Abstract
The title compound has been prepared in four steps starting from ethyl 2-fluorobenzoate. The final product as well as the intermediates are fully characterised by spectroscopic methods with the 1H and 13C NMR spectra, featuring coupling to 19F being particularly informative.
1. Introduction
Among simple heterocyclic auxiliary groups, the 4,5-dihydrooxazole (or 2-oxazoline) has proved to be one of the most useful, directing ortho-metalation on an aryl ring and facilitating a wide range of subsequent synthetic transformations, many of them in an asymmetric fashion [1,2,3]. In our own work, we have studied 2-(2-benzyloxyphenyl)oxazolines and observed that, upon treatment with a strong base, they may undergo either cyclisation to afford 3-aminobenzofurans or 1,2-Wittig rearrangement. While treatment with n-BuLi/KOt-Bu generally led to direct cyclisation to benzofuran products, using n-BuLi on its own gave the Wittig rearrangement [4]. It was later found that in isomeric thiophene-based systems, products derived from either direct cyclisation or Wittig rearrangement could be obtained depending on the distance between the oxazoline and benzyloxy functions [5]. In the meantime, it was discovered that the N-butylamide group, CONHBu, is more effective in directing the Wittig rearrangement in such systems [6]. In an attempt to broaden the scope of such transformations, various alternatives to the benzyloxy substituent have been examined including trifluoroethoxy, OCH2CF3, and this was prepared in the N-butylbenzamide series but did not react on treatment with a strong base. As described in a review article [7], the formation of α-trifluoromethyl anions may result in a range of different reaction outcomes depending on the other groups present. In this paper, we describe the stepwise synthesis of the corresponding 2-(2-trifluoroethoxyphenyl)oxazoline 4 and full characterisation of the final product and the intermediates for the first time.
2. Results
The starting point for our synthesis was 2-fluorobenzoyl chloride which is commercially available at a reasonable cost. Simply adding this to an excess of ethanol and heating for 3 h followed by evaporation gave the ethyl ester, ethyl 2-fluorobenzoate. We have previously reported the preparation and use of this compound in studies on pyrolysis of 2-aminophenyl-substituted stabilised phosphonium ylides [8]. To introduce the 2,2,2-trifluoroethoxy group [9], a patent procedure was followed [10] involving deprotonation of 2,2,2-trifuoroethanol with potassium tert-butoxide in THF followed by addition of the ethyl 2-fluorobenzoate to afford 1 in good yield (Scheme 1). Remarkably for such a simple compound, this has not been characterised before, and so we report here its full characterisation including mp, HRMS, IR and 1H, 13C and 19F NMR spectra (see Section 3 and Supplementary Materials). The NMR spectra were particularly informative with the trifluoroethoxy group giving rise to signals at δF −74.0, a 2H quartet at δH 4.41 (3JH-F 8.0 Hz) and quartets at δC 123.2 (1JC-F 276 Hz) and 67.5 (2JC-F 35.5 Hz). The corresponding methyl ester has been described in a patent [11] wherein it was prepared by a different method and only characterised by a melting point and elemental analysis. Alkaline hydrolysis of ester 1 using sodium hydroxide in aqueous dioxane [12] gave the corresponding carboxylic acid 2 which had a melting point in good agreement with the literature value [11] but is also characterised spectroscopically for the first time. The preparation of 2 by a different method is also described in a patent [13] but there it is only characterised by LCMS.
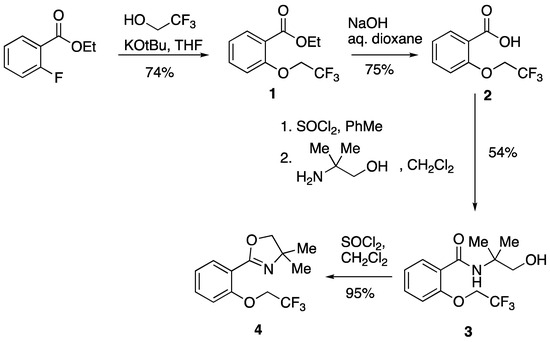
Scheme 1.
Stepwise construction of compound 4.
In order to prepare the target oxazoline 4, the acid 2 was first converted into its acid chloride using thionyl chloride, and this was reacted directly with 2-amino-2-methylpropan-1-ol to give the previously unknown hydroxy amide 3 in moderate yield. This gave 1H, 13C and 9F NMR spectra in agreement with expectation featuring the usual signals for OCH2CF3 and a broad singlet at δH 7.60 (NH) together with a 1H triplet at δH 5.20 and 2H doublet (3J 6.4 Hz) at δH 3.69 for CH2OH. The final cyclisation to give the oxazoline 4 was accomplished in excellent yield by treatment with thionyl chloride, and this previously unknown compound was fully characterised. Disappointingly in terms of the original aim of the work, treatment of compound 4 with a variety of strong bases under a range of conditions led to recovery of unchanged starting material. As we previously described for the corresponding amide, 2-(2,2,2-trifluoroethoxy)-N-butylbenzamide [6], trifluoroethoxy does not undergo either direct cyclisation or Wittig rearrangement in these systems.
Interestingly, after our work had been completed and while this paper was in preparation, the 6-methyl analogue of compound 4 was reported [14], prepared by direct palladium-catalysed oxidative alkoxylation of the 2-(2-methylphenyl)oxazoline. Its spectroscopic data are in good agreement with those of 4 although the fluorine coupling has not been recognised in their interpretation.
In summary, we have described an efficient four-step synthesis of the oxazoline 4 with full spectroscopic characterisation of the final product and the intermediates 1–3 for the first time.
3. Experimental Section
3.1. General Experimental Details
Melting points were recorded on a Reichert hot-stage microscope (Reichert, Vienna, Austria) and are uncorrected. IR spectra were recorded using the ATR technique on a Shimadzu IRAffinity 1S instrument. NMR spectra were obtained using Bruker AVII 400 or AVIII 500 instruments (Bruker, Billerica, MA, USA). Spectra were run in CDCl3 with internal Me4Si as the reference for 1H and 13C and external CFCl3 for 19F. Chemical shifts are reported in ppm to high frequency of the reference, and coupling constants are in Hz. 19F spectra were run with 1H decoupling. NMR spectra were processed using iNMR reader, version 6.3.3 (Mestrelab Research, Santiago de Compostela, Spain). Ethyl 2-fluorobenzoate was prepared by cautious addition of 2-fluorobenzoyl chloride (Fluorochem) to an excess of ethanol followed by evaporation.
3.2. Ethyl 2-(2,2,2-Trifluoroethoxy)benzoate 1
Following a literature procedure [10], potassium tert-butoxide (6.77 g, 60.3 mmol) was added to a stirred 0 °C solution of 2,2,2-trifluoroethanol (6.02 g, 60.2 mmol) in dry THF (150 mL). After stirring at 0 °C for 30 min, a solution of ethyl 2-fluorobenzoate (5.05 g, 30.0 mmol) in dry THF (20 mL) was added dropwise and the reaction mixture was allowed to warm to rt for 6 h. Further 2,2,2-trifluoroethanol (3.02 g, 30.2 mmol) and potassium tert-butoxide (3.44 g, 30.7 mmol) were added and, after stirring at rt for a further 16 h, the reaction mixture was concentrated in vacuo. The residue was partitioned between water (100 mL) and Et2O (100 mL), the two layers were separated, and the aqueous layer was re-extracted with Et2O (2 × 100 mL). The combined organic layers were dried and evaporated to give 1 (5.54 g, 74%) as a colourless solid which was used without further purification; mp 39–42 °C; IR (ATR): νmax/cm−1 1686, 1645, 1603, 1452, 1385, 1277, 1152, 1101, 1061, 968, 866, 752 and 691; 1H NMR (500 MHz): δ 7.85 (1 H, dd, J 7.5, 2.0, 6-H), 7.48 (1 H, ddd, J 8.5, 7.5, 1.8, 4-H), 7.13 (1 H, td, J 7.5, 1.0, 5-H), 6.97 (1 H, dd, J 8.5, 1.0, 3-H), 4.41 (2 H, q, JHF 8.0, CH2CF3), 4.37 (2 H, q, J 7.0, CH2CH3) and 1.38 (3 H, t, J 7.0, CH3); 13C NMR (125 MHz): δ 165.9 (C, C=O), 156.7 (C, C–O), 133.4 (CH, Ar), 132.0 (CH, Ar), 123.2 (C, q, JCF 276, CF3), 122.8 (CH, Ar), 122.3 (C, Ar), 115.5 (CH, Ar), 67.5 (CH2, q, JCF 35.5, CH2CF3), 61.2 (CH2CH3) and 14.1 (CH3); 19F NMR (470 MHz): δF −74.0; HRMS (NSI): found, 249.0732. C11H12F3O3 (M + H) requires 249.0733.
3.3. 2-(2,2,2-Trifluoroethoxy)benzoic acid 2
A solution of sodium hydroxide (2.23 g, 55.8 mmol) in water (40 mL) was added to a stirred solution of ethyl 2-(2,2,2-trifluoroethoxy) benzoate 1 (9.21 g, 37.1 mmol) in 1,4-dioxane (90 mL), and the reaction mixture was heated under reflux for 2 h. After cooling to rt, the reaction mixture was concentrated in vacuo, and the residue was dissolved in water (150 mL) and washed with Et2O (50 mL). The aqueous layer was acidified to pH 1 by the addition of 2 M HCl before being extracted with Et2O (2 × 100 mL). After drying and evaporation of the combined organic layers, the residue was recrystallised (EtOAc/hexane) to give 2 (6.15 g, 75%) as colourless crystals; mp 83–85 °C (lit. [11] 85–86.5 °C); IR (ATR): νmax/cm−1 1682, 1603, 1487, 1462, 1414, 1269, 1225, 1152, 1059, 961, 883, 858, 768, 702, 669 and 648; 1H NMR (400 MHz): δ 