
Thymosin Beta-4 Modulates Cardiac Remodeling by Regulating ROCK1 Expression in Adult Mammals
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
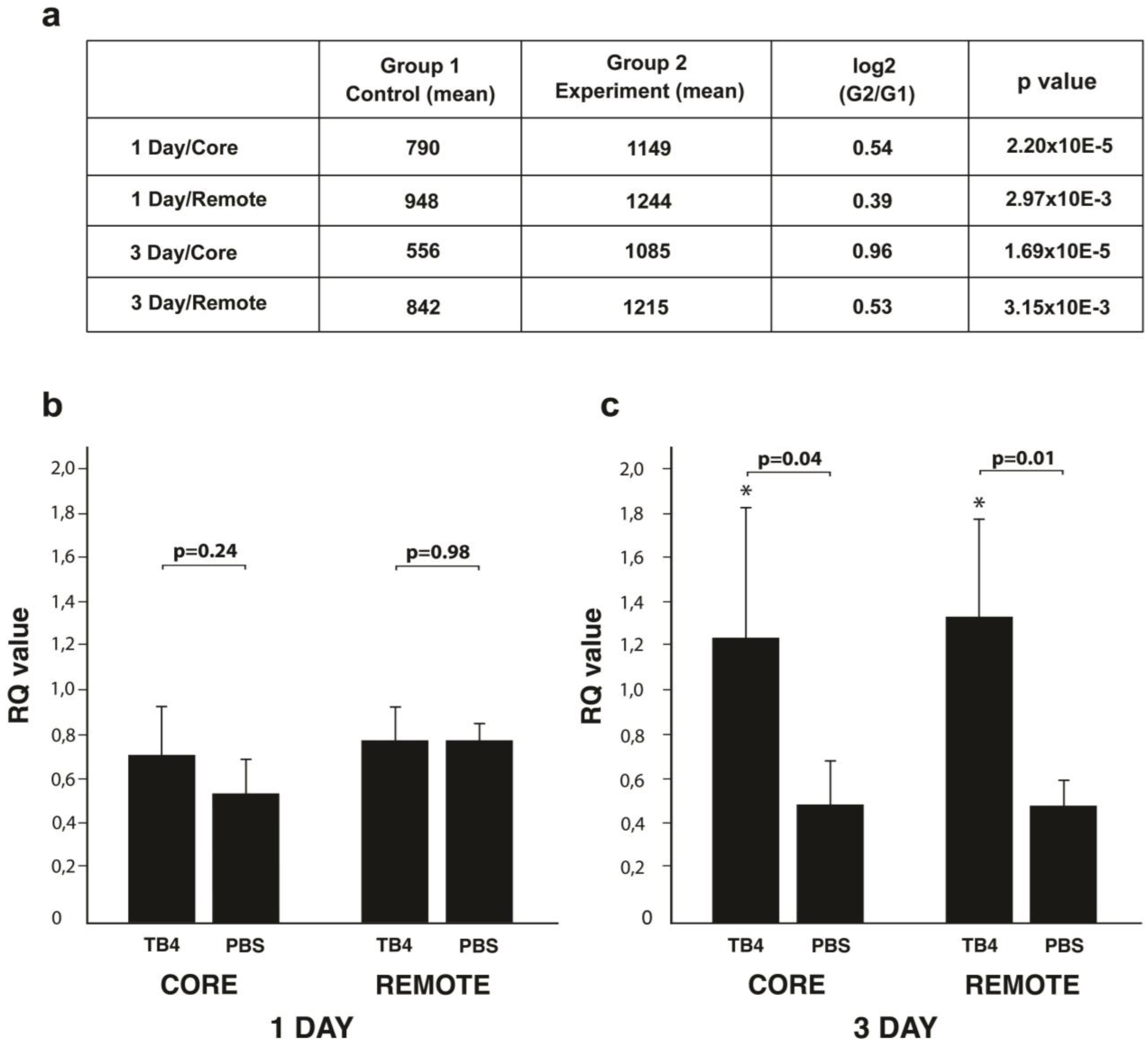
2.1. TB4 Alters miR139-5p Expression in Adult Infarcted Mammalian Hearts
2.2. miR139-5p Target ROCK1 Decreases Following TB4 Treatment in Adult Infarcted Mammalian Hearts
