
Transient Inflammation of Pancreatic Exocrine Tissue in Autoimmune Diabetes Follows Onset of Islet Damage and Utilizes Heparanase-1
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
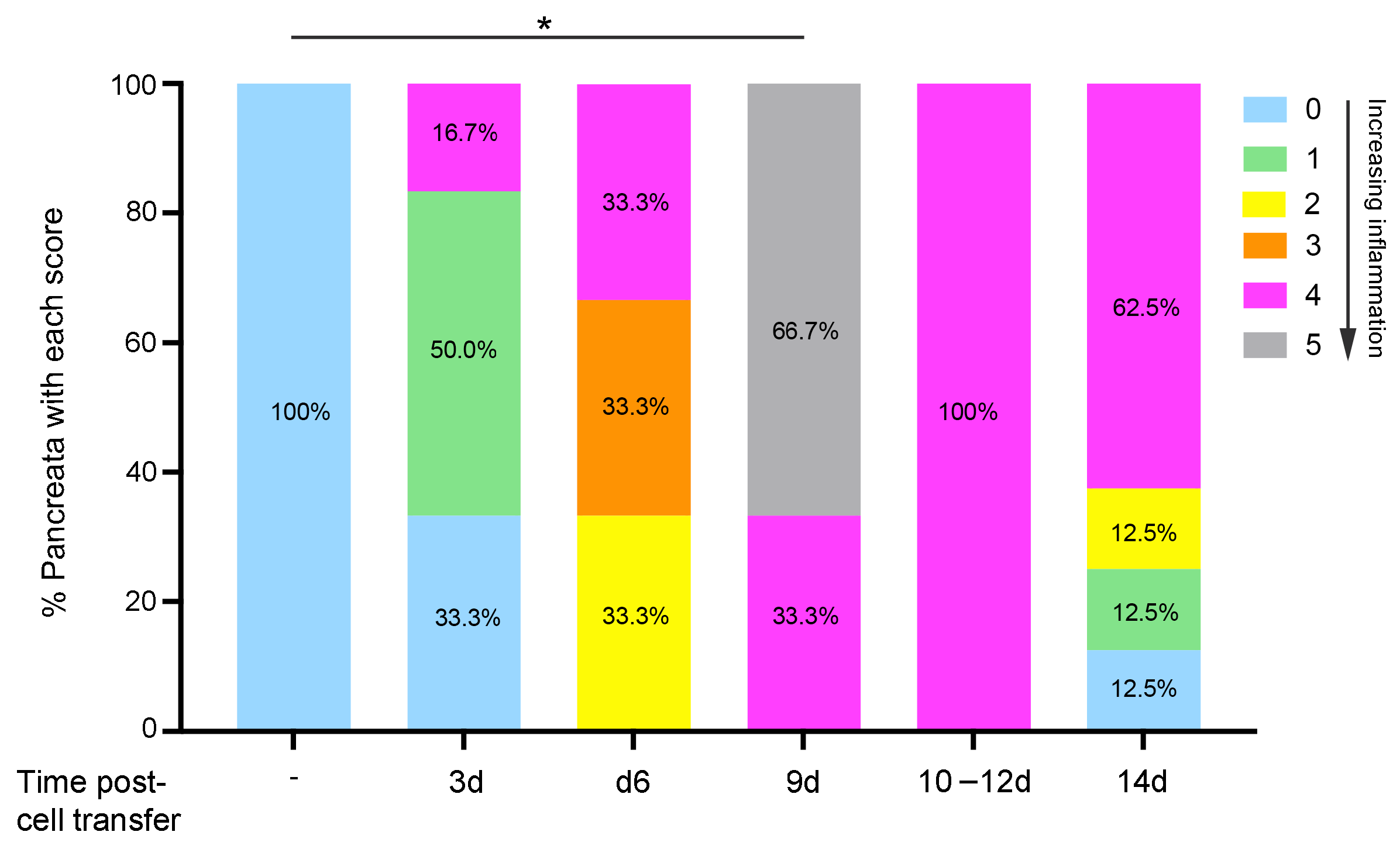
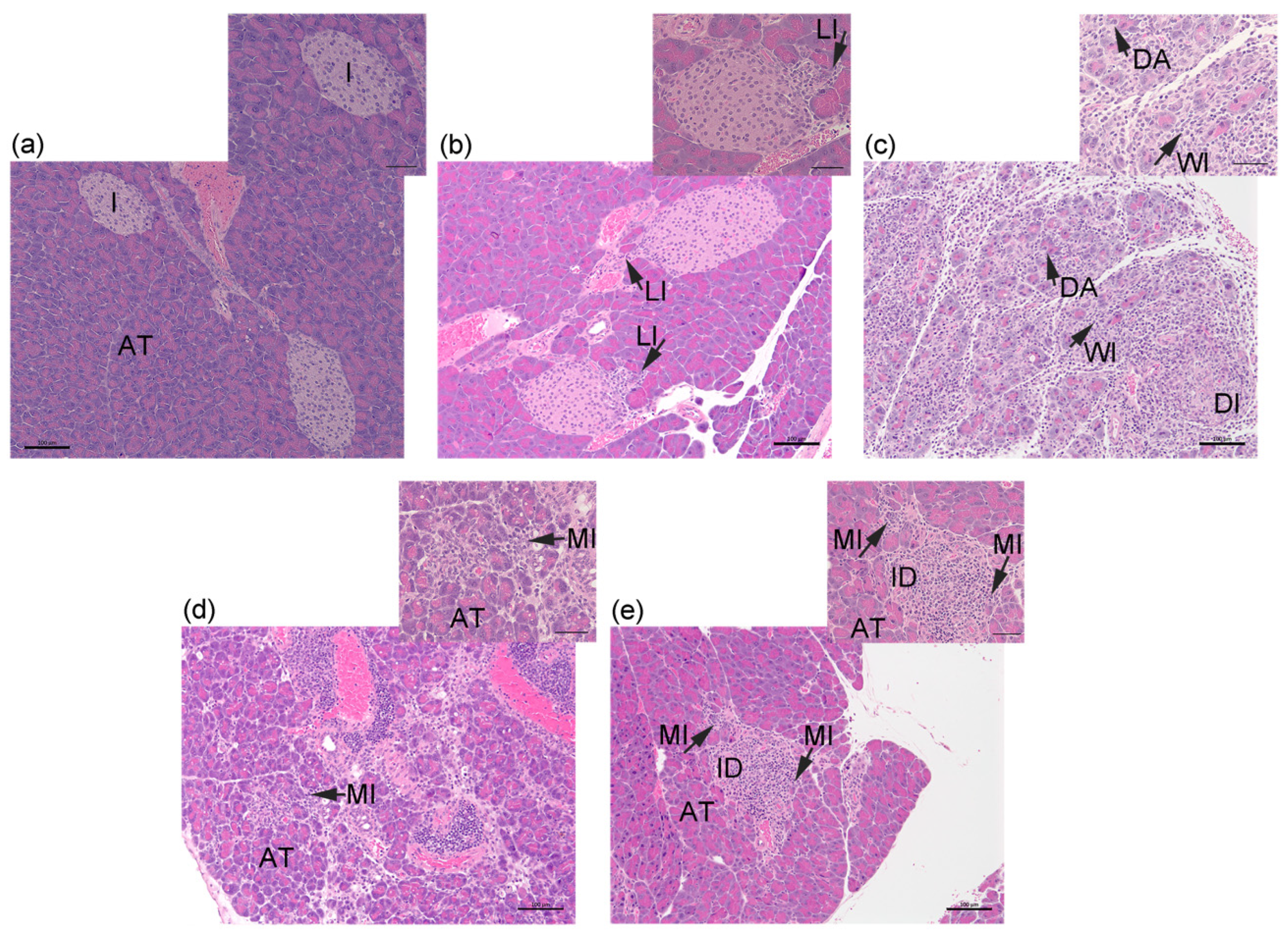
2.1. Transfer of Islet Antigen-Specific T Cells Induces Acute Inflammation in the Exocrine Pancreas of RIP-OVAhi Mice
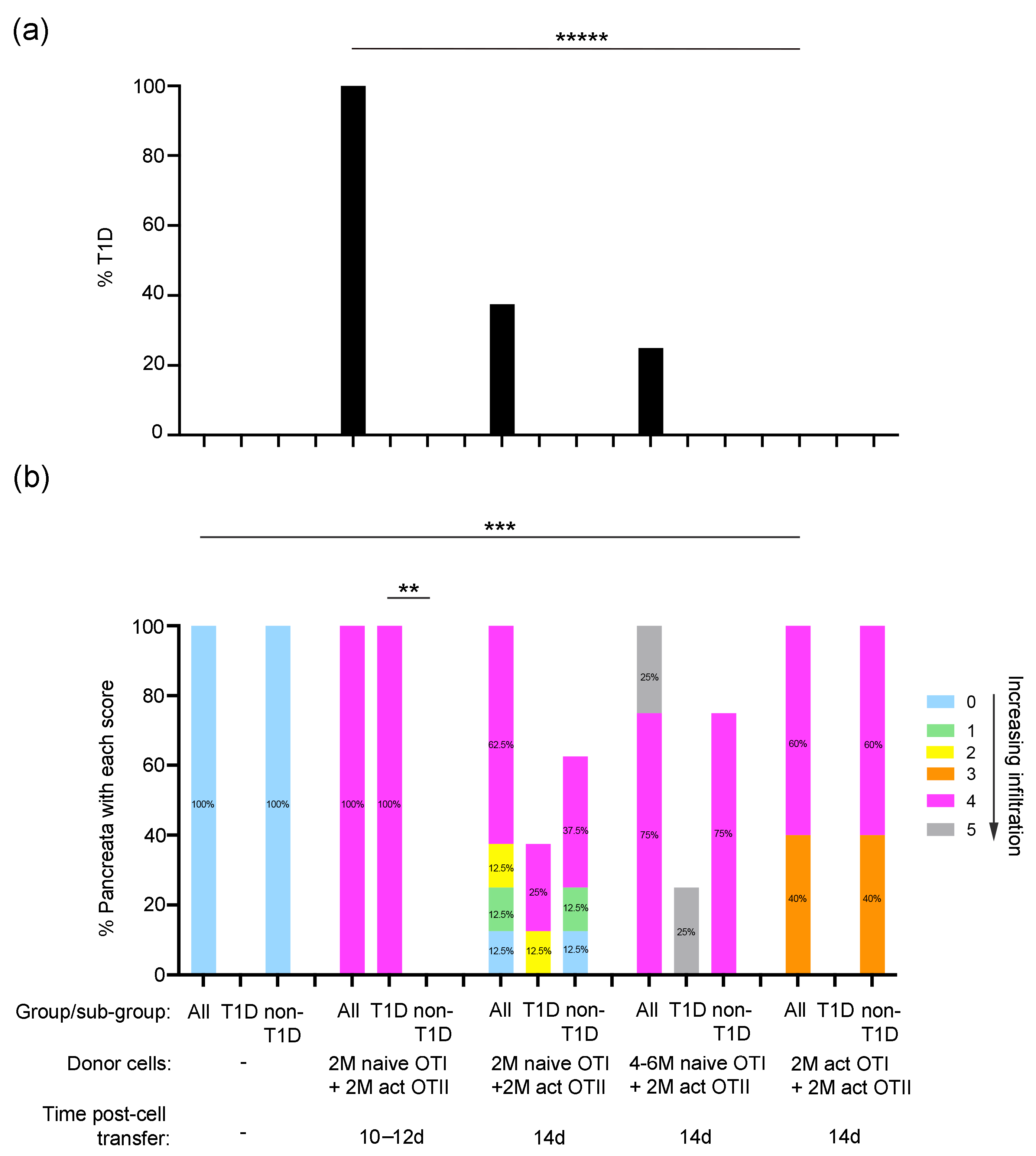
2.2. Properties of Donor Islet Antigen-Specific T Cells That Induce Pancreas Inflammation and Diabetes in RIP-OVAhi Mice
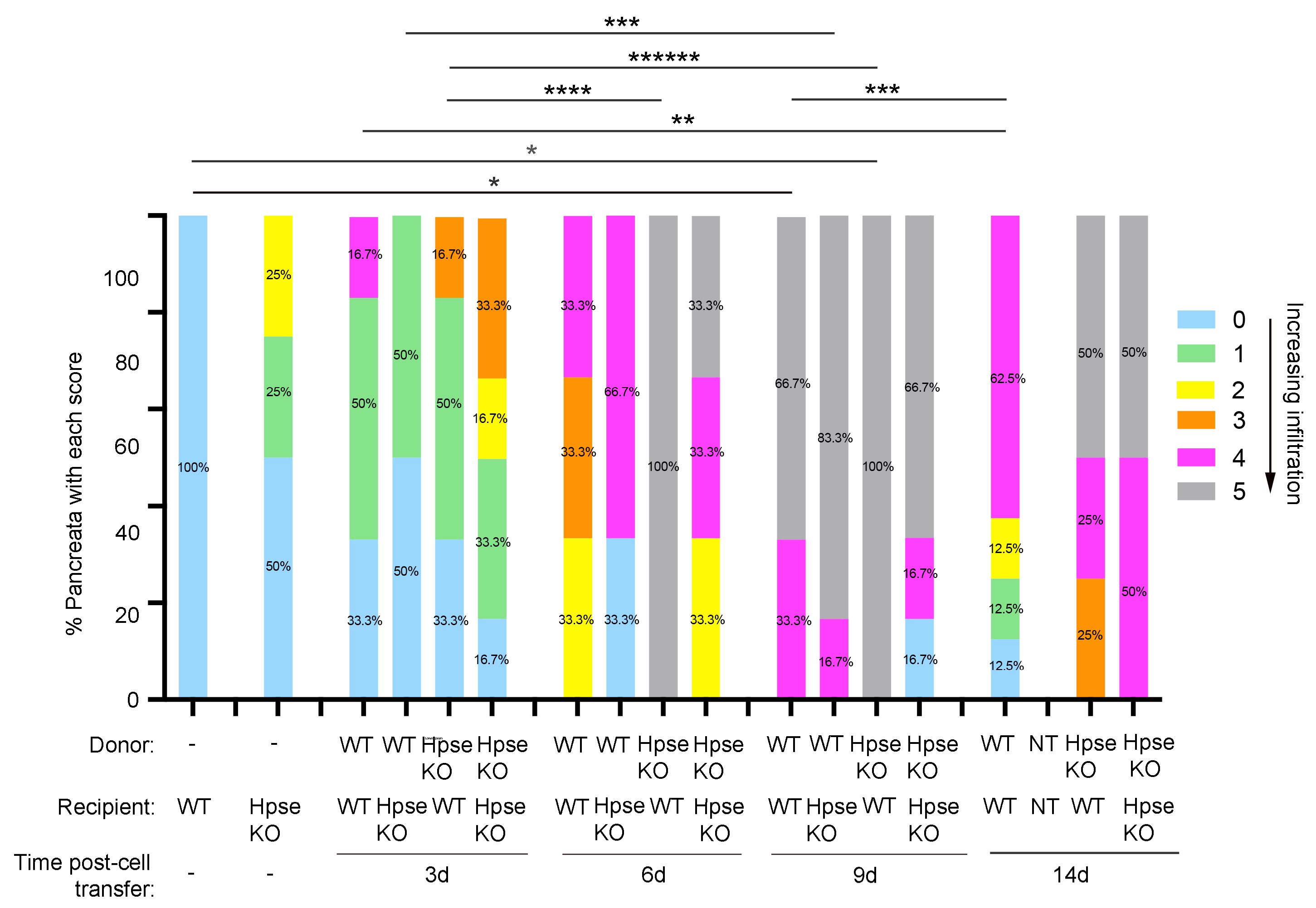
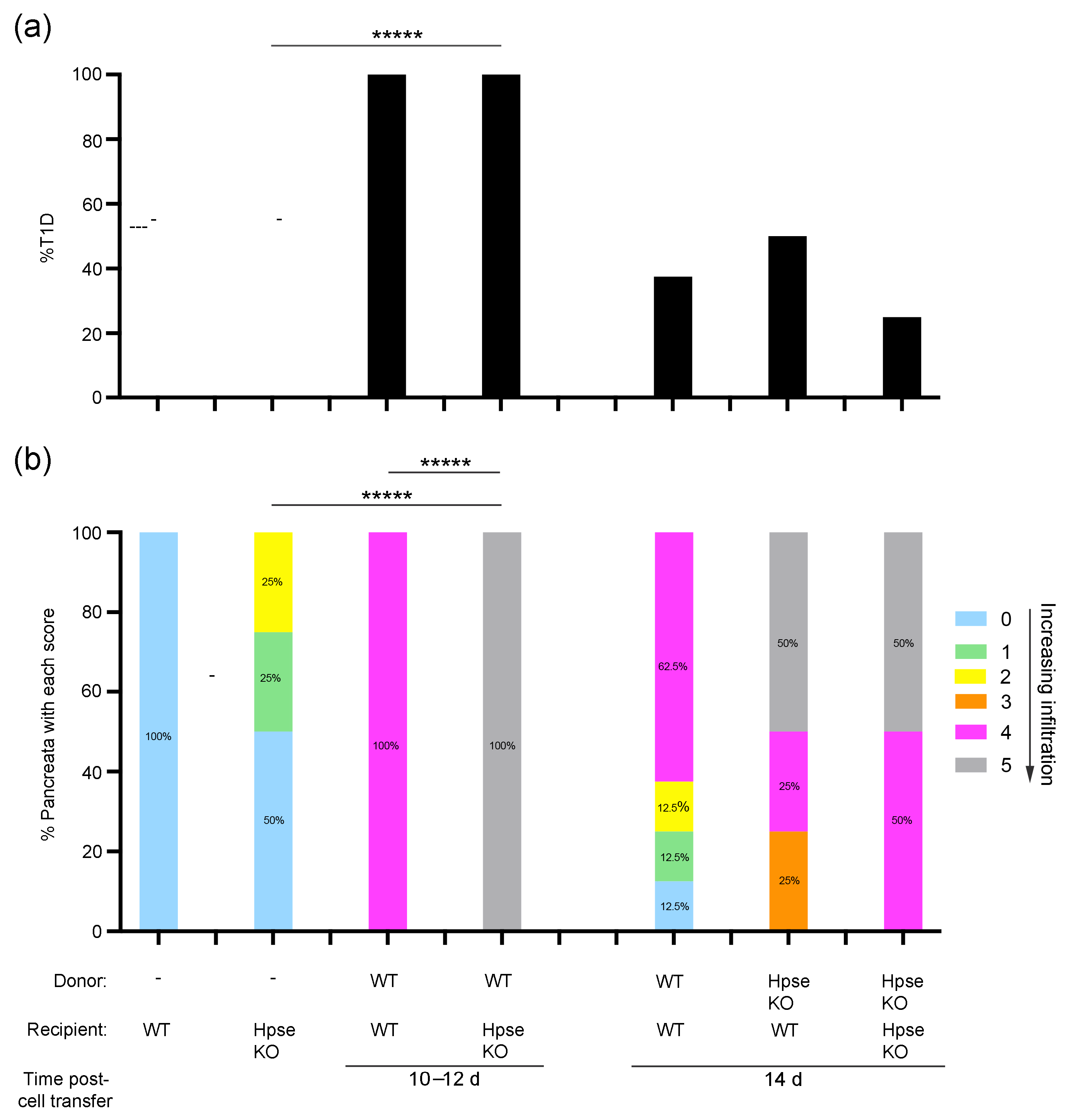
2.3. Destructive Exocrine Tissue Inflammation in RIP-OVAhi Mice Preferentially Utilizes Recipient HPSE-1
2.4. Donor HPSE-1 Plays a Dominant Role in the Induction of Diabetes in RIP-OVAhi Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Diabetes Induction
4.3. Flow Cytometry
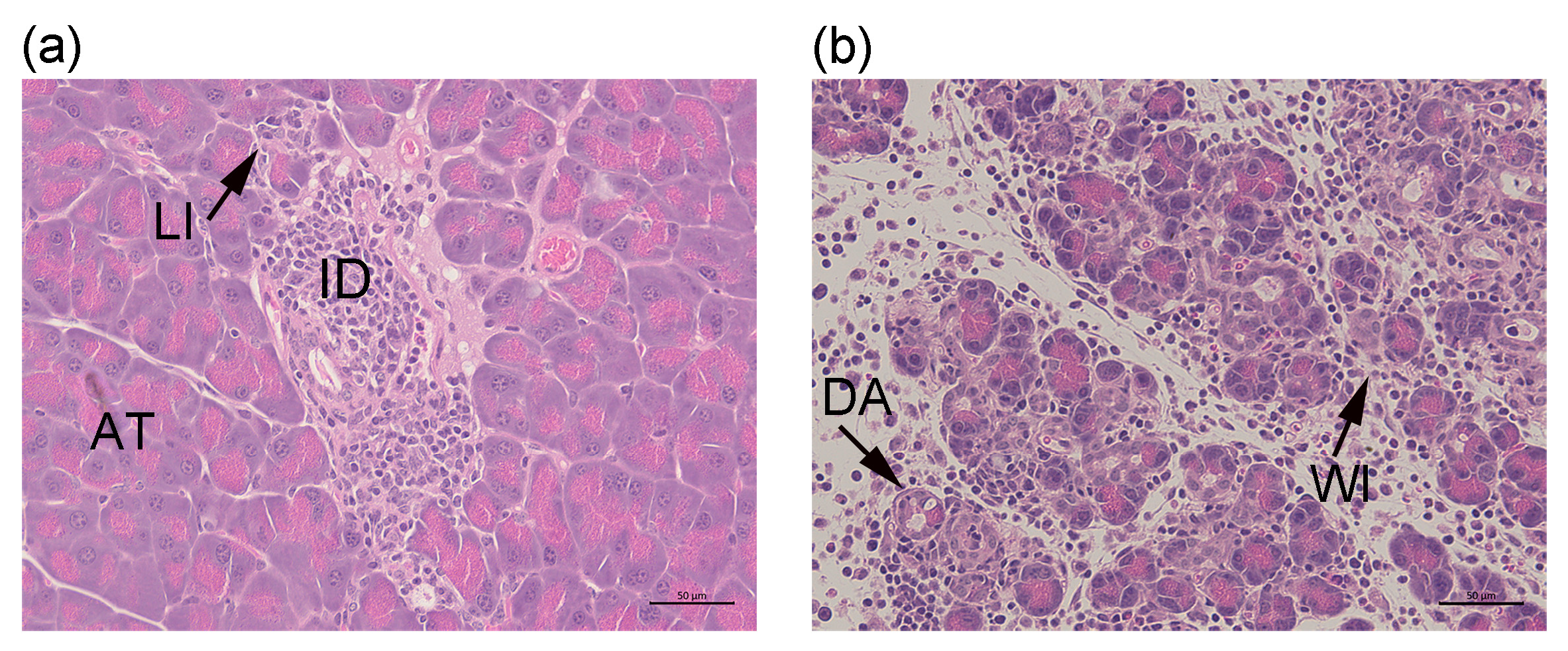
4.4. Histology and Assessment of Pancreas Inflammation
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foster, T.P.; Bruggeman, B.; Campbell-Thompson, M.; Atkinson, M.A.; Haller, M.J.; Schatz, D.A. Exocrine pancreas dysfunction in Type 1 diabetes. Endocr. Pract. 