [35], ICELLNET [36], singlecellsignalR [37], Omnipath [38], DLRP [39], CCIDB [40], Cell-Cell Interaction Database [41], Reactome [42], connectomeDB [43], IUPHAR-DB [44], CITEdb [45], Cellcall [46], CellCommuNet [47], IUPHAR/BPS Guide to Pharmacology [48], A draft network of ligand–receptor-mediated multicellular signalling in human [49], PlantPhoneDB [50], FlyPhoneDB [51], InterCellDB [52] | We will provide detailed key features and innovative points, as well as website information, for each database in Supplementary Table S1, respectively. |
| Protein–protein interaction database | HPRD [53], HPMR [54], PICKLE [55], APID [56], IntAct [57], Pathway Commons [58], The Human Protein Atlas(HPA) [59], UniProt [60], STRING [61], BioGRID [62], Mapping the human membrane proteome [63], GPS-prot [64], Wiki-pi [65], iHOP [66] | |
| Metabolite database | MACC [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.; Liang, Y.; Xi, Q.; Zuo, Y. New Insights and Implications of Cell–Cell Interactions in Developmental Biology. Int. J. Mol. Sci. 2025, 26, 3997. https://doi.org/10.3390/ijms26093997
Wu G, Liang Y, Xi Q, Zuo Y. New Insights and Implications of Cell–Cell Interactions in Developmental Biology. International Journal of Molecular Sciences. 2025; 26(9):3997. https://doi.org/10.3390/ijms26093997
Chicago/Turabian StyleWu, Guanhao, Yuchao Liang, Qilemuge Xi, and Yongchun Zuo. 2025. "New Insights and Implications of Cell–Cell Interactions in Developmental Biology" International Journal of Molecular Sciences 26, no. 9: 3997. https://doi.org/10.3390/ijms26093997
APA StyleWu, G., Liang, Y., Xi, Q., & Zuo, Y. (2025). New Insights and Implications of Cell–Cell Interactions in Developmental Biology. International Journal of Molecular Sciences, 26(9), 3997. https://doi.org/10.3390/ijms26093997