
MicroRNA-142-3p Overcomes Drug Resistance in Hepatocellular Carcinoma by Targeting YES1 and TWF1

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
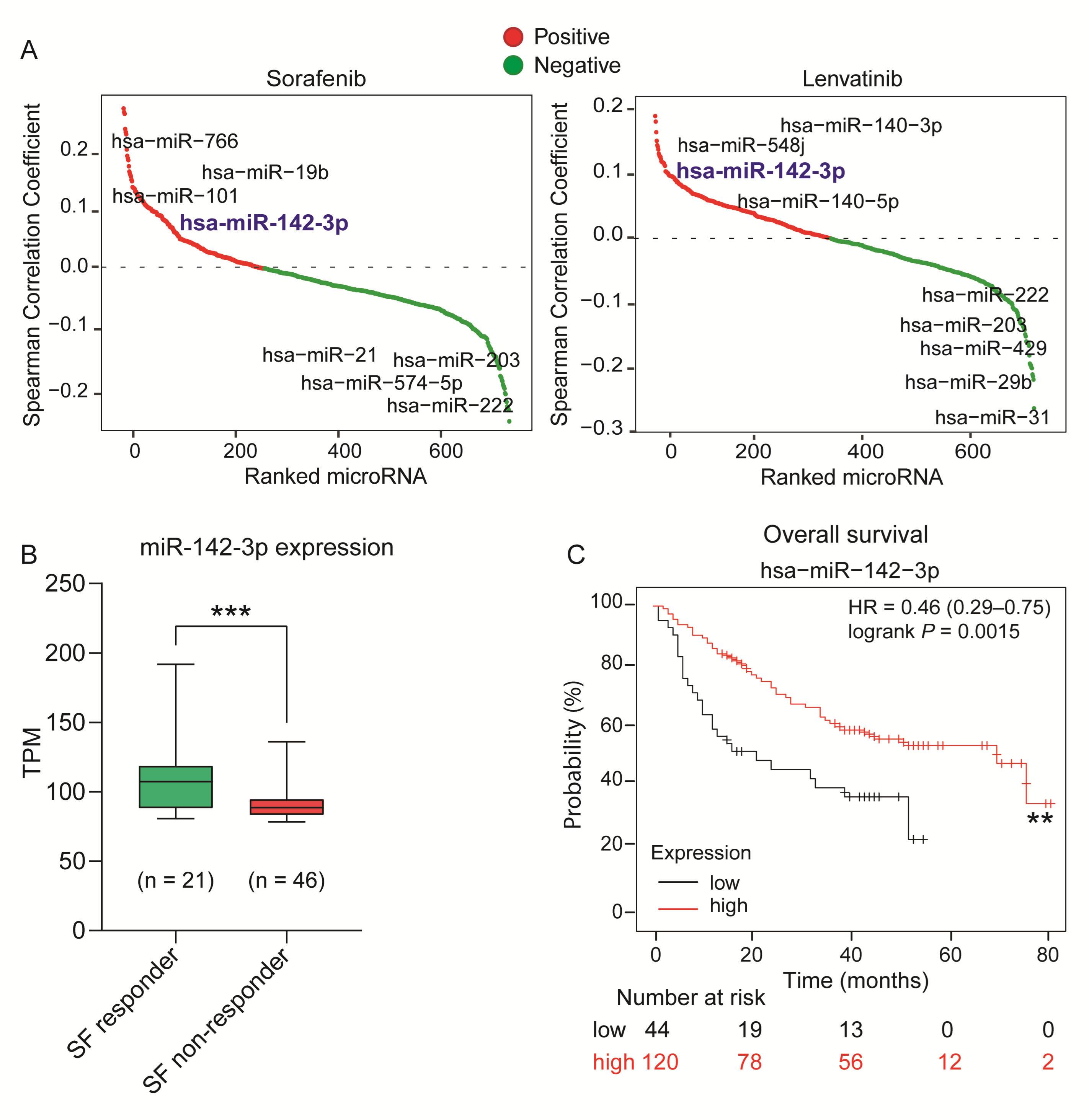
2.1. Identification of miR-142-3p as a Potential Tumor Suppressor miRNA in HCC
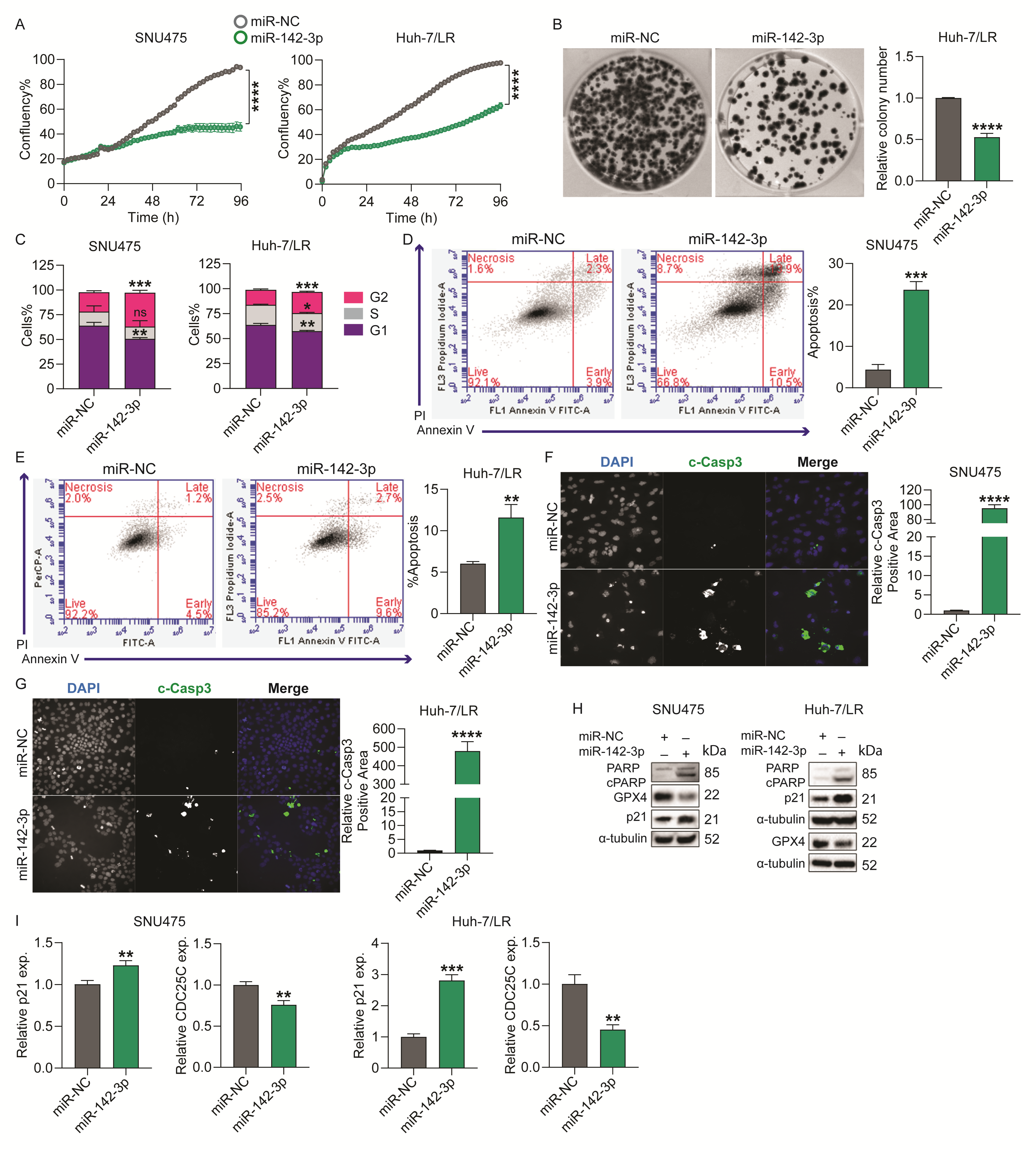
2.2. MicroRNA-142-3p Inhibits HCC Cell Survival by Regulating Cell Cycle and Apoptosis
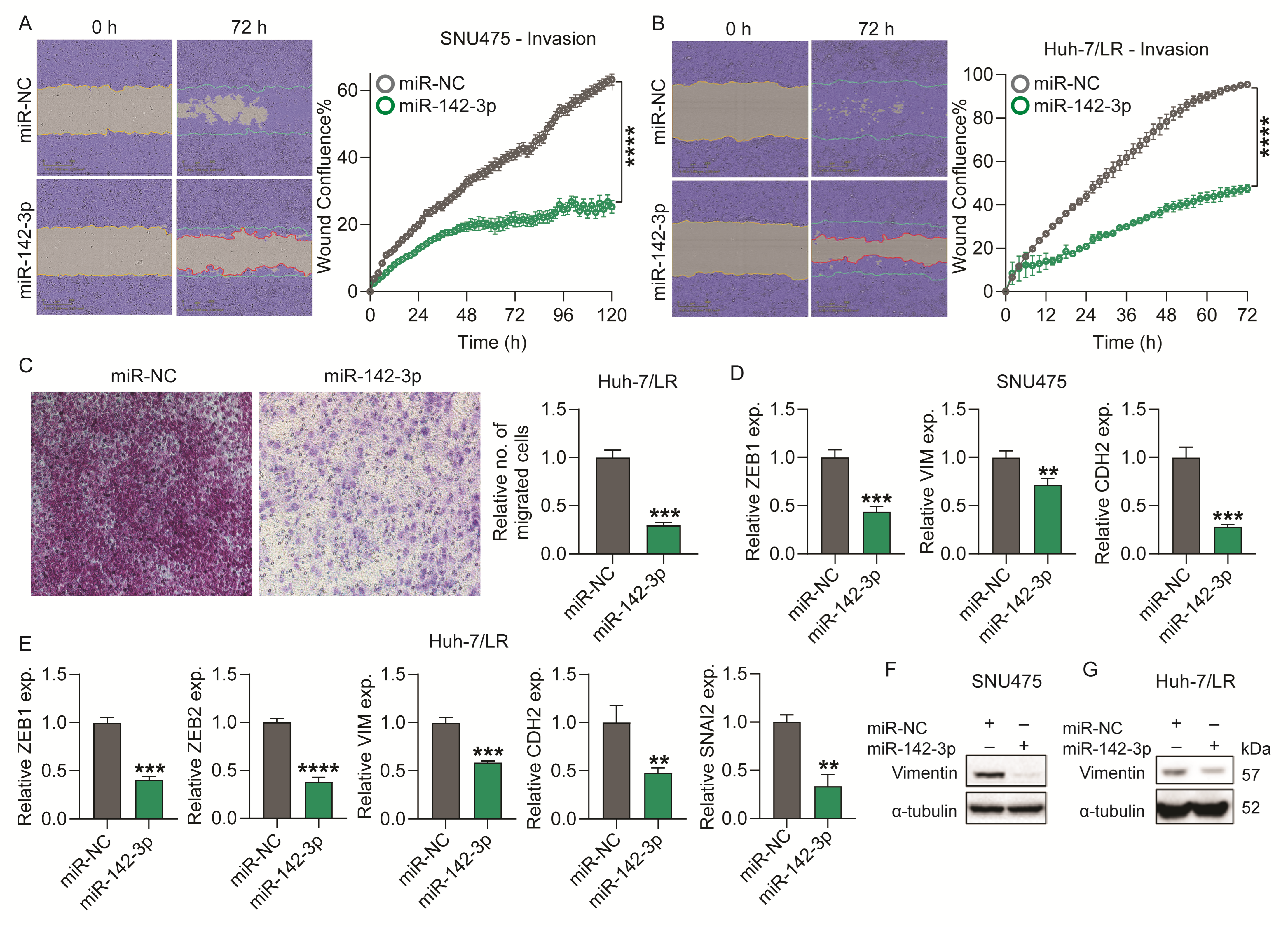
2.3. Overexpression of miR-142-3p Suppresses HCC Cell Chemotaxis and Invasion
2.4. Identification of miR-142-3p Downstream Gene Targets and Molecular Pathways
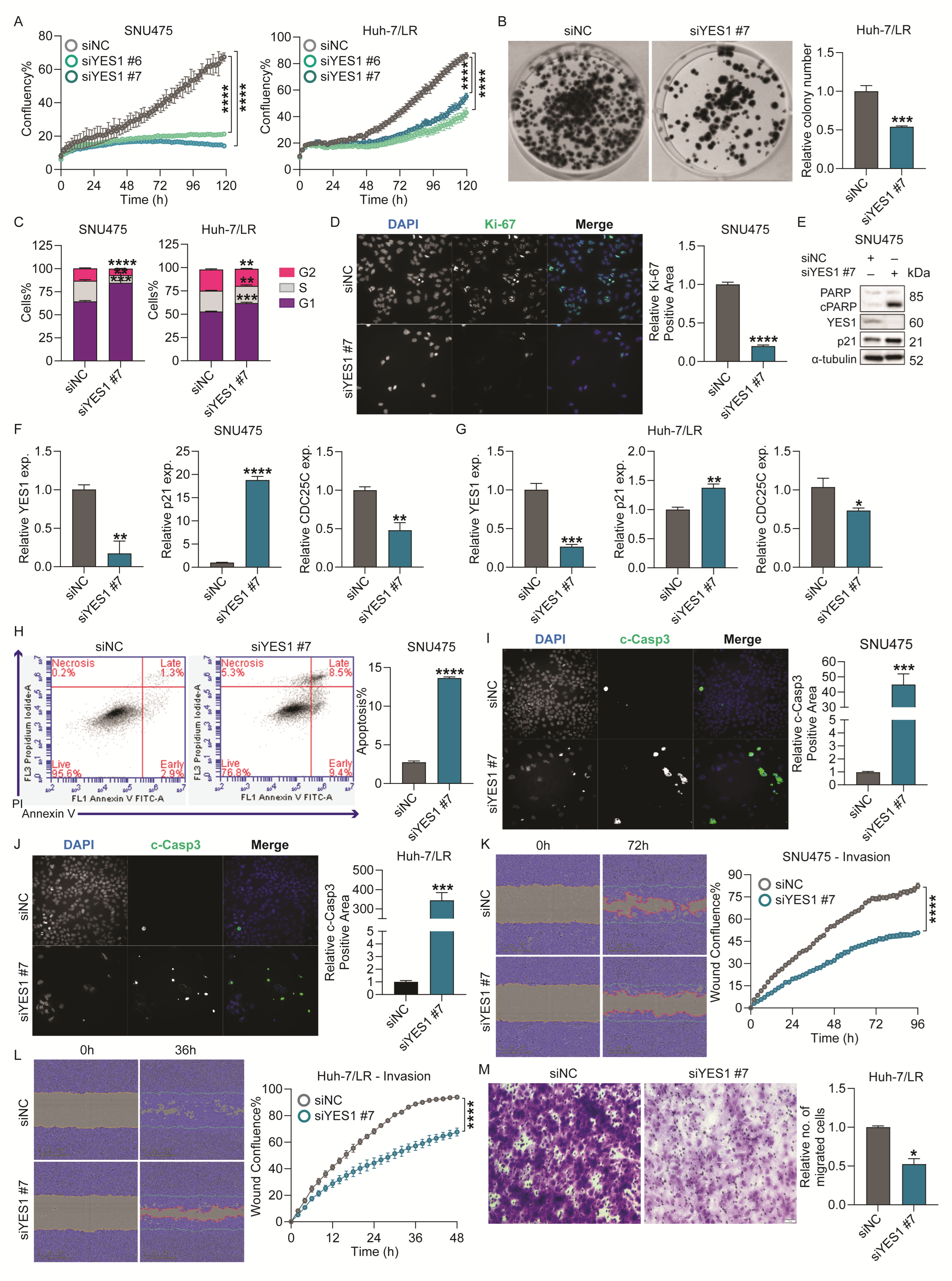
2.5. YES1 Knockdown Reduced Survival, Colony Formation and Migration/Invasion of TKI-Resistant HCC Cells In Vitro
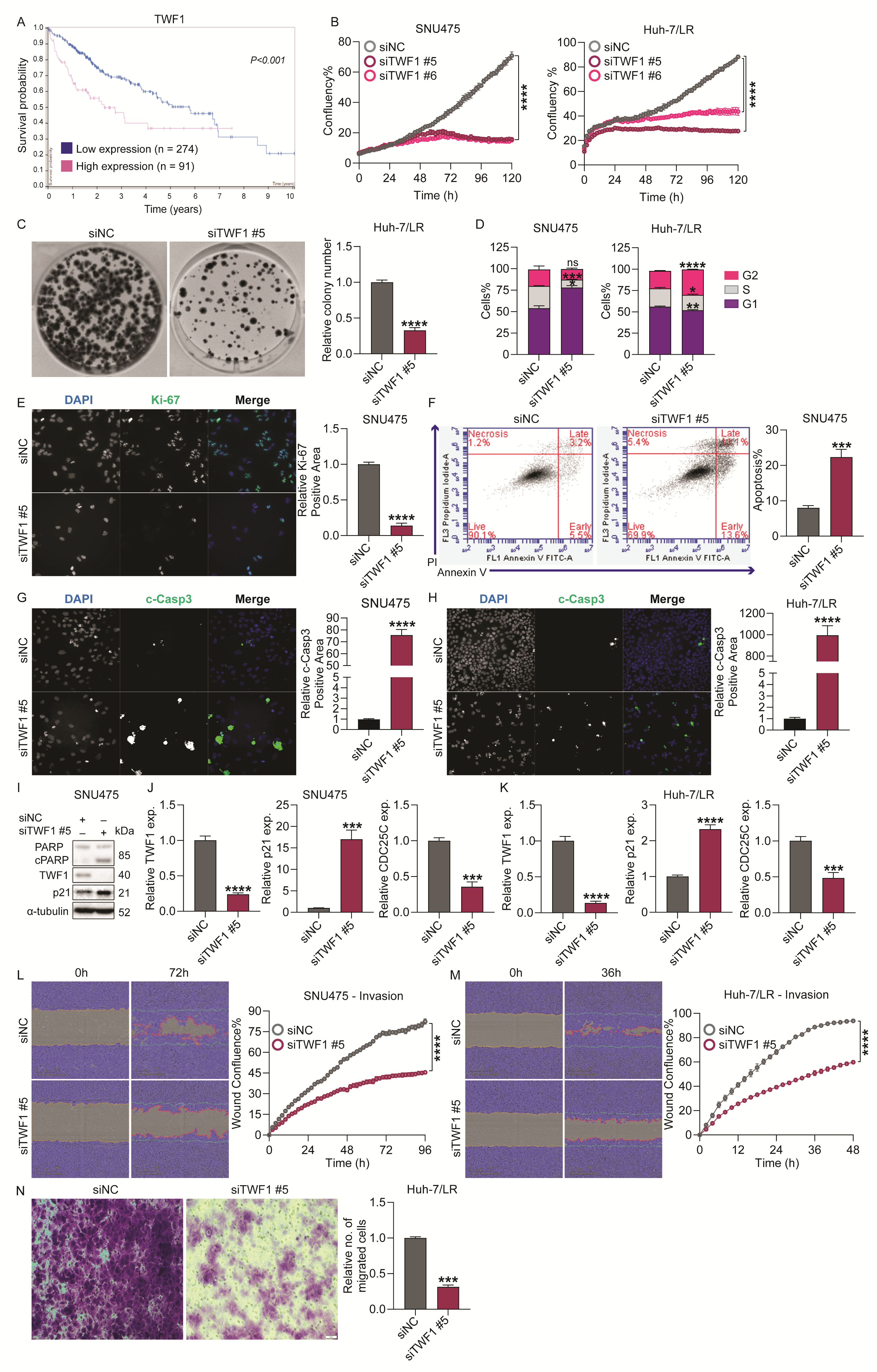
2.6. TWF1 Knockdown Reduced Survival, Colony Formation, and Migration/Invasion Capabilities of TKI-Resistant HCC Cells In Vitro
2.7. MicroRNA-142-3p Restores Lenvatinib Sensitivity in TKI-Resistant HCC Cells Partially via Targeting YES1 and TWF1
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Data Retrieval from Publicly Available Databases
4.3. Cell Culture
4.4. Transfection of miRNA and siRNA Molecules
4.5. RNA Isolation and Real-Time-Quantitative Polymerase Chain Reaction (RT-qPCR) Assay
4.6. Protein Isolation and Western Immunoblotting
4.7. Incucyte Proliferation Assay
4.8. Cell Cycle and Annexin V-FITC/PI Apoptosis Assays
4.9. Incucyte Wound Healing Assay
4.10. Transwell Chemotaxis Assay
4.11. Colony Formation Assay
4.12. Immunofluorescence Assay
4.13. LysoTracker Deep Red and DQ Red BSA Staining
4.14. RNA Sequencing and Bioinformatic Analysis
4.15. Luciferase Reporter Assay
4.16. Dose–Response Curves, Determination of IC50 Values, and Combinational Synergy Assays
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.K.; Tsui, Y.M.; Ho, D.W.; Ng, I.O. Cellular heterogeneity and plasticity in liver cancer. Semin. Cancer Biol. 2022, 82, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Ladd, A.D.; Duarte, S.; Sahin, I.; Zarrinpar, A. Mechanisms of drug resistance in HCC. Hepatology 2024, 79, 926–940. [Google Scholar] [CrossRef]