2.3. ROCK1 mRNA Expression Following Infarction
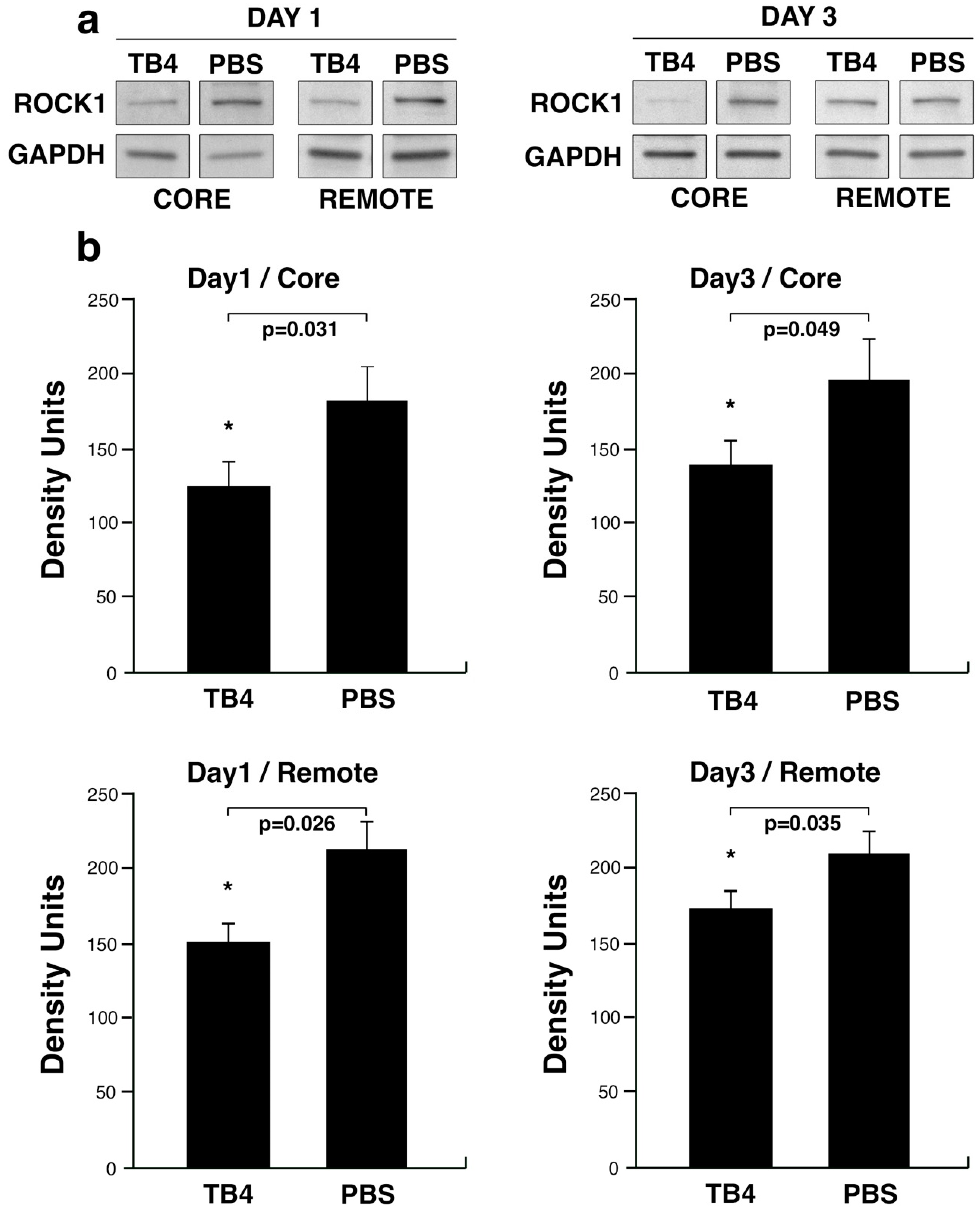
2.4. TB4 Decreases ROCK1 Protein Levels in the Hypoxic Adult Mammalian Mouse Heart In Vivo—Immunohistochemistry
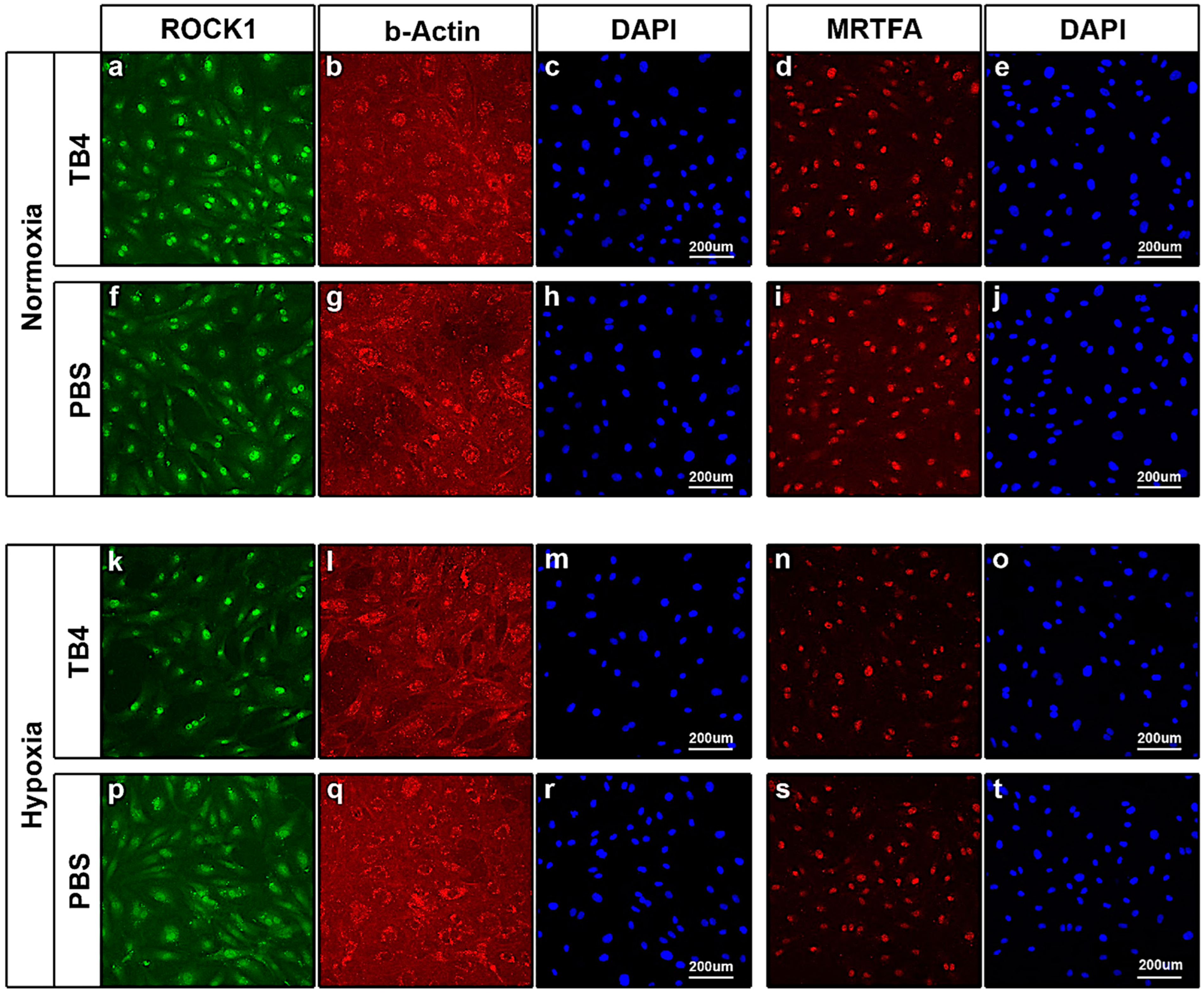
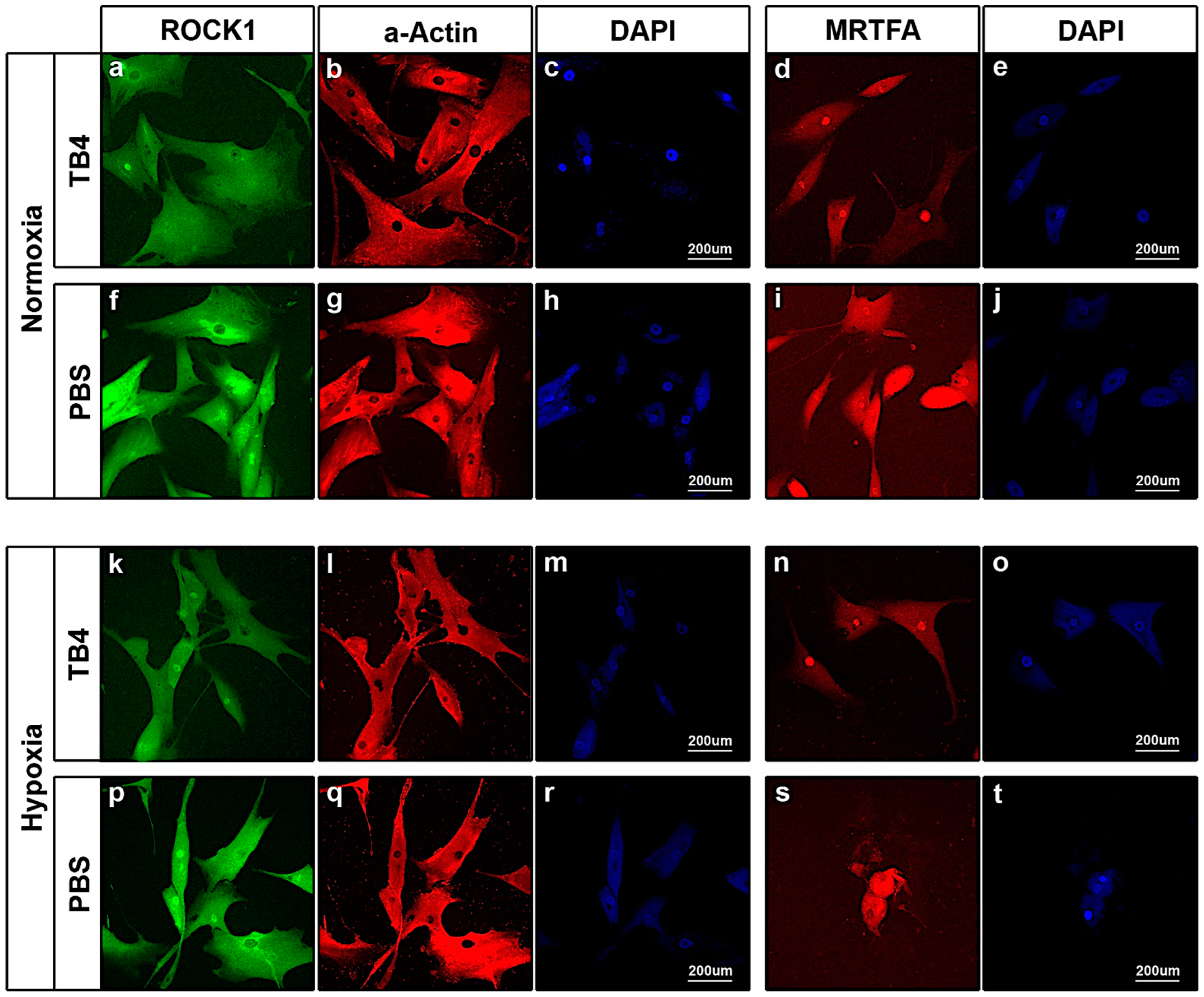
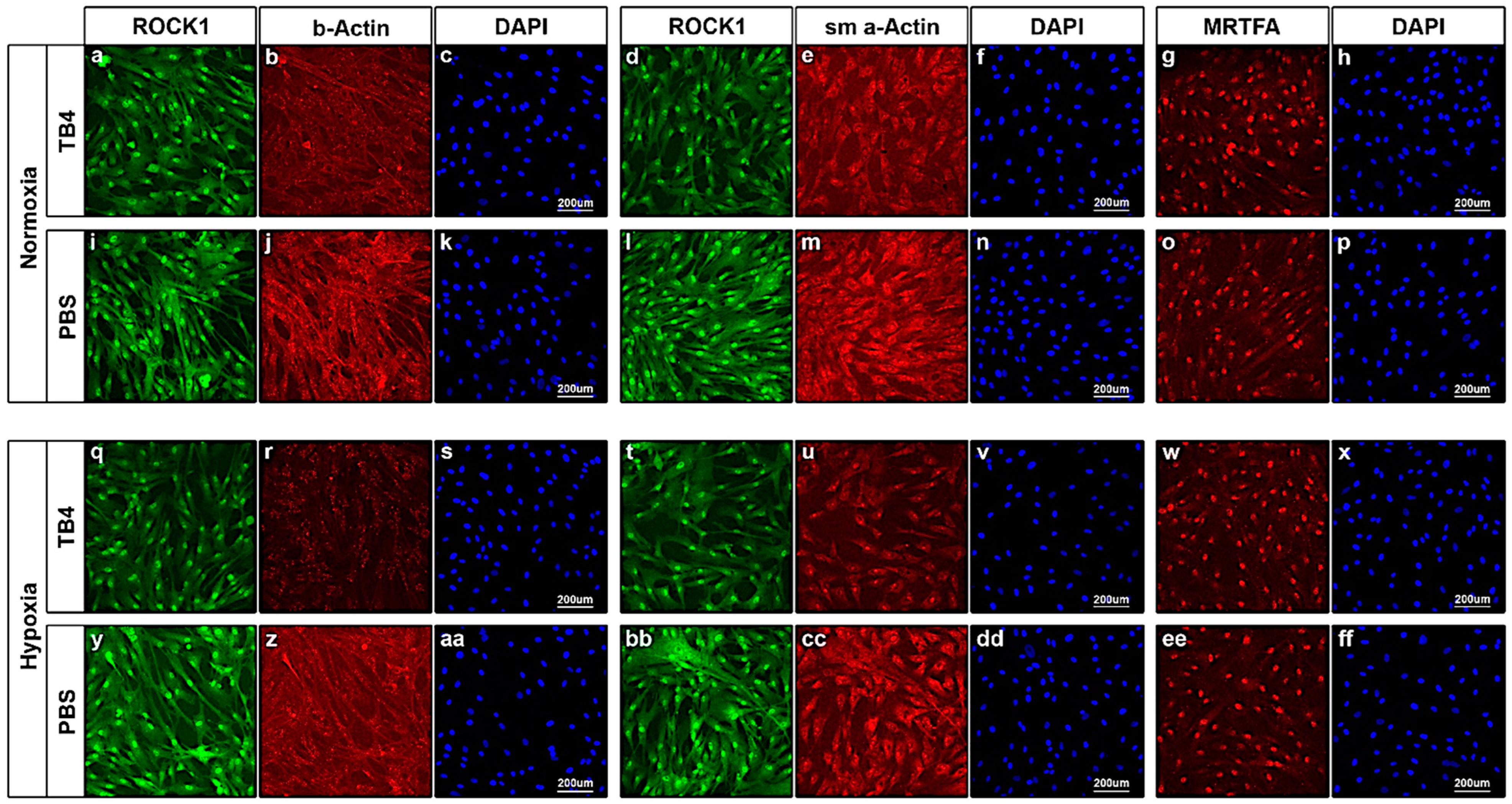
2.5. In Vitro Effect of TB4 on ROCK1, Actin and MRTFA Levels in Adult Human Cardiac Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animal Procedures
5.2. miRNA Microarray
5.3. Real-Time Quantitative PCR—miRNA
5.4. Real-Time Quantitative PCR—mRNA
5.5. Western Blot
5.6. Cell Culturing—Normoxic and Hypoxic Conditions
5.7. Immunocytochemistry
5.8. Immunohistochemistry—Cryopreserved Sections
5.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Reveals Leading Causes of Death and Disability Worldwide: 2000–2019; World Health Organization: Geneva, Switzerland, 2020; Available online: https://www.who.int/news/item/09-12-2020-who-reveals-leading-causes-of-death-and-disability-worldwide-2000-2019 (accessed on 30 January 2024).