2020, 26, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Alexandre-Heymann, L.; Mallone, R.; Boitard, C.; Scharfmann, R.; Larger, E. Structure and function of the exocrine pancreas in patients with type 1 diabetes. Rev. Endocr. Metab. Disord. 2019, 20, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.; Rodriguez-Calvo, T.; Battaglia, M. Abnormalities of the exocrine pancreas in Type 1 diabetes. Curr. Diabetes Rep. 2015, 15, 79. [Google Scholar] [CrossRef]
- Dozio, N.; Indirli, R.; Giamporcaro, G.M.; Frosio, L.; Mandelli, A.; Laurenzi, A.; Bolla, A.M.; Stabilini, A.; Valle, A.; Locatelli, M.; et al. Impaired exocrine pancreatic function in different stages of type 1 diabetes. BMJ Open Diabetes Res. Care 2021, 9, e001158. [Google Scholar] [CrossRef] [PubMed]
- Giovenzana, A.; Vecchio, F.; Cugnata, F.; Nonis, A.; Mandelli, A.; Stabilini, A.; Mazzi, B.A.; De Pellegrin, M.; Laurenzi, A.; Bonfanti, R.; et al. Exocrine pancreas function is impaired in adult relatives of patients with type 1 diabetes. Acta Diabetol. 2022, 59, 473–479. [Google Scholar] [CrossRef]
- Valikangas, T.; Lietzen, N.; Jaakkola, M.K.; Krogvold, L.; Eike, M.C.; Kallionpaa, H.; Tuomela, S.; Mathews, C.; Gerling, I.C.; Oikarinen, S.; et al. Pancreas whole tissue transcriptomics highlights the role of the exocrine pancreas in patients with recently diagnosed Type 1 diabetes. Front. Endocrinol. 2022, 13, 861985. [Google Scholar] [CrossRef]