- Duan, B.; Huang, C.; Bai, J.; Zhang, Y.L.; Wang, X.; Yang, J.; Li, J. Multidrug Resistance. In Hepatocellular Carcinoma; Exon Publications: Brisbane, Australia, 2019; pp. 141–158. [Google Scholar]
- Rani, V.; Sengar, R.S. Biogenesis and mechanisms of microRNA-mediated gene regulation. Biotechnol. Bioeng. 2022, 119, 685–692. [Google Scholar] [CrossRef]
- Zhou, K.; Liu, M.; Cao, Y. New insight into microRNA functions in cancer: Oncogene–microRNA–tumor suppressor gene network. Front. Mol. Biosci. 2017, 4, 46. [Google Scholar] [CrossRef]
- Roy, B.; Ghose, S.; Biswas, S. Therapeutic strategies for miRNA delivery to reduce hepatocellular carcinoma. Semin. Cell Dev. Biol. 2022, 124, 134–144. [Google Scholar] [CrossRef]
- Li, J.; Bao, H.; Huang, Z.; Liang, Z.; Wang, M.; Lin, N.; Ni, C.; Xu, Y. Little things with significant impact: miRNAs in hepatocellular carcinoma. Front. Oncol. 2023, 13, 1191070. [Google Scholar] [CrossRef]
- Iwamoto, H.; Suzuki, H.; Masuda, A.; Sakaue, T.; Nakamura, T.; Tanaka, T.; Sakai, M.; Imamura, Y.; Yano, H.; Torimura, T.; et al. A tumor endothelial cell-specific microRNA replacement therapy for hepatocellular carcinoma. iScience 2024, 27, 108797. [Google Scholar] [CrossRef]
- Szczepanek, J.; Skorupa, M.; Tretyn, A. MicroRNA as a Potential Therapeutic Molecule in Cancer. Cells 2022, 11, 1008. [Google Scholar] [CrossRef]
- Oliver, F.J.; de la Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.C.; de Murcia, G.; Murcia, J.M.d. Importance of Poly(ADP-ribose) Polymerase and Its Cleavage in Apoptosis: Lesson From An Uncleavable Mutant*. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Lan, G.; Zhu, L.; Guo, Q. Breaking the Barriers of Therapy Resistance: Harnessing Ferroptosis for Effective Hepatocellular Carcinoma Therapy. J. Hepatocell. Carcinoma 2024, 11, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Lossaint, G.; Horvat, A.; Gire, V.; Bačević, K.; Mrouj, K.; Charrier-Savournin, F.; Georget, V.; Fisher, D.; Dulić, V. Reciprocal regulation of p21 and Chk1 controls the cyclin D1-RB pathway to mediate senescence onset after G2 arrest. J. Cell Sci. 2022, 135, jcs259114. [Google Scholar] [CrossRef]
- Xu, T.; He, B.S.; Pan, B.; Pan, Y.Q.; Sun, H.L.; Liu, X.X.; Xu, X.N.; Chen, X.X.; Zeng, K.X.; Xu, M. MiR-142-3p functions as a tumor suppressor by targeting RAC1/PAK1 pathway in breast cancer. J. Cell. Physiol. 2020, 235, 4928–4940. [Google Scholar] [CrossRef]
