Abstract
Ab initio B3LYP/6-311++G** calculations have been carried out for the benzoic acid dimer for the stable and saddle point structures. The energy barrier for the proton tunneling amounts to 6.5 kcal/mol. The normal mode frequencies have been computed including modes coupled to the proton tunneling mode. Two-dimensional model potentials, formed from symmetric mode coupling potential and squeezed double well potential, have been fitted to the calculated energy barrier, geometries and frequencies, and used to analyze proton dynamics. The calculated proton tunneling energy splitting in the vibrationally ground states of the low-frequency modes is ~230 cm-1. The two-dimensional model PES predict monotonic increase of the tunneling splitting with the excitation of the planar modes. Depending of the sign of the coupling parameter out-of-plane modes can either suppress or promote the splittings.
Introduction
The importance of proton tunneling in chemical and biological systems is well known, e.g. for the DNA base pairing, as discussed by Löwdin [1]. The phenomenon of potential barrier penetration plays an important role in many branches of physics: quantum field theory, fission of atomic nuclei, scanning tunneling microscopy and solid state physics [2]. Theoretical studies of proton tunneling require the knowledge of multi-dimensional potential energy surfaces (PES’s) which are difficult to obtain from ab initio calculations, especially for electronically excited states. From the theoretical point of view proton transfer in the ground electronic state is more easily tractable. Extensive experimental and theoretical studies have been reported for multidimensional proton tunneling in tropolone [3,4,5,6,7,8,9,10,11,12,13]. Vener et al. [10] studied theoretically multidimensional proton tunneling in tropolone by using adiabatic separation of variables. Smedarchina et al. [11] used the instanton approach to account for tunnelling splittings. Takada and Nakamura [12] on the base of high accuracy ab initio calculations proposed a model potential energy surface (PES) for the electronically ground state and employed it to analyze the proton tunnelling dynamics. Wójcik et al. [13] reported the results of the high accuracy ab initio MO calculations of the potential energy surfaces in the excited à state of tropolone, and by fitting the two- and three-dimensional analytical model potentials to these surfaces and solving the multidimensional vibrational problems, interpreted the existing experimental data. Other systems for which tunnelling have been studied include malonaldehyde [14,15,16,17], formic acid [18], hydrogen-oxalate anion [16], substituted tropolone [19,20] and 5-methyl-9-hydroxyphenalenone (OH and OD) [21], methanol tetramer [22] and hydrogen carbonate dimer ion [23]. Recently a mixed quantum-classical approach has been used to study dynamics of hydrogen-bonded systems [24,25].