- Singh, A.; Museedi, A.S.; Grossman, S.A. Acute Coronary Syndrome. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Vaartjes, I.; Hendrix, A.; Hertogh, E.M.; Grobbee, D.E.; Doevendans, P.A.; Mosterd, A.; Bots, M.L. Sudden death in persons younger than 40 years of age: Incidence and causes. Eur. J. Prev. Cardiol. 2009, 16, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Ambrose, J.A. Understanding myocardial infarction. F1000Research 2018, 7, F1000 Faculty Rev-1378. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling—Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Maar, K.; Hetenyi, R.; Maar, S.; Faskerti, G.; Hanna, D.; Lippai, B.; Takatsy, A.; Bock-Marquette, I. Utilizing Developmentally Essential Secreted Peptides Such as Thymosin Beta-4 to Remind the Adult Organs of Their Embryonic State—New Directions in Anti-Aging Regenerative Therapies. Cells 2021, 10, 1343. [Google Scholar] [CrossRef]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Bock-Marquette, I.; Saxena, A.; White, M.D.; Michael DiMaio, J.; Srivastava, D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature 2004, 432, 466–472. [Google Scholar] [CrossRef]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar]
- Wang, H.; Haeger, S.M.; Kloxin, A.M.; Leinwand, L.A.; Anseth, K.S. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS ONE 2012, 7, e39969. [Google Scholar] [CrossRef]
- Wang, H.; Tibbitt, M.W.; Langer, S.J.; Leinwand, L.A.; Anseth, K.S. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 19336–19341. [Google Scholar] [CrossRef]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef]
- Stempien-Otero, A.; Kim, D.-H.; Davis, J. Molecular networks underlying myofibroblast fate and fibrosis. J. Mol. Cell. Cardiol. 2016, 97, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Dees, C.; Pileckyte, M.; Szucs, G.; Spriewald, B.M.; Zwerina, J.; Distler, O.; Schett, G.; Distler, J.H.W. Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2008, 58, 2553–2564. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; DiMaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Haudek, S.B.; Gupta, D.; Dewald, O.; Schwartz, R.J.; Wei, L.; Trial, J.; Entman, M.L. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc. Res. 2009, 83, 511–518. [Google Scholar] [CrossRef]
- Goldstein, A.L.; Slater, F.D.; White, A. Preparation, assay, and partial purification of a thymic lymphocytopoietic factor (thymosin). Proc. Natl. Acad. Sci. USA 1966, 56, 1010–1017. [Google Scholar] [CrossRef]
- Yu, F.; Lin, S.; Morrison-Bogorad, M.; Atkinson, M.; Yin, H. Thymosin beta 10 and thymosin beta 4 are both actin monomer sequestering proteins. J. Biol. Chem. 1993, 268, 502–509. [Google Scholar] [CrossRef]
- Yu, F.X.; Lin, S.C.; Morrison--Bogorad, M.; Yin, H.L. Effects of thymosin beta 4 and thymosin beta 10 on actin structures in living cells. Cell Motil. Cytoskelet. 1994, 27, 13–25. [Google Scholar] [CrossRef]
- Carlier, M.-F.; Didry, D.; Erk, I.; Lepault, J.; Van Troys, M.L.; Vandekerckhove, J.; Perelroizen, I.; Yin, H.; Doi, Y.; Pantaloni, D. Tbeta 4 is not a simple G-actin sequestering protein and interacts with F-actin at high concentration. J. Biol. Chem. 1996, 271, 9231–9239. [Google Scholar] [CrossRef]
- Ehrlich, H.P.; Hazard, S.W., 3rd. Thymosin beta4 enhances repair by organizing connective tissue and preventing the appearance of myofibroblasts. Ann. N. Y. Acad. Sci. 2010, 1194, 118–124. [Google Scholar] [CrossRef]
- Frohm, M.; Gunne, H.; Bergman, A.; Agerberth, B.; Bergman, T.; Boman, A.; Lidén, S.; Jörnvall, H.; Boman, H.G. Biochemical and antibacterial analysis of human wound and blister fluid. Eur. J. Biochem. 1996, 237, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, H.K.; Kulik, V.; Goldstein, A.L. Thymosin β4 and the anti-fibrotic switch. Int. Immunopharmacol. 2023, 115, 109628. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.C.; Zhang, Z.G.; Zhang, J.; Xiong, Y.; Zhang, L.; Chopp, M. Treatment of neurological injury with thymosin β4. Ann. N. Y. Acad. Sci. 2012, 1269, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Pardon, M.C. Anti-inflammatory potential of thymosin β4 in the central nervous system: Implications for progressive neurodegenerative diseases. Expert Opin. Biol. Ther. 2018, 18 (Suppl. S1), 165–169. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Meng, Y.; Zhang, Y.; Zhang, Z.G.; Morris, D.C.; Chopp, M. Treatment of traumatic brain injury with thymosin β4 in rats. J. Neurosurg. 2011, 114, 102–115. [Google Scholar] [CrossRef]
- Mason, W.J.; Vasilopoulou, E. The Pathophysiological Role of Thymosin β4 in the Kidney Glomerulus. Int. J. Mol. Sci. 2023, 24, 7684. [Google Scholar] [CrossRef]
- Vasilopoulou, E.; Riley, P.R.; Long, D.A. Thymosin-β4: A key modifier of renal disease. Expert Opin. Biol. Ther. 2018, 18, 185–192. [Google Scholar] [CrossRef]