- Virostko, J.; Williams, J.; Hilmes, M.; Bowman, C.; Wright, J.J.; Du, L.; Kang, H.; Russell, W.E.; Powers, A.C.; Moore, D.J. Pancreas volume declines during the first year after diagnosis of Type 1 diabetes and exhibits altered diffusion at disease onset. Diabetes Care 2019, 42, 248–257. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, T.; Ekwall, O.; Amirian, N.; Zapardiel-Gonzalo, J.; von Herrath, M.G. Increased immune cell infiltration of the exocrine pancreas: A possible contribution to the pathogenesis of Type 1 diabetes. Diabetes 2014, 63, 3880–3890. [Google Scholar] [CrossRef]
- Garciafigueroa, Y.; Phillips, B.E.; Engman, C.; Trucco, M.; Giannoukakis, N. Neutrophil-associated inflammatory changes in the pre-diabetic pancreas of early-age NOD mice. Front. Endocrinol. 2021, 12, 565981. [Google Scholar] [CrossRef]
- Papaccio, G.; Chieffi-Baccari, G.; Mezzogiorno, V.; Esposito, V. Extraislet infiltration in NOD mouse pancreas: Observations after immunomodulation. Pancreas 1993, 8, 459–464. [Google Scholar] [CrossRef]
- Behrens, G.M.; Li, M.; Davey, G.M.; Allison, J.; Flavell, R.A.; Carbone, F.R.; Heath, W.R. Helper requirements for generation of effector CTL to islet beta cell antigens. J. Immunol. 2004, 172, 5420–5426. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sweet, R.A.; Hoyne, G.F.; Simeonovic, C.J.; Parish, C.R. Acute T-cell-driven inflammation requires the endoglycosidase heparanase-1 from multiple cell types. Int. J. Mol. Sci. 2022, 23, 4625. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, A.; Riitano, G.; Recalchi, S.; Manganelli, V.; Costi, R.; Saccoliti, F.; Pulcinelli, F.; Garofalo, T.; Misasi, R.; Longo, A.; et al. Effect of heparanase inhibitor on tissue factor overexpression in platelets and endothelial cells induced by anti-beta2-GPI antibodies. J. Thromb. Haemost. 2021, 19, 2302–2313. [Google Scholar] [CrossRef]
- Edovitsky, E.; Lerner, I.; Zcharia, E.; Peretz, T.; Vlodavsky, I.; Elkin, M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood 2006, 107, 3609–3616. [Google Scholar] [CrossRef]
- Parish, C.R. The role of heparan sulphate in inflammation. Nat. Rev. Immunol. 2006, 6, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowski, A.F.; Popp, S.K.; Freeman, C.; Parish, C.R.; Simeonovic, C.J. Heparan sulfate and heparanase play key roles in mouse beta cell survival and autoimmune diabetes. J. Clin. Investig. 2012, 122, 132–141. [Google Scholar] [CrossRef]