System which draws our attention in the present paper is benzoic acid dimer. It is present in the structure of the crystal [26,27,28]. Its proton transfer has been recently studied by inelastic neutron scattering by Plazanet et al. [29] and Fillaux et al. [30]. Vibrational spectra of benzoic acid have been reported in Refs. [31,32]. In this article we present the results of high accuracy ab initio MO and DFT calculations of the potential energy surfaces for the ground state of benzoic acid dimer in the stable and saddle point structures, and by fitting the two-dimensional analytical model potentials to these surfaces and solving the multidimensional vibrational problems, we predict the effects of excitations of the low-frequency in- and out-of-plane modes on the proton tunnelling splittings.
This paper is organized as follows. The results of our quantum chemical calculations for the ground state of benzoic acid dimer are presented in Sec. II. Model studies of the tunnelling are discussed in Sec. III. Concluding remarks are given in Sec. IV.
Quantum Chemical Calculations
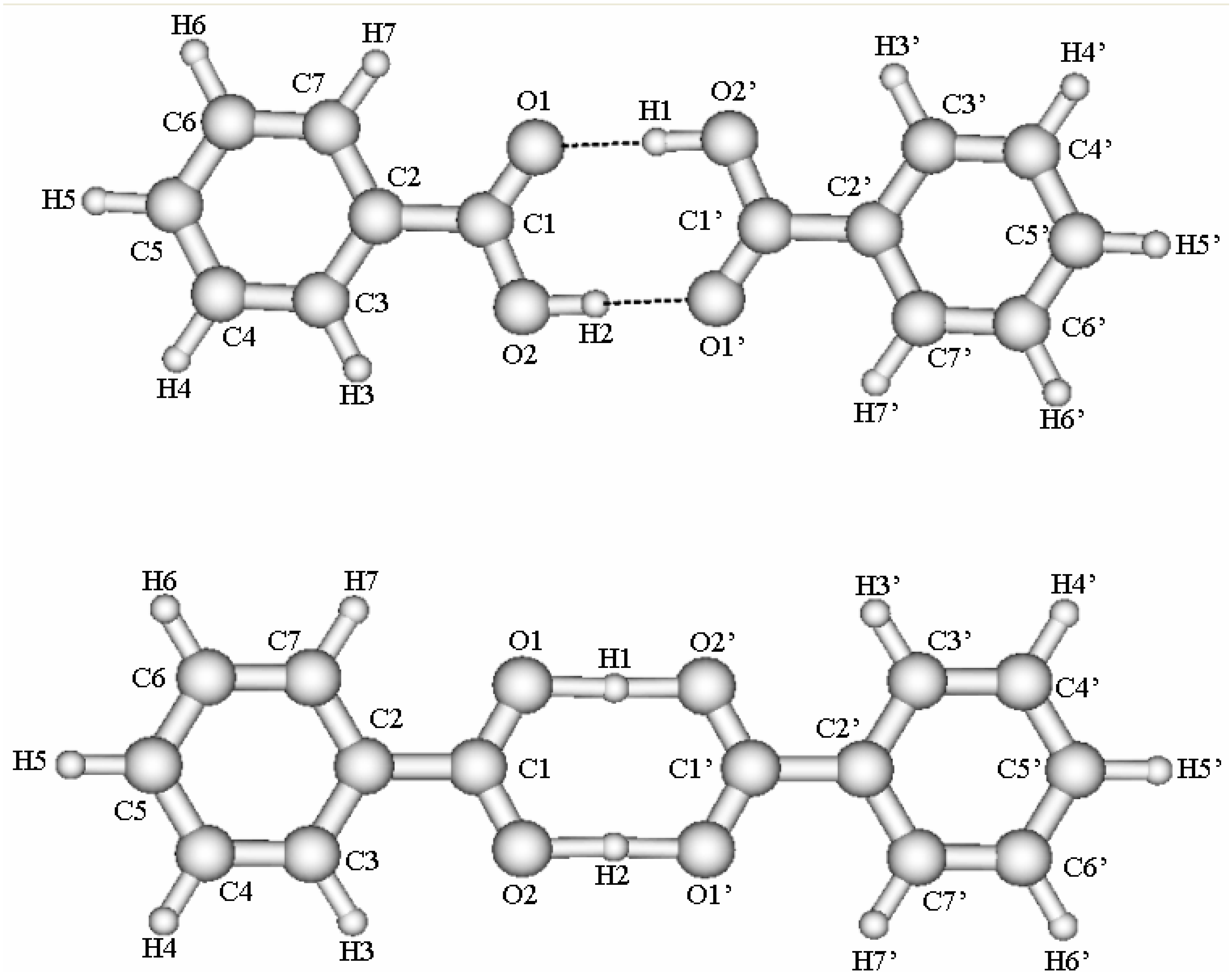
We performed ab initio B3LYP/6-311++G** calculations for the ground state of benzoic acid dimer using the Gaussian 98 program package [33]. The calculations have been done in the Computer Center of Hokkaido University in Sapporo, Japan and the Department of Materials Chemistry, Uppsala University in Sweden. The calculations have been performed for the stable and the saddle point structures. The geometry of benzoic acid dimer in the stable and the saddle point structures, calculated by the B3LYP/6-311++G** method is shown in Fig. 1. Optimized geometry is summarized in Table 1. The complex is planar in the stable and the saddle point structures. The calculated values reproduce the experimental bond lengths and angles reasonably well. There are small discrepancies for C1-O2 and some C-C distances. Experimental C-H distances taken from Ref. [26] are only estimates. The angles are well reproduced except O2C1C2. The influence of the saddle point structure on geometry in hydrogen-bonded cyclic structure is considerable.
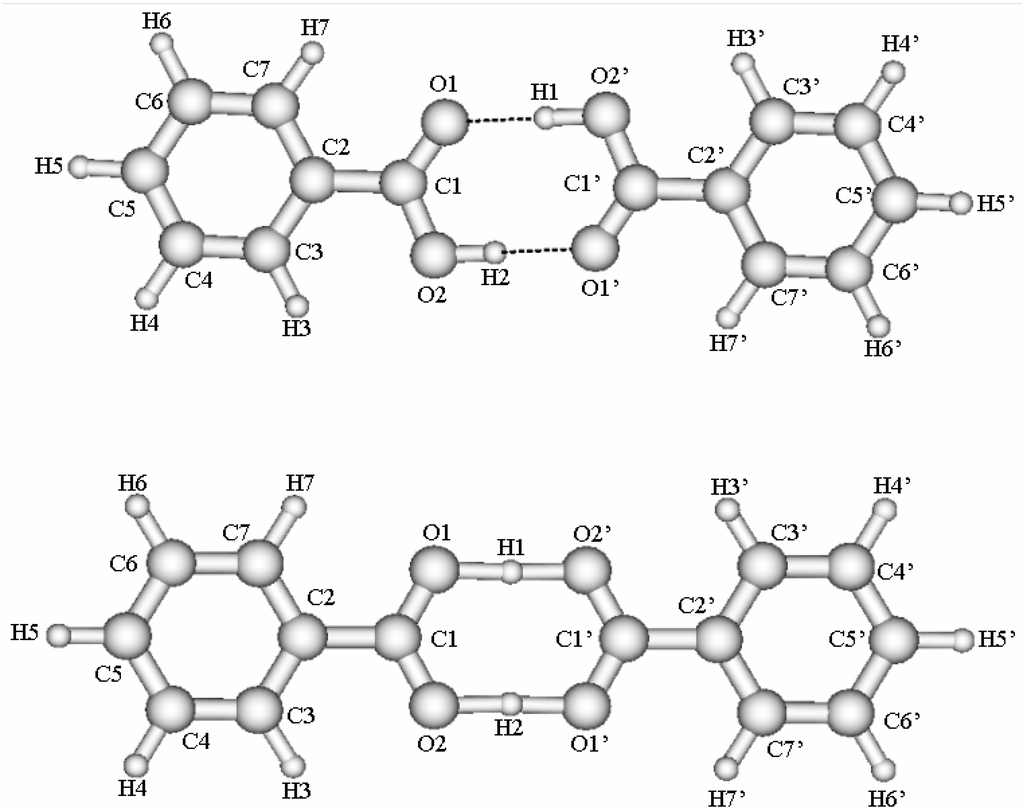
Figure 1.