- Vasilopoulou, E.; Winyard, P.J.; Riley, P.R.; A Long, D. The role of thymosin-β4 in kidney disease. Expert Opin. Biol. Ther. 2015, 15 (Suppl. S1), 187–190. [Google Scholar] [CrossRef]
- Tian, Z.; Yao, N.; Wang, F.; Ruan, L. Thymosin β4 Suppresses LPS-Induced Murine Lung Fibrosis by Attenuating Oxidative Injury and Alleviating Inflammation. Inflammation 2022, 45, 59–73. [Google Scholar] [CrossRef]
- Yu, R.; Gao, D.; Bao, J.; Sun, R.; Cui, M.; Mao, Y.; Li, K.; Hu, E.; Zhai, Y.; Liu, Y.; et al. Exogenous Thymosin Beta 4 Suppresses IPF-Lung Cancer in Mice: Possibly Associated with Its Inhibitory Effect on the JAK2/STAT3 Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 3818. [Google Scholar] [CrossRef]
- Sosne, G. Thymosin beta 4 and the eye: The journey from bench to bedside. Expert Opin. Biol. Ther. 2018, 18 (Suppl. S1), 99–104. [Google Scholar] [CrossRef]
- Sosne, G.; Qiu, P.; Ousler, G.W.; Dunn, S.P.; Crockford, D. Thymosin β4: A potential novel dry eye therapy. Ann. N. Y. Acad. Sci. 2012, 1270, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Sosne, G.; Rimmer, D.; Kleinman, H.K.; Ousler, G. Thymosin Beta 4: A Potential Novel Therapy for Neurotrophic Keratopathy, Dry Eye, and Ocular Surface Diseases. Vitam. Horm. 2016, 102, 277–306. [Google Scholar]
- Bako, P.; Lippai, B.; Nagy, J.; Kramer, S.; Kaszas, B.; Tornoczki, T.; Bock-Marquette, I. Thymosin beta-4—A potential tool in healing middle ear lesions in adult mammals. Int. Immunopharmacol. 2023, 116, 109830. [Google Scholar] [CrossRef]
- Hong, Y.; Yao, Q.; Zheng, L. Thymosin β4 attenuates liver fibrosis via suppressing Notch signaling. Biochem. Biophys. Res. Commun. 2017, 493, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Cheng, M.; Liu, Y.; Yao, Y.; Zhu, Z.; Zhang, B.; Mou, Q.; Cheng, Y. Thymosin-β4 inhibits proliferation and induces apoptosis of hepatic stellate cells through PI3K/AKT pathway. Oncotarget 2017, 8, 68847–68853. [Google Scholar] [CrossRef]
- Bock-Marquette, I.; Maar, K.; Maar, S.; Lippai, B.; Faskerti, G.; Gallyas, F., Jr.; Olson, E.N.; Srivastava, D. Thymosin beta-4 denotes new directions towards developing prosperous anti-aging regenerative therapies. Int. Immunopharmacol. 2023, 116, 109741. [Google Scholar] [CrossRef] [PubMed]
- Bock-Marquette, I.; Shrivastava, S.; Pipes, G.T.; Thatcher, J.E.; Blystone, A.; Shelton, J.M.; Galindo, C.L.; Melegh, B.; Srivastava, D.; Olson, E.N.; et al. Thymosin β4 mediated PKC activation is essential to initiate the embryonic coronary developmental program and epicardial progenitor cell activation in adult mice in vivo. J. Mol. Cell. Cardiol. 2009, 46, 728–738. [Google Scholar] [CrossRef]
- Shrivastava, S.; Srivastava, D.; Olson, E.N.; DiMaio, J.M.; Bock-Marquette, I. Thymosin β4 and cardiac repair. In Thymosins in Health and Disease: 2nd International Symposium; Annals of the New York Academy of Sciences; Garaci, E., Goldstein, A.L., Eds.; The New York Academy of Sciences: New York, NY, USA, 2010; pp. 87–96. [Google Scholar]
- Srivastava, D.; Saxena, A.; Michael Dimaio, J.; Bock-Marquette, I. Thymosin β4 is cardioprotective after myocardial infarction. In Thymosins in Health and Disease: First International Symposium; Annals of the New York Academy of Sciences; Goldstein, A.L., Garaci, E., Eds.; The New York Academy of Sciences: New York, NY, USA, 2007; pp. 161–170. [Google Scholar]
- Bollini, S.; Riley, P.R.; Smart, N. Thymosin β4: Multiple functions in protection, repair and regeneration of the mammalian heart. Expert Opin. Biol. Ther. 2015, 15 (Suppl. S1), 163–174. [Google Scholar] [CrossRef]
- Crockford, D. Development of thymosin beta4 for treatment of patients with ischemic heart disease. Ann. N. Y. Acad. Sci. 2007, 1112, 385–395. [Google Scholar] [CrossRef]
- Gladka, M.M.; Kohela, A.; Molenaar, B.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; Kremer, V.; Vos, H.R.; Huibers, M.M.H.; Haigh, J.J.; et al. Cardiomyocytes stimulate angiogenesis after ischemic injury in a ZEB2-dependent manner. Nat. Commun. 2021, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.L.; Kleinman, H.K. Advances in the basic and clinical applications of thymosin β4. Expert. Opin. Biol. Ther. 2015, 15 (Suppl. S1), 139–145. [Google Scholar] [CrossRef]
- Marks, E.D.; Kumar, A. Thymosin β4: Roles in Development, Repair, and Engineering of the Cardiovascular System. Vitam. Horm. 2016, 102, 227–249. [Google Scholar] [PubMed]
- Peng, H.; Xu, J.; Yang, X.-P.; Dai, X.; Peterson, E.L.; Carretero, O.A.; Rhaleb, N.-E.; Nawaito, S.A.; Sahadevan, P.; Clavet-Lanthier, M.; et al. Thymosin-β4 prevents cardiac rupture and improves cardiac function in mice with myocardial infarction. Am. J. Physiol. Circ. Physiol. 2014, 307, H741–H751. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.-D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Tan, S.H.; Loo, S.J.; Gao, Y.; Tao, Z.H.; Su, L.P.; Wang, C.X.; Zhang, S.L.; Mu, Y.H.; Cui, Y.H.; Abdurrachim, D.; et al. Thymosin β4 increases cardiac cell proliferation, cell engraftment, and the reparative potency of human induced-pluripotent stem cell-derived cardiomyocytes in a porcine model of acute myocardial infarction. Theranostics 2021, 11, 7879–7895. [Google Scholar] [CrossRef]