- Coppieters, K.; Amirian, N.; von Herrath, M. Intravital imaging of CTLs killing islet cells in diabetic mice. J. Clin. Investig. 2012, 122, 119–131. [Google Scholar] [CrossRef]
- Espinosa-Carrasco, G.; Le Saout, C.; Fontanaud, P.; Michau, A.; Mollard, P.; Hernandez, J.; Schaeffer, M. Integrin beta1 optimizes diabetogenic T cell migration and function in the pancreas. Front. Immunol. 2018, 9, 1156. [Google Scholar] [CrossRef]
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 2013, 19, 65–73. [Google Scholar] [CrossRef]
- Popp, S.K.; Vecchio, F.; Brown, D.J.; Fukuda, R.; Suzuki, Y.; Takeda, Y.; Wakamatsu, R.; Sarma, M.A.; Garrett, J.; Giovenzana, A.; et al. Circulating platelet-neutrophil aggregates characterize the development of type 1 diabetes in humans and NOD mice. JCI Insight 2022, 7, e153993. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Beckhove, P.; Lerner, I.; Pisano, C.; Meirovitz, A.; Ilan, N.; Elkin, M. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012, 5, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Larbi, K.Y.; Young, R.E.; Nourshargh, S. Migration of leukocytes through the vessel wall and beyond. Thromb. Haemost. 