Benzoic acid in the stable and saddle point structures.
Figure 1.
Benzoic acid in the stable and saddle point structures.
The calculated frequencies and symmetries of the normal modes at the stable and the saddle point structure are summarized in Table 2. The modes calculated by the B3LYP/6-311++G** method and used in the model calculations are pictured in Fig. 2.. The frequencies have not been scaled. The agreement between experimental and calculated frequencies is generally good, especially in the low and medium-frequency regions. The calculated frequencies in the region of O-H and C-H stretchings are overestimated by about 100-150 cm-1 and the frequency of the O-H symmetric stretching mode ν73 is overestimated by ~500 cm-1. This is partly due to harmonic approximation and effects of intermolecular interactions on this mode in the crystal.

Table 1.
Optimized geometries of the stable and saddle point structures of benzoic acid by the B3LYP/6-311++G** (DFT) method.
| Stable structure | Saddle point structure | |||
| DFT | EXP. | DFT | ||
| Bond lengths (Å) | ||||
| O1··· O2’ | 2,663 | 2,64a; 2,629b; 2,633c | 2,410 | |
| O1 – H1 | 1,663 | 1,64b | 1,205 | |
| O2 – H2 | 1,000 | 0,988b | 1,205 | |
| C1 – O1 | 1,230 | 1,24a; 1,268b; 1,263 c | 1,272 | |
| C1 – O2 | 1,323 | 1,29a; 1,275b; 1,275 c | 1,271 | |
| C2 – C1 | 1,486 | 1,48a; 1,484 c | 1,486 | |
| C2 – C3 | 1,400 | 1,39a; 1,390 c | 1,400 | |
| C3 – C4 | 1,391 | 1,42a; 1,387 c | 1,391 | |
| C4 – C5 | 1,395 | 1,36a; 1,379 c | 1,395 | |
| C5 – C6 | 1,395 | 1,37a; 1,384 c | 1,395 | |
| C6 – C7 | 1,390 | 1,41a; 1,401 c | 1,391 | |
| C7 – C2 | 1,400 | 1,39a; 1,392 c | 1,400 | |
| C3 – H3 | 1,082 | 0,79a | 1,082 | |
| C4 – H4 | 1,084 | 0,96a | 1,084 | |
| C5 – H5 | 1,084 | 0,91a | 1,084 | |
| C6 – H6 | 1,084 | 0,96a | 1,084 | |
| C7 – H7 | 1,083 | 0,79a | 1,082 | |
| Bond angles (o) | ||||
| O1H1O2’ | 177,14 | 179,63 | ||
| C1O1H1 | 126,84 | 116,66 | ||
| C1O2H2 | 110,27 | 116,71 | ||
| O1C1O2 | 123,26 | 122 a; 123,2c | 123,78 | |
| O2C1C2 | 114,50 | 118 a; 119,9 c | 118,12 | |
| C2C1O1 | 122,24 | 122 a; 120,2 c | 118,11 | |
| C1C2C3 | 121,40 | 122 a; 118,0 c | 120,04 | |
| C3C2C7 | 119,90 | 119 a; 119,9 c | 119,92 | |
| C7C2C1 | 118,70 | 119 a; 118,8 c | 120,04 | |
| C2C3C4 | 119,86 | 118 a; 120,1 c | 119,94 | |
| C3C4C5 | 120,02 | 123 a; 119,9 c | 120,03 | |
| C4C5C6 | 120,15 | 118 a; 120,3 c | 120,16 | |
| C5C6C7 | 119,98 | 122 a; 119,7 c | 120,03 | |
| C6C7C2 | 120,02 | 120 a; 119,8 c | 119,94 | |
| C2C3H3 | 119,48 | 119,11 | ||
| H3C3C4 | 120,66 | 120,95 | ||
| C3C4H4 | 119,85 | 119,89 | ||
| H4C4C5 | 120,07 | 120,08 | ||
| C4C5H5 | 119,91 | 119,92 | ||
| H5C5C6 | 119,94 | 119,92 | ||
| C5C6H6 | 120,09 | 120,08 | ||
| H6C6C7 | 119,93 | 119,89 | ||
| C6C7H7 | 121,12 | 120,95 | ||
| H7C7C2 | 118,86 | 119,12 | ||
a Ref. 26, b Ref. 27, c Ref. 28.