- Zhu, J.; Song, J.; Yu, L.; Zheng, H.; Zhou, B.; Weng, S.; Fu, G. Safety and efficacy of autologous thymosin β4 pre-treated endothelial progenitor cell transplantation in patients with acute ST segment elevation myocardial infarction: A pilot study. Cytotherapy 2016, 18, 1037–1042. [Google Scholar] [CrossRef]
- Pipes, G.; Yang, J. Cardioprotection by Thymosin Beta 4. Vitam. Horm. 2016, 102, 209–226. [Google Scholar]
- Gupta, S.; Kumar, S.; Sopko, N.; Qin, Y.; Wei, C.; Kim, I. Thymosin β4 and cardiac protection: Implication in inflammation and fibrosis. Ann. N. Y. Acad. Sci. 2012, 1269, 84–91. [Google Scholar] [CrossRef]
- Renga, G.; Oikonomou, V.; Stincardini, C.; Pariano, M.; Borghi, M.; Costantini, C.; Bartoli, A.; Garaci, E.; Goldstein, A.L.; Romani, L. Thymosin β4 limits inflammation through autophagy. Expert Opin. Biol. Ther. 2018, 18, 171–175. [Google Scholar] [CrossRef]
- Sosne, G.; Qiu, P.; Kurpakus-Wheater, M. Thymosin beta 4: A novel corneal wound healing and anti-inflammatory agent. Clin. Ophthalmol. 2007, 1, 201–207. [Google Scholar] [PubMed]
- Hinkel, R.; El Aouni, C.; Bock-Marquette, I.; Hatzopoulos, A.; Boekstegers, P.; Kupatt, C. Cardioprotective Potential of Thymosin s4 in a Preclinical Pig Model of Ischemia/Reperfusion Injury. J. Stem Cells Regen. Med. 2007, 2, 98–99. [Google Scholar] [PubMed]
- Hinkel, R.; Ball, H.L.; DiMaio, J.M.; Shrivastava, S.; Thatcher, J.E.; Singh, A.N.; Sun, X.; Faskerti, G.; Olson, E.N.; Kupatt, C.; et al. C-terminal variable AGES domain of Thymosin β4: The molecule’s primary contribution in support of post-ischemic cardiac function and repair. J. Mol. Cell. Cardiol. 2015, 87, 113–125. [Google Scholar] [CrossRef]
- Ehrlich, H.P.; Hazard, S.W. Thymosin β4 affecting the cytoskeleton organization of the myofibroblasts. Ann. N. Y. Acad. Sci. 2012, 1269, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, M.; Sun, Z.; Zhuang, S.; Zhang, W.; Yang, Z.; Han, X.; Nie, S. MicroRNA-139-5p improves sepsis-induced lung injury by targeting Rho-kinase1. Exp. Ther. Med. 2021, 22, 1059. [Google Scholar] [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef]
- Hinkel, R.; Trenkwalder, T.; Petersen, B.; Husada, W.; Gesenhues, F.; Lee, S.; Hannappel, E.; Bock-Marquette, I.; Theisen, D.; Leitner, L.; et al. MRTF-A controls vessel growth and maturation by increasing the expression of CCN1 and CCN2. Nat. Commun. 2014, 5, 3970. [Google Scholar] [CrossRef]
- Kupatt, C.; Ziegler, T.; Bähr, A.; Le Noble, F. Thymosin ß4 and MRTF-A mitigate vessel regression despite cardiovascular risk factors. Int. Immunopharmacol. 2023, 117, 109786. [Google Scholar] [CrossRef]
- Blomme, B.; Deroanne, C.; Hulin, A.; Lambert, C.; Defraigne, J.O.; Nusgens, B.; Radermecker, M.; Colige, A. Mechanical strain induces a pro-fibrotic phenotype in human mitral valvular interstitial cells through RhoC/ROCK/MRTF-A and Erk1/2 signaling pathways. J. Mol. Cell. Cardiol. 2019, 135, 149–159. [Google Scholar] [CrossRef]
- Tarbit, E.; Singh, I.; Peart, J.N.; Rose’meyer, R.B. Biomarkers for the identification of cardiac fibroblast and myofibroblast cells. Heart Fail. Rev. 2018, 24, 1–15. [Google Scholar] [CrossRef]
- Majesky, M.W.; Dong, X.R.; Hoglund, V.; Mahoney, W.M., Jr.; Daum, G. The adventitia: A dynamic interface containing resident progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Rikitake, Y.; Oyama, N.; Wang, C.Y.C.; Noma, K.; Satoh, M.; Kim, H.H.; Liao, J.K. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation 2005, 112, 2959–2965. [Google Scholar] [CrossRef]
- Shimizu, T.; Liao, J.K. Rho Kinases and Cardiac Remodeling. Circ. J. 2016, 80, 1491–1498. [Google Scholar] [CrossRef]
- Schofield, A.V.; Bernard, O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Asp, P.; Wihlborg, M.; Karlén, M.; Farrants, A.-K.O. Expression of BRG1, a human SWI/SNF component, affects the organisation of actin filaments through the RhoA signalling pathway. J. Cell Sci. 2002, 115 Pt 13, 2735–2746. [Google Scholar] [CrossRef]
- Ishizaki, T.; Naito, M.; Fujisawa, K.; Maekawa, M.; Walanabe, N.; Saito, Y.; Narumiya, S. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 1997, 404, 118–124. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The Cardiac Fibroblast: Therapeutic Target in Myocardial Remodeling and Failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Hieu, T.B.; Ma, F.; Yu, Y.; Cao, Z.; Wang, M.; Wu, W.; Mao, Y.; Rose, P.; Law, B.Y.-K.; et al. ZYZ-168 alleviates cardiac fibrosis after myocardial infarction through inhibition of ERK1/2-dependent ROCK1 activation. Sci. Rep. 2017, 7, 43242. [Google Scholar] [CrossRef]