- Xie, N.; Meng, Q.; Zhang, Y.; Luo, Z.; Xue, F.; Liu, S.; Li, Y.; Huang, Y. MicroRNA-142-3p suppresses cell proliferation, invasion and epithelial-to-mesenchymal transition via RAC1-ERK1/2 signaling in colorectal cancer. Mol. Med. Rep. 2021, 24, 568. [Google Scholar] [CrossRef]
- Zeng, Y.; He, H.; Zhang, Y.; Wang, X.; Yang, L.; An, Z. CCNB2, TOP2A, and ASPM Reflect the Prognosis of Hepatocellular Carcinoma, as Determined by Weighted Gene Coexpression Network Analysis. Biomed. Res. Int. 2020, 2020, 4612158. [Google Scholar] [CrossRef]
- Meng, Z.; Wu, J.; Liu, X.; Zhou, W.; Ni, M.; Liu, S.; Guo, S.; Jia, S.; Zhang, J. Identification of potential hub genes associated with the pathogenesis and prognosis of hepatocellular carcinoma via integrated bioinformatics analysis. J. Int. Med. Res. 2020, 48, 0300060520910019. [Google Scholar] [CrossRef]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Kurisu, S.; Takenawa, T. WASP and WAVE family proteins: Friends or foes in cancer invasion? Cancer Sci. 2010, 101, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Wiza, C.; Nascimento, E.B.; Ouwens, D.M. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1453–E1460. [Google Scholar] [CrossRef]
- Huo, G.; Wang, Y.; Chen, J.; Song, Y.; Zhang, C.; Guo, H.; Zuo, R.; Zhu, F.; Cui, J.; Chen, W.; et al. A Pan-Cancer Analysis of the Oncogenic Role of Twinfilin Actin Binding Protein 1 in Human Tumors. Front. Oncol. 2021, 11, 692136. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, N.; Nakanishi, Y.; Mizuno, T.; Horiguchi-Takei, K.; Akiyama, N.; Tanimura, H.; Hasegawa, M.; Satoh, Y.; Tachibana, Y.; Fujii, T.; et al. YES1 Is a Targetable Oncogene in Cancers Harboring YES1 Gene Amplification. Cancer Res. 2019, 79, 5734–5745. [Google Scholar] [CrossRef]
- Du, Y.; Wu, S.; Xi, S.; Xu, W.; Sun, L.; Yan, J.; Gao, H.; Wang, Y.; Zheng, J.; Wang, F.; et al. ASH1L in Hepatoma Cells and Hepatic Stellate Cells Promotes Fibrosis-Associated Hepatocellular Carcinoma by Modulating Tumor-Associated Macrophages. Adv. Sci. 2024, 11, e2404756. [Google Scholar] [CrossRef]