Table 2.
Calculated normal mode frequencies for the stable and saddle point structures of benzoic acid by the B3LYP/6-311++G** (DFT) method.
| No. | Symmetry | Frequency (cm-1) | ||
| Stable structure | Saddle point structure | Literature Infrared | ||
| 1 | AU | 22 | 22 | 25b |
| 2 | AU | 33 | 35 | 35b |
| 3 | BG | 41 | 17 | 41b |
| 4 | BU | 60 | 75 | 71b |
| 5 | BG | 64 | 62 | 79b |
| 6 | AU | 87 | 79 | 94a |
| 7 | AG | 106 | 120 | 110b |
| 8 | AG | 114 | 201 | 127b |
| 9 | AU | 172 | 174 | 146a |
| 10 | BG | 179 | 177 | |
| 11 | AG | 259 | 294 | |
| 12 | BU | 283 | 363 | |
| 13 | BU | 389 | 385 | |
| 14 | BG | 414 | 413 | |
| 15 | AU | 415 | 415 | |
| 16 | AG | 423 | 588 | |
| 17 | AU | 446 | 448 | 421a |
| 18 | BG | 448 | 451 | |
| 19 | AG | 512 | 529 | |
| 20 | BU | 546 | 723 | 491a |
| 21 | AG | 632 | 632 | |
| 22 | BU | 632 | 631 | 615a |
| 23 | AG | 669 | 729 | |
| 24 | BU | 679 | 698 | 669a |
| 25 | BG | 694 | 699 | |
| 26 | AU | 696 | 697 | 687a |
| 27 | BG | 707 | 680 | |
| 28 | AU | 724 | 729 | 711a |
| 29 | BG | 804 | 788 | |
| 30 | AG | 804 | 824 | |
| 31 | BU | 816 | 866 | 767a |
| 32 | AU | 831 | 838 | 813a |
| 33 | AU | 866 | 865 | |
| 34 | BG | 866 | 865 | 856a |
| 35 | BG | 886 | 1203 | |
| 36 | BG | 961 | 961 | |
| 37 | AU | 963 | 965 | 937a |
| 38 | AU | 982 | 1327 | 960a |
| 39 | BG | 1000 | 999 | |
| 40 | AU | 1000 | 999 | 974a |
| 41 | BG | 1010 | 1009 | 998a |
| 42 | AU | 1011 | 1009 | |
| 43 | BU | 1018 | 1015 | 1002a |
| 44 | AG | 1018 | 1018 | |
| 45 | BU | 1046 | 1044 | 1027a |
| 46 | AG | 1046 | 1047 | |
| 47 | AG | 1101 | 1101 | |
| 48 | BU | 1101 | 1101 | 1066a |
| 49 | BU | 1146 | 1121 | 1027a |
| 50 | AG | 1150 | 1173 | |
| 51 | BU | 1184 | 1184 | 1164a |
| 52 | AG | 1184 | 1184 | |
| 53 | BU | 1198 | 1185 | 1185a |
| 54 | AG | 1199 | 1202 | |
| 55 | AG | 1309 | 1436 | 1316a |
| 56 | BU | 1317 | 1563 | 1322a |
| 57 | AG | 1344 | 1337 | |
| 58 | BU | 1346 | 1338 | 1297a |
| 59 | AG | 1350 | 1350 | |
| 60 | BU | 1350 | 1350 | 1380a |
| 61 | BU | 1453 | 1449 | 1430a |
| 62 | AG | 1474 | 1477 | |
| 63 | BU | 1480 | 1478 | 1456a |
| 64 | AG | 1486 | 1697 | |
| 65 | AG | 1524 | 1523 | |
| 66 | BU | 1524 | 1526 | 1496a |
| 67 | BU | 1620 | 1616 | 1590a |
| 68 | AG | 1621 | 1614 | |
| 69 | BU | 1643 | 1642 | 1606a |
| 70 | AG | 1643 | 1641 | |
| 71 | AG | 1686 | 1696 | 1699a |
| 72 | BU | 1731 | 1690 | 1738a |
| 73 | AG | 3102 | 1194 i | 2605a |
| 74 | BU | 3166 | 3167 | 3012a |
| 75 | AG | 3166 | 3167 | |
| 76 | BU | 3179 | 3179 | 3041a |
| 77 | AG | 3179 | 3179 | |
| 78 | BU | 3187 | 3189 | 3068a |
| 79 | AG | 3188 | 3189 | |
| 80 | BU | 3197 | 1216 | 3312a |
| 81 | AG | 3203 | 3206 | |
| 82 | BU | 3203 | 3206 | 3079a |
| 83 | BU | 3210 | 3207 | 3098a |
| 84 | AG | 3210 | 3207 | |
a Ref. 30, b Ref. 31.
In Table 3 we present the calculated energies and potential barriers. The energy barrier for the proton transfer amounts to 6.497 kcal/mol, no ZPE and only 1.867 kcal/mol, with ZPE.