2003, 90, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Irving-Rodgers, H.F.; Ziolkowski, A.F.; Parish, C.R.; Sado, Y.; Ninomiya, Y.; Simeonovic, C.J.; Rodgers, R.J. Molecular composition of the peri-islet basement membrane in NOD mice: A barrier against destructive insulitis. Diabetologia 2008, 51, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Korpos, E.; Kadri, N.; Kappelhoff, R.; Wegner, J.; Overall, C.M.; Weber, E.; Holmberg, D.; Cardell, S.; Sorokin, L. The peri-islet basement membrane, a barrier to infiltrating leukocytes in Type 1 diabetes in mouse and human. Diabetes 2013, 62, 531–542. [Google Scholar] [CrossRef]
- Simeonovic, C.J.; Popp, S.K.; Starrs, L.M.; Brown, D.J.; Ziolkowski, A.F.; Ludwig, B.; Bornstein, S.R.; Wilson, J.D.; Pugliese, A.; Kay, T.W.H.; et al. Loss of intra-islet heparan sulfate is a highly sensitive marker of type 1 diabetes progression in humans. PLoS ONE 2018, 13, e0191360. [Google Scholar] [CrossRef]
- Matzner, Y.; Bar-Ner, M.; Yahalom, J.; Ishai-Michaeli, R.; Fuks, Z.; Vlodavsky, I. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J. Clin. Investig. 1985, 76, 1306–1313. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Eldor, A.; Haimovitz-Friedman, A.; Matzner, Y.; Ishai-Michaeli, R.; Lider, O.; Naparstek, Y.; Cohen, I.R.; Fuks, Z. Expression of heparanase by platelets and circulating cells of the immune system: Possible involvement in diapedesis and extravasation. Invasion Metastasis 1992, 12, 112–127. [Google Scholar]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef]