- Harvey, K.A.; Paranavitana, C.N.; Zaloga, G.P.; Siddiqui, R.A. Diverse signaling pathways regulate fibroblast differentiation and transformation through Rho kinase activation. J. Cell. Physiol. 2007, 211, 353–363. [Google Scholar] [CrossRef]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2012, 229, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-I.; Lee, Y.-H.; Chen, P.-H.; Lin, Y.-C.; Chou, M.-H.; Kao, Y.-H. Cobalt chloride induces RhoA/ROCK activation and remodeling effect in H9c2 cardiomyoblasts: Involvement of PI3K/Akt and MAPK pathways. Cell. Signal. 2017, 36, 25–33. [Google Scholar] [CrossRef]
- Loirand, G. Rho Kinases in Health and Disease: From Basic Science to Translational Research. Pharmacol. Rev. 2015, 67, 1074–1095. [Google Scholar]
- Noma, K.; Oyama, N.; Liao, J.K. Physiological role of ROCKs in the cardiovascular system. Am. J. Physiol. Physiol. 2006, 290, C661–C668. [Google Scholar] [CrossRef]
- Santos, G.L.; Hartmann, S.; Zimmermann, W.-H.; Ridley, A.; Lutz, S. Inhibition of Rho-associated kinases suppresses cardiac myofibroblast function in engineered connective and heart muscle tissues. J. Mol. Cell. Cardiol. 2019, 134, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Lighthouse, J.K.; Small, E.M. Transcriptional control of cardiac fibroblast plasticity. J. Mol. Cell. Cardiol. 2016, 91, 52–60. [Google Scholar] [CrossRef]
- Mokalled, M.H.; Carroll, K.J.; Cenik, B.K.; Chen, B.; Liu, N.; Olson, E.N.; Bassel-Duby, R. Myocardin-related transcription factors are required for cardiac development and function. Dev. Biol. 2015, 406, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M. The Actin–MRTF–SRF Gene Regulatory Axis and Myofibroblast Differentiation. J. Cardiovasc. Transl. Res. 2012, 5, 794–804. [Google Scholar] [CrossRef]
- Knipe, R.S.; Nurunnabi, M.; Probst, C.K.; Spinney, J.J.; Abe, E.; Bose, R.J.C.; Ha, K.; Logue, A.; Nguyen, T.; Servis, R.; et al. Myofibroblast-specific inhibition of the Rho kinase-MRTF-SRF pathway using nanotechnology for the prevention of pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2023, 324, L190–L198. [Google Scholar] [CrossRef]
- Dai, Y.; Luo, W.; Chang, J. Rho kinase signaling and cardiac physiology. Curr. Opin. Physiol. 2018, 1, 14–20. [Google Scholar] [CrossRef]
- Sisson, T.H.; Ajayi, I.O.; Subbotina, N.; Dodi, A.E.; Rodansky, E.S.; Chibucos, L.N.; Kim, K.K.; Keshamouni, V.G.; White, E.S.; Zhou, Y.; et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am. J. Pathol. 2015, 185, 969–986. [Google Scholar] [CrossRef] [PubMed]
- Young, K.M.; Reinhart-King, C.A. Cellular mechanosignaling for sensing and transducing matrix rigidity. Curr. Opin. Cell Biol. 2023, 83, 102208. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-D.; Jeong, S.-J.; Lee, H.-Y.; Lim, D.-S.; Lee, B.-H.; Bae, C.-S.; Jeong, M.-J. The Effect of Thymosin β4 for Osteoblast Adhesion on Titanium Surface. J. Nanosci. Nanotechnol. 2015, 15, 5663–5667. [Google Scholar] [CrossRef]
- Hattori, T.; Shimokawa, H.; Higashi, M.; Hiroki, J.; Mukai, Y.; Tsutsui, H.; Kaibuchi, K.; Takeshita, A. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation 2004, 109, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Kim, H.-H.; Huang, Z.; Seto, M.; Yano, K.; Asano, T.; Moskowitz, M.A.; Liao, J.K. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005, 36, 2251–2257. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar]
- López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse myocardial fibrosis: Mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 2021, 18, 479–498. [Google Scholar]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maar, K.; Thatcher, J.E.; Karpov, E.; Rendeki, S.; Gallyas, F., Jr.; Bock-Marquette, I. Thymosin Beta-4 Modulates Cardiac Remodeling by Regulating ROCK1 Expression in Adult Mammals. Int. J. Mol. Sci. 2025, 26, 4131. https://doi.org/10.3390/ijms26094131
Maar K, Thatcher JE, Karpov E, Rendeki S, Gallyas F Jr., Bock-Marquette I. Thymosin Beta-4 Modulates Cardiac Remodeling by Regulating ROCK1 Expression in Adult Mammals. International Journal of Molecular Sciences. 2025; 26(9):4131. https://doi.org/10.3390/ijms26094131
Chicago/Turabian StyleMaar, Klaudia, Jeffrey E. Thatcher, Egor Karpov, Szilard Rendeki, Ferenc Gallyas, Jr., and Ildiko Bock-Marquette. 2025. "Thymosin Beta-4 Modulates Cardiac Remodeling by Regulating ROCK1 Expression in Adult Mammals" International Journal of Molecular Sciences 26, no. 9: 4131. https://doi.org/10.3390/ijms26094131
APA StyleMaar, K., Thatcher, J. E., Karpov, E., Rendeki, S., Gallyas, F., Jr., & Bock-Marquette, I. (2025). Thymosin Beta-4 Modulates Cardiac Remodeling by Regulating ROCK1 Expression in Adult Mammals. International Journal of Molecular Sciences, 26(9), 4131. https://doi.org/10.3390/ijms26094131