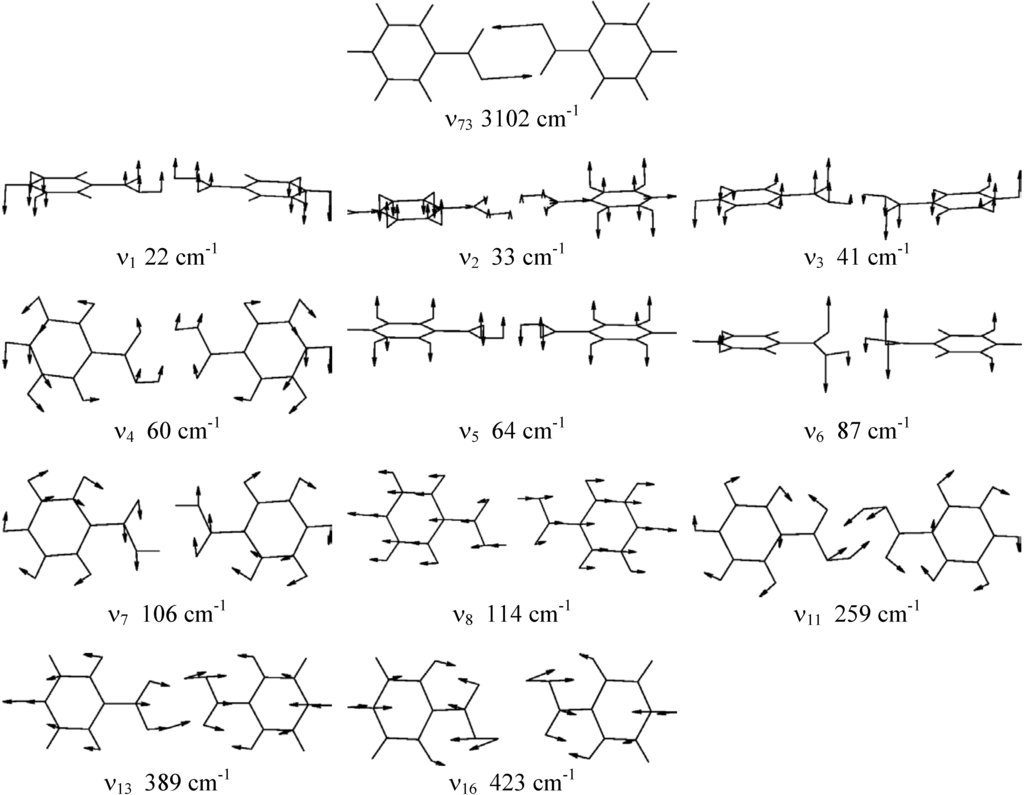
Figure 2.
Normal modes of benzoic acid calculated by the B3LYP/6-311++G** method.
Figure 2.
Normal modes of benzoic acid calculated by the B3LYP/6-311++G** method.
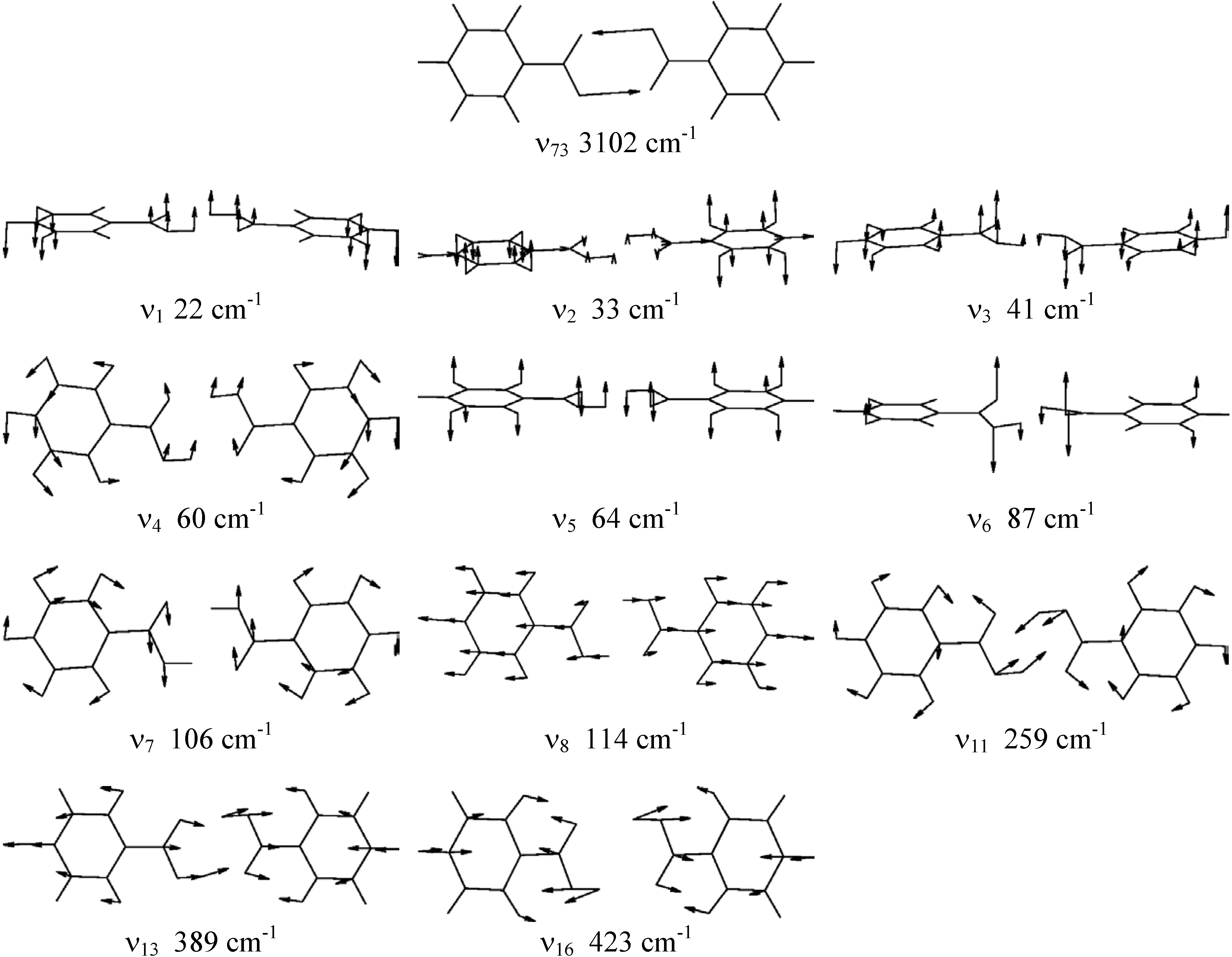
Table 3.
Calculated energies and barrier for benzoic acid by the B3LYP/6-311++G** (DFT) method.
| DFT | ||
| Energy, stable structure | No ZPE, a.u. | -841,923 |
| With ZPE, a.u. | -841,691 | |
| Energy, saddle point structure | No ZPE, a.u. | -841,912 |
| With ZPE, a.u. | -841,688 | |
| Energy barrier | No ZPE, kcal/mol | 6,497 |
| No ZPE, cm-1 | 2272 | |
| With ZPE, kcal/mol | 1,867 | |
| With ZPE, cm-1 | 653 | |
Model Calculations
On the basis of the ab initio calculations we constructed two-dimensional model PES for the proton tunnelling ν73 coupled to chosen low-frequency modes of benzoic acid dimer which largely affect the tunnelling. These are the low-frequency planar hydrogen-bond stretching and bending modes ν4, ν7, ν8, ν11, ν13 and ν16 and the lowest-frequency out-of-plane modes ν1, ν2, ν3, ν5 and ν6. They are shown in Fig. 2 and their calculated vibrational frequencies are listed in Table 2. We assumed the symmetric (synchronous) double proton transfer in benzoic acid dimer in accordance with the results of ref. [18].
The two-dimensional model potentials used to simulate the couplings are [12,13]:
(a) the symmetric mode coupling potential (SMC) describing couplings of the proton tunneling mode ν1 with the planar modes ν8, ν9 and ν21:
and