- Miftah, H.; Naji, O.; Ssi, S.A.; Ghouzlani, A.; Lakhdar, A.; Badou, A. NR2F6, a new immune checkpoint that acts as a potential biomarker of immunosuppression and contributes to poor clinical outcome in human glioma. Front. Immunol. 2023, 14, 1139268. [Google Scholar] [CrossRef]
- Metelli, A.; Wu, B.X.; Fugle, C.W.; Rachidi, S.; Sun, S.; Zhang, Y.; Wu, J.; Tomlinson, S.; Howe, P.H.; Yang, Y.; et al. Surface Expression of TGFβ Docking Receptor GARP Promotes Oncogenesis and Immune Tolerance in Breast Cancer. Cancer Res. 2016, 76, 7106–7117. [Google Scholar] [CrossRef]
- Hu, L.; Bai, Z.G.; Ma, X.M.; Bai, N.; Zhang, Z.T. MRFAP1 plays a protective role in neddylation inhibitor MLN4924-mediated gastric cancer cell death. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8273–8280. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, D.; Tao, J.; Xu, M.; Zhang, X.; Hou, H. Role of YES1 signaling in tumor therapy resistance. Cancer Innov. 2023, 2, 210–218. [Google Scholar] [CrossRef]
- Kook, E.; Lee, J.; Kim, D.H. YES1 as a potential target to overcome drug resistance in EGFR-deregulated non-small cell lung cancer. Arch. Toxicol. 2024, 98, 1437–1455. [Google Scholar] [CrossRef] [PubMed]
- Bockhorn, J.; Dalton, R.; Nwachukwu, C.; Huang, S.; Prat, A.; Yee, K.; Chang, Y.-F.; Huo, D.; Wen, Y.; Swanson, K.E.; et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat. Commun. 2013, 4, 1393. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, S.; Vartiainen, M.; Lappalainen, P. Twinfilin, a molecular mailman for actin monomers. J. Cell Sci. 2002, 115 Pt 5, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.B.; Collins, A.; Goode, B.L. High-speed depolymerization at actin filament ends jointly catalysed by Twinfilin and Srv2/CAP. Nat. Cell Biol. 2015, 17, 1504–1511. [Google Scholar] [CrossRef]
- Dong, Y.; Quan, C. NPFs-mediated actin cytoskeleton: A new viewpoint on autophagy regulation. Cell Commun. Signal. 2024, 22, 111. [Google Scholar] [CrossRef]
- Wang, Y.; Zuo, R.; Huo, G.; Han, Z.; He, Y.; Luo, Y.; Chen, L.; Li, G.; Cui, J.; Zhu, F.; et al. TWF1 induces autophagy and accelerates malignant phenotype in lung adenocarcinoma via inhibiting the cAMP signaling pathway. FASEB J. 2023, 37, e23051. [Google Scholar] [CrossRef]
- Hua, Y.Q.; Zhu, Y.D.; Xie, G.Q.; Zhang, K.; Sheng, J.; Zhu, Z.F.; Ning, Z.Y.; Chen, H.; Chen, Z.; Meng, Z.Q.; et al. Long non-coding SBF2-AS1 acting as a competing endogenous RNA to sponge microRNA-142-3p to participate in gemcitabine resistance in pancreatic cancer via upregulating TWF1. Aging 2019, 11, 8860–8878. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Garmendia, I.; Redin, E.; Montuenga, L.M.; Calvo, A. Yes1: A novel therapeutic target and biomarker in cancer. Mol. Cancer Ther. 2022, 21, 1371–1380. [Google Scholar] [CrossRef]
- Schwartz-Roberts, J.L.; Clarke, R. Blockage of Lysosomal Degradation Is Detrimental to Cancer Cell Survival: Role of Autophagy Activation. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2014; pp. 269–281. [Google Scholar]
- Tarailo-Graovac, M.; Drögemöller, B.I.; Wasserman, W.W.; Ross, C.J.; van den Ouweland, A.M.; Darin, N.; Kollberg, G.; Van Karnebeek, C.D.; Blomqvist, M. Identification of a large intronic transposal insertion in SLC17A5 causing sialic acid storage disease. Orphanet J. Rare Dis. 2017, 12, 28. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Z.; Sun, D.; Sweeney, S.T.; Gao, F.-B. Syntaxin 13, a genetic modifier of mutant CHMP2B in frontotemporal dementia, is required for autophagosome maturation. Mol. Cell 2013, 52, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Dong, J.; Zhao, Y.; Peng, X.; Tang, J.; Chen, X.; Wang, L.; Hu, D.N.; Reinach, P.S.; Qu, J.; et al. miR-142-3p suppresses uveal melanoma by targeting CDC25C, TGFβR1, GNAQ, WASL, and RAC1. Cancer Manag. Res. 2019, 11, 4729–4742. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Pang, D.; Yu, S. Serum level of miR-142-3p predicts prognostic outcome for colorectal cancer following curative resection. J. Int. Med. Res. 2019, 47, 2116–2125. [Google Scholar] [CrossRef]
- Mansoori, B.; Duijf, P.H.G.; Mohammadi, A.; Safarzadeh, E.; Ditzel, H.J.; Gjerstorff, M.F.; Cho, W.C.; Baradaran, B. MiR-142-3p targets HMGA2 and suppresses breast cancer malignancy. Life Sci. 2021, 276, 119431. [Google Scholar] [CrossRef]
- Li, J.; Yue, H.; Li, W.; Zhu, G.; Zhu, T.; Chen, R.; Lu, X. Bevacizumab confers significant improvements in survival for ovarian cancer patients with low miR-25 expression and high miR-142 expression. J. Ovarian Res. 2021, 14, 166. [Google Scholar] [CrossRef]
- Sharma, P.; Yadav, P.; Jain, R.P.; Bera, A.K.; Karunagaran, D. miR-142-3p simultaneously targets HMGA1, HMGA2, HMGB1, and HMGB3 and inhibits tumorigenic properties and in-vivo metastatic potential of human cervical cancer cells. Life Sci. 2022, 291, 120268. [Google Scholar] [CrossRef]
- Jin, C.; Xiao, L.; Zhou, Z.; Zhu, Y.; Tian, G.; Ren, S. MiR-142-3p suppresses the proliferation, migration and invasion through inhibition of NR2F6 in lung adenocarcinoma. Hum. Cell 2019, 32, 437–446. [Google Scholar] [CrossRef]
- Hua, S.; Liu, C.; Liu, L.; Wu, D. miR-142-3p inhibits aerobic glycolysis and cell proliferation in hepatocellular carcinoma via targeting LDHA. Biochem. Biophys. Res. Commun. 2018, 496, 947–954. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, L.Q.; Huang, Y.; Quan, J.; Hu, X.; Tang, D.; Kang, R.; Li, N.; Fan, X.G. miR-142-3p Inhibits the Metastasis of Hepatocellular Carcinoma Cells by Regulating HMGB1 Gene Expression. Curr. Mol. Med. 2018, 18, 135–141. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Liu, W.; Yin, Y.; Jiang, J.; Yan, C.; Wang, Y.; Li, L. Mechanism of HBV-positive liver cancer cell exosomal miR-142-3p by inducing ferroptosis of M1 macrophages to promote liver cancer progression. Transl. Cancer Res. 2022, 11, 1173–1187. [Google Scholar] [CrossRef]
- He, C.; Liu, Z.; Jin, L.; Zhang, F.; Peng, X.; Xiao, Y.; Wang, X.; Lyu, Q.; Cai, X. lncRNA TUG1-Mediated Mir-142-3p Downregulation Contributes to Metastasis and the Epithelial-to-Mesenchymal Transition of Hepatocellular Carcinoma by Targeting ZEB1. Cell. Physiol. Biochem. 2018, 48, 1928–1941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, J.; Zhou, H.; Chen, Y.; Zhi, Y.; Zhang, B.; Chen, L.; Chu, X.; Wang, R.; Zhang, C. PU.1/microRNA-142-3p targets ATG5/ATG16L1 to inactivate autophagy and sensitize hepatocellular carcinoma cells to sorafenib. Cell Death Dis. 2018, 9, 312. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- This Phase III Trial Is Trying to Find Out If a New Targeted Therapy (Ipatasertib) Can Slow the Growth of Advanced Breast Cancer When Added to Standard Hormone Therapy (Fulvestrant). NCT0465058. 2024. Available online: https://trials.cancervic.org.au/details/feed-cta-trial311 (accessed on 21 August 2024).
- Mariya Rozenblit, A.M.K. A Phase Ib/III, Open-label, Randomised Study of Capivasertib Plus CDK4/6 Inhibitors and Fulvestrant Versus CDK4/6 Inhibitors and Fulvestrant in Hormone Receptor-Positive and Human Epidermal Growth Factor Receptor 2-Negative Locally Advanced, Unresectable or Metastatic Breast Cancer (CAPItello-292). Study HIC#:2000036257. 2024. Available online: https://clinicaltrials.gov/study/NCT04862663 (accessed on 30 September 2024).
- Vo, K.T.; Sabnis, A.J.; Williams, P.M.; Roy-Chowdhuri, S.; Patton, D.R.; Coffey, B.; Reid, J.M.; Piao, J.; Saguilig, L.; Alonzo, T.A.; et al. Phase II Study of Ulixertinib in Children and Young Adults With Tumors Harboring Activating Mitogen-Activated Protein Kinase Pathway Alterations: APEC1621J of the National Cancer Institute-Children’s Oncology Group Pediatric MATCH Trial. JCO Precis. Oncol. 2024, 8, e2400103. [Google Scholar] [CrossRef]
- Tolcher, A.; Flaherty, K.; Shapiro, G.I.; Berlin, J.; Witzig, T.; Habermann, T.; Bullock, A.; Rock, E.; Elekes, A.; Lin, C.; et al. A First-in-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients with Advanced Cancers. Oncologist 2018, 23, 658-e72. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Kook, E.; Chun, K.S.; Kim, D.H. Emerging Roles of YES1 in Cancer: The Putative Target in Drug Resistance. Int. J. Mol. Sci. 2024, 25, 1450. [Google Scholar] [CrossRef]
- Ingles, N.; Piemonte, K.; Singh, S.; Weber-Bonk, K.; Keri, R. Abstract P1-13-11: YES1 is a targetable vulnerability for improving taxane response in triple-negative breast cancer. Cancer Res. 2023, 83, P1-13. [Google Scholar] [CrossRef]
- Lindauer, M.; Hochhaus, A. Dasatinib. Recent. Results Cancer Res. 2018, 212, 29–68. [Google Scholar] [CrossRef]
- Perez, M.; Lucena-Cacace, A.; Marín-Gómez, L.M.; Padillo-Ruiz, J.; Robles-Frias, M.J.; Saez, C.; Garcia-Carbonero, R.; Carnero, A. Dasatinib, a Src inhibitor, sensitizes liver metastatic colorectal carcinoma to oxaliplatin in tumors with high levels of phospho-Src. Oncotarget 2016, 7, 33111–33124. [Google Scholar] [CrossRef]
- Kadota, H.; Yuge, R.; Shimizu, D.; Miyamoto, R.; Otani, R.; Hiyama, Y.; Takigawa, H.; Hayashi, R.; Urabe, Y.; Kitadai, Y.; et al. Anti-Programmed Cell Death-1 Antibody and Dasatinib Combination Therapy Exhibits Efficacy in Metastatic Colorectal Cancer Mouse Models. Cancers 2022, 14, 6146. [Google Scholar] [CrossRef] [PubMed]
- Guégan, J.P.; Lapouge, M.; Voisin, L.; Saba-El-Leil, M.K.; Tanguay, P.L.; Lévesque, K.; Brégeon, J.; Mes-Masson, A.M.; Lamarre, D.; Haibe-Kains, B.; et al. Signaling by the tyrosine kinase Yes promotes liver cancer development. Sci. Signal. 2022, 15, eabj4743. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, A.; Seike, T.; Danjo, T.; Nakajima, T.; Otsubo, N.; Yamaguchi, D.; Tsuji, Y.; Hamaguchi, K.; Yasunaga, M.; Nishiya, Y.; et al. The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am. J. Cancer Res. 2020, 10, 4399–4415. [Google Scholar]
- Song, X.; Kong, F.; Zong, Z.; Ren, M.; Meng, Q.; Li, Y.; Sun, Z. miR-124 and miR-142 enhance cisplatin sensitivity of non-small cell lung cancer cells through repressing autophagy via directly targeting SIRT1. RSC Adv. 2019, 9, 5234–5243. [Google Scholar] [CrossRef]
- Duan, B.; Zhang, H.; Zhu, Z.; Yan, X.; Ji, Z.; Li, J. LncRNA LINC01871 sponging miR-142-3p to modulate ZYG11B promotes the chemoresistance of colorectal cancer cells by inducing autophagy. Anticancer. Drugs 2023, 34, 827–836. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, J.; Tao, S.; Zhang, S.; Mao, L.; Shi, X.; Wang, W.; Cheng, C.; Shi, Y.; Yang, Q. miR-142-3p improves paclitaxel sensitivity in resistant breast cancer by inhibiting autophagy through the GNB2-AKT-mTOR pathway. Cell. Signal. 2023, 103, 110566. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Role of Tumor Cell Intrinsic and Host Autophagy in Cancer. Cold Spring Harb. Perspect. Med. 2024, 14, a041539. [Google Scholar] [CrossRef]
- O’ Hara, M.H.; Karasic, T.B.; Vasilevskaya, I.; Redlinger, M.; Loaiza-Bonilla, A.; Teitelbaum, U.R.; Giantonio, B.J.; Damjanov, N.; Reiss, K.A.; Rosen, M.A.; et al. Phase II Trial of the Autophagy Inhibitor Hydroxychloroquine with FOLFOX and Bevacizumab in Front Line Treatment of Metastatic Colorectal Cancer; American Society of Clinical Oncology: Alexandria, VA, USA, 2017. [Google Scholar]
- Pasquier, B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef]
- Deng, J.; Thennavan, A.; Dolgalev, I.; Chen, T.; Li, J.; Marzio, A.; Poirier, J.T.; Peng, D.H.; Bulatovic, M.; Mukhopadhyay, S.; et al. ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1-mutant lung cancer. Nat. Cancer 2021, 2, 503–514. [Google Scholar] [CrossRef]
- Han, Y.H.; Wang, Y.Q.; Bo, J.Q.; Guo, Y.N.; Liu, L.X. ABCC1 is a predictive biomarker for prognosis and therapy in hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 3597. [Google Scholar]
- Branch, M.R.; Hsu, C.L.; Ohnishi, K.; Shen, W.C.; Lee, E.; Meisenhelder, J.; Winborn, B.; Sopher, B.L.; Taylor, J.P.; Hunter, T.; et al. MAP4K3 inhibits Sirtuin-1 to repress the LKB1-AMPK pathway to promote amino acid-dependent activation of the mTORC1 complex. Life Sci. Alliance 2023, 6, e202201525. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-P.; Chuang, H.-C.; Lee, M.-C.; Tsou, H.-H.; Lee, L.-W.; Li, J.-P.; Tan, T.-H. GLK/MAP4K3 overexpression associates with recurrence risk for non-small cell lung cancer. Oncotarget 2016, 7, 41748. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N. Compensatory angiogenesis and tumor refractoriness. Oncogenesis 2015, 4, e153. [Google Scholar] [CrossRef]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Wyser Rmili, C.; Leow, C.C.; De Palma, M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef]
- Spina, A.; De Pasquale, V.; Cerulo, G.; Cocchiaro, P.; Della Morte, R.; Avallone, L.; Pavone, L.M. HGF/c-MET Axis in Tumor Microenvironment and Metastasis Formation. Biomedicines 2015, 3, 71–88. [Google Scholar] [CrossRef]
- You, W.K.; McDonald, D.M. The hepatocyte growth factor/c-Met signaling pathway as a therapeutic target to inhibit angiogenesis. BMB Rep. 2008, 41, 833–839. [Google Scholar] [CrossRef]
- Tan, P.H.; Chia, S.S.; Toh, S.L.; Goh, J.C.; Nathan, S.S. The dominant role of IL-8 as an angiogenic driver in a three-dimensional physiological tumor construct for drug testing. Tissue Eng. Part A 2014, 20, 1758–1766. [Google Scholar] [CrossRef]
- Petreaca, M.L.; Yao, M.; Liu, Y.; Defea, K.; Martins-Green, M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol. Biol. Cell 2007, 18, 5014–5023. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, L.; Wang, X.; Jin, H. RNA-based therapeutics: An overview and prospectus. Cell Death Dis. 2022, 13, 644. [Google Scholar] [CrossRef]
- Roy, R.; Winteringham, L.N.; Lassmann, T.; Forrest, A.R.R. Expression Levels of Therapeutic Targets as Indicators of Sensitivity to Targeted Therapeutics. Mol. Cancer Ther. 2019, 18, 2480–2489. [Google Scholar] [CrossRef]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.-Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N.; et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Kabir, T.D.; Ganda, C.; Brown, R.M.; Beveridge, D.J.; Richardson, K.L.; Chaturvedi, V.; Candy, P.; Epis, M.; Wintle, L.; Kalinowski, F.; et al. A microRNA-7/growth arrest specific 6/TYRO3 axis regulates the growth and invasiveness of sorafenib-resistant cells in human hepatocellular carcinoma. Hepatology 2018, 67, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahboobnia, K.; Kabir, T.D.; Hou, R.; Liu, P.; Forrest, A.; Beveridge, D.J.; Richardson, K.L.; Stuart, L.M.; Yeoh, G.C.; Leedman, P.J. MicroRNA-142-3p Overcomes Drug Resistance in Hepatocellular Carcinoma by Targeting YES1 and TWF1. Int. J. Mol. Sci. 2025, 26, 4161. https://doi.org/10.3390/ijms26094161
Mahboobnia K, Kabir TD, Hou R, Liu P, Forrest A, Beveridge DJ, Richardson KL, Stuart LM, Yeoh GC, Leedman PJ. MicroRNA-142-3p Overcomes Drug Resistance in Hepatocellular Carcinoma by Targeting YES1 and TWF1. International Journal of Molecular Sciences. 2025; 26(9):4161. https://doi.org/10.3390/ijms26094161
Chicago/Turabian StyleMahboobnia, Khadijeh, Tasnuva D. Kabir, Rui Hou, Peiwen Liu, Alistair Forrest, Dianne J. Beveridge, Kirsty L. Richardson, Lisa M. Stuart, George C. Yeoh, and Peter J. Leedman. 2025. "MicroRNA-142-3p Overcomes Drug Resistance in Hepatocellular Carcinoma by Targeting YES1 and TWF1" International Journal of Molecular Sciences 26, no. 9: 4161. https://doi.org/10.3390/ijms26094161
APA StyleMahboobnia, K., Kabir, T. D., Hou, R., Liu, P., Forrest, A., Beveridge, D. J., Richardson, K. L., Stuart, L. M., Yeoh, G. C., & Leedman, P. J. (2025). MicroRNA-142-3p Overcomes Drug Resistance in Hepatocellular Carcinoma by Targeting YES1 and TWF1. International Journal of Molecular Sciences, 26(9), 4161. https://doi.org/10.3390/ijms26094161