- Grossmann, J. Molecular mechanisms of “detachment-induced apoptosis—Anoikis”. Apoptosis 2002, 7, 247–260. [Google Scholar] [CrossRef]
- Loka, R.S.; Song, Z.; Sletten, E.T.; Kayal, Y.; Vlodavsky, I.; Zhang, K.; Nguyen, H.M. Heparan sulfate mimicking glycopolymer prevents pancreatic beta cell destruction and suppresses inflammatory cytokine expression in islets under the challenge of upregulated heparanase. ACS Chem. Biol. 2022, 17, 1387–1400. [Google Scholar] [CrossRef]
- Kurts, C.; Sutherland, R.M.; Davey, G.; Li, M.; Lew, A.M.; Blanas, E.; Carbone, F.R.; Miller, J.F.A.P.; Heath, W.R. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc. Natl. Acad. Sci. USA 1999, 96, 12703–12707. [Google Scholar] [CrossRef] [PubMed]
- Zcharia, E.; Jia, J.; Zhang, X.; Baraz, L.; Lindahl, U.; Peretz, T.; Vlodavsky, I.; Li, J.P. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS ONE 2009, 4, e5181. [Google Scholar] [CrossRef] [PubMed]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T-cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Barnden, M.J.; Allison, J.; Heath, W.R.; Carbone, F.R. Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998, 76, 34–40. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simeonovic, C.J.; Wu, Z.; Popp, S.K.; Hoyne, G.F.; Parish, C.R. Transient Inflammation of Pancreatic Exocrine Tissue in Autoimmune Diabetes Follows Onset of Islet Damage and Utilizes Heparanase-1. Int. J. Mol. Sci. 2025, 26, 4120. https://doi.org/10.3390/ijms26094120
Simeonovic CJ, Wu Z, Popp SK, Hoyne GF, Parish CR. Transient Inflammation of Pancreatic Exocrine Tissue in Autoimmune Diabetes Follows Onset of Islet Damage and Utilizes Heparanase-1. International Journal of Molecular Sciences. 2025; 26(9):4120. https://doi.org/10.3390/ijms26094120
Chicago/Turabian StyleSimeonovic, Charmaine J., Zuopeng Wu, Sarah K. Popp, Gerard F. Hoyne, and Christopher R. Parish. 2025. "Transient Inflammation of Pancreatic Exocrine Tissue in Autoimmune Diabetes Follows Onset of Islet Damage and Utilizes Heparanase-1" International Journal of Molecular Sciences 26, no. 9: 4120. https://doi.org/10.3390/ijms26094120
APA StyleSimeonovic, C. J., Wu, Z., Popp, S. K., Hoyne, G. F., & Parish, C. R. (2025). Transient Inflammation of Pancreatic Exocrine Tissue in Autoimmune Diabetes Follows Onset of Islet Damage and Utilizes Heparanase-1. International Journal of Molecular Sciences, 26(9), 4120. https://doi.org/10.3390/ijms26094120