(b) the squeezed double well potential (SQZ) describing couplings of the proton tunneling mode ν1 with the out-of-plane modes ν12, ν13 and ν24:
where x and y and z, denote the coordinates of the proton tunneling and the low-frequency modes, respectively, ωx, ωy and ωz are the angular frequencies, 2xo the distance between the two minima, α and γ the coupling strengths. In the formulas (1) and (2) the potentials are expressed in the units of the quantum ћωx and the coordinates x, y and z are dimensionless:
and denote the dimensional coordinates and mx, my and mz are the effective masses.
The parameters xo, α and γ of the potentials (1) and (2) were estimated from the formulas:
where ΔE, ys and denote the energy barrier, the value of the normal coordinate y of the coupled mode of the SMC potential at the saddle point structure and the angular frequency of the mode z at the saddle point structure, respectively. We used no ZPE energy barriers. The parameters used in subsequent calculations of the energy splittings have been calculated from the results of the B3LYP/6-311++G** data and are listed in Table 4. The parameter α describing coupling between the O-H stretching mode ν73 and the mode ν4, ν7, ν8, ν11, ν13 or ν16 represent an analogue of linear distortion parameters b used for theoretical reproduction of the X-H infrared band shapes of tropolone [34], salicylaldehyde [35] or aspirin [36]. Both different spectroscopic facts thus have the same origin, the anharmonic coupling in the potential energy between the two X-H and X...Y vibrations.
Tunnelling energy splittings have been calculated variationally. The convergence has been confirmed by reproducing exactly the energy splittings reported by Takada and Nakamura [12] and calculated by the DVR method [37,38]. The results are presented in Table 5 for the two-dimensional model potentials (1) and (2). The proton tunnelling energy splitting in the vibrationally ground states of the low-frequency modes is ca. 230 cm-1. This compares reasonably well with the experimental value 172 cm-1 obtained from the inelastic neutron scattering measurements for benzoic acid - D5H and recently reported by Fillaux et al. [30]. The two-dimensional model PES qualitatively explain increase of the tunnelling splitting with the excitation of the planar ν4, ν7, ν8,ν11, ν13 and ν16 modes. For the out-of-plane modes the monotonic change depend on a sign of the coupling parameter γ. When the sign is positive (for modes ν1 and ν2) one obtains monotonic decrease of the splitting with the excitation, and when it is negative (for the ν3, ν5 and ν6 mode) the excitations cause increase of the splittings. This remains in agreement with our previous theoretical results for the excited state of tropolone [13] and hydrogen carbonate dimer [23]. Experimentally observed in tropolone promotion of the tunnelling by the excitation of the planar modes and its suppression by the excitation of the out-of-plane modes [7,8] are qualitatively confirmed in the present two-dimensional model calculations. Our previous results on the excited state of tropolone [13], hydrogen carbonate dimer ion [23] and the present ones do not require any adjustments and present pure quantum-mechanical approach to the problem of tunnelling splittings.
Table 4.
Parameters of the two-dimensional models.
| Mode | xo | α | γ | |
| Stable | Saddle point | |||
| ν4 | 2,42 | 0,178 | ||
| ν7 | 2,42 | 0,096 | ||
| ν8 | 2,42 | 0,122 | ||
| ν11 | 2,42 | 0,040 | ||
| ν13 | 2,42 | 0,056 | ||
| ν16 | 2,42 | 0,138 | ||
| ν1 | 2,42 | 0,00000017 | ||
| ν2 | 2,42 | 0,00000597 | ||
| ν3 | 2,42 | -0,00010000 | ||
| ν5 | 2,42 | -0,00000807 | ||
| ν6 | 2,42 | -0,00007099 | ||
Table 5.
Energy splittings (cm-1) calculated for two-dimensional model potentials.
| Quantum number | SMC | SQZ | ||||||||||
| ν4 | ν7 | ν8 | ν11 | ν13 | ν16 | ν1 | ν2 | ν3 | ν5 | ν6 | ||
| 0 | 229,24 | 229,50 | 229,36 | 229,67 | 229,65 | 229,54 | 229,71 | 229,63 | 230,99 | 229,78 | 230,10 | |
| 1 | 229,43 | 229,68 | 229,69 | 229,86 | 230,52 | 235,50 | 229,71 | 229,47 | 233,54 | 229,89 | 230,87 | |
| 2 | 229,62 | 229,86 | 230,02 | 230,05 | 231,38 | 241,32 | 229,70 | 229,30 | 236,12 | 230,01 | 231,64 | |
| 3 | 229,82 | 230,04 | 230,35 | 230,25 | 232,23 | 247,01 | 229,69 | 229,14 | 238,74 | 230,13 | 232,41 | |
| 4 | 230,01 | 230,22 | 230,68 | 230,44 | 233,09 | 252,57 | 229,69 | 228,97 | 241,39 | 230,25 | 233,18 | |
| 5 | 230,20 | 230,40 | 231,01 | 230,63 | 233,94 | 258,02 | 229,68 | 228,81 | 244,08 | 230,37 | 233,96 | |
| 6 | 230,39 | 230,57 | 231,34 | 230,82 | 234,79 | 263,36 | 229,67 | 228,64 | 246,80 | 230,49 | 234,74 | |
| 7 | 230,58 | 230,75 | 231,67 | 231,02 | 235,63 | 268,59 | 229,66 | 228,48 | 249,55 | 230,60 | 235,52 | |
| 8 | 230,77 | 230,93 | 231,99 | 231,21 | 236,48 | 273,72 | 229,66 | 228,31 | 252,34 | 230,72 | 236,30 | |
| 9 | 230,96 | 231,11 | 232,32 | 231,40 | 237,31 | 278,76 | 229,65 | 228,15 | 255,16 | 230,84 | 237,09 | |
| 10 | 231,15 | 231,29 | 232,65 | 231,59 | 238,15 | 283,70 | 229,64 | 227,98 | 258,01 | 230,96 | 237,88 | |
Conclusions
The proton tunnelling dynamics in benzoic acid dimer in the ground electronic state has been studied by performing quantum mechanical calculations of the potential energy surface and fitting it by two-dimensional model potentials. The tunnelling energy splittings for different vibrationally excited states have been calculated. The calculated proton tunnelling energy splitting in the vibrationally ground states of the low-frequency modes (~230 cm-1) compares reasonably well with the experimental value 172 cm-1 obtained from the inelastic neutron scattering measurements for benzoic acid - D5H [29]. Our model calculations predict the promotion of the proton tunnelling by the excitation of the planar modes. Depending of the sign of the coupling parameter γ, out-of-plane modes can either supress or promote the tunnelling. This results are confirmed by existing experimental data [7,8]. Our model presents a pure quantum-mechanical approach to the problem of the proton tunnelling splittings.
References
- Löwdin, P.O. Rev. Mod. Phys. 1963, 35, 724. [CrossRef]
- Nakamura, H. Ann. Rev. Phys. Chem. 1997, 48, 299. [CrossRef]
- Alves, A.C.P.; Hollas, J.M. Mol. Phys. 1973, 25, 1305.
- Redington, R.L.; Redington, T.E. J. Mol. Spectr. 1979, 78, 229. [CrossRef]
- Tomioka, Y.; Ito, M.; Mikami, N. J. Phys. Chem. 1983, 87, 4401. [CrossRef]
- Redington, R.L.; Chen, Y.; Scherer, G.J.; Field, R.W. J. Chem. Phys. 1988, 88, 627. [CrossRef]
- Sekiya, H.; Nagashima, Y.; Nishimura, Y. J. Chem. Phys. 1990, 92, 5761.
- Sekiya, H.; Nagashima, Y.; Tsuji, T.; Nishimura, Y.; Mori, A.; Takeshita, H. J. Phys. Chem. 1991, 95, 10311. [CrossRef]
- Nishi, K.; Sekiya, H.; Kawakami, H.; Mori, A.; Nishimura, Y. J. Chem. Phys. 1998, 109, 1589.
- Vener, M.V.; Scheiner, S.; Sokolov, N.D. J. Chem. Phys. 1994, 101, 9755.
- Smedarchina, Z.; Siebrand, W.; Zgierski, M.Z. J. Chem. Phys. 1996, 104, 1203.
- Takada, S.; Nakamura, H. J. Chem. Phys. 1995, 102, 3977.
- Wójcik, M.J.; Nakamura, H.; Iwata, S.; Tatara, W. J. Chem. Phys. 2000, 112, 6322.
- Carrington, T. Jr.; Miller, W.H. J. Chem. Phys. 1986, 84, 4364.
- Shida, N.; Barbara, P.F.; Almlof, J.E. J. Chem. Phys. 1989, 91, 4061.
- Smedarchina, Z.; Siebrand, W.; Zgierski, M.Z. J. Chem. Phys. 1995, 103, 5326.
- Sewell, T.D.; Guo, Y.; Thompson, D.L. J. Chem. Phys. 1995, 103, 8557.
- Shida, N.; Barbara, P.; Almlof, J.E. J. Chem. Phys. 1991, 94, 3633.
- Nishi, K.; Sekiya, H.; Kawakami, H.; Mori, A.; Nishimura, Y. J. Chem. Phys. 1998, 109, 1589.
- Nishi, K.; Sekiya, H.; Kawakami, H.; Mori, A.; Nishimura, Y. J. Chem. Phys. 1999, 111, 3961.
- Nishi, K.; Sekiya, H.; Mochida, T.; Sugawara, T.; Nishimura, Y. J. Chem. Phys. 2000, 112, 5002.
- Vener, M.V.; Sauer, J. J. Chem. Phys. 2001, 114, 2623.
- Wójcik, M.J.; Tatara, W.; Ikeda, S. J. Mol. Struct. 2002, 614, 109. [CrossRef]
- Decornez, M.; Drukker, H.; Hammes-Schiffer, S. J. Phys. Chem. A 1999, 103, 2891. [CrossRef]
- Mavri, S.; Grdadolnik, J. J. Phys. Chem. A 2001, 105, 2045. [CrossRef]
- Sim, G.A.; Monteath Robertson, J.; Goodwin, T.H. Acta Cryst. 1955, 8, 157.
- Wilson, C.C.; Shanklandnd, N.; Florence, A.J. Chem. Phys. Lett. 1996, 253, 103.
- Bruno, G.; Randaccio, L. Acta Cryst. 1980, B36, 1711.
- Plazanet, M.; Fukushima, N.; Johnson, M.R.; Horsewill, A.J.; Trommsdorf, H.P. J. Chem. Phys. 2001, 115, 3241.
- Fillaux, F.; Limage, M.H.; Romain, F. Chem. Phys. 2002, 276, 181.
- Stepanian, S.G.; Reva, I.D.; Radchenko, E.D.; Sheina, G.G. Vibrat. Spectrosc. 1996, 11, 123. [CrossRef]
- Zelsmann, H.R.; Mielke, Z. Chem. Phys. Lett. 1991, 186, 501. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; Dapprich, S.; Millam, J.M.; Daniels, A.D.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Ortiz, J.V.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, W.; Johnson, B.; Chen, W.; Wong, M.W.; Andres, J.L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian 98, Revision A.6; Gaussian: Pittsburgh PA, 1998. [Google Scholar]
- Wójcik, M.J.; Boczar, M.; Stoma, M. Int. J. Quant. Chem. 1999, 73, 275.
- Boczar, M.; Wójcik, J.M.; Szczeponek, K.; Jamróz, D.; Ikeda, S. Int. J. Quant. Chem. 2002, 90, 689. [CrossRef]
- Boczar, M.; Wójcik, M.J.; Szczeponek, K.; Jamróz, D.; Zieba, A.; Kawałek, B. Chem. Phys. 2002, 286, 63. [CrossRef]
- Light, J.C.; Hamilton, I.P.; Lill, J.V. J. Chem. Phys. 1985, 82, 1400.
- Whitnell, R.M.; Light, J.C. J. Chem. Phys. 1989, 90, 1774.
© 2003 by MDPI (http://www.mdpi.org). Reproduction for noncommercial purposes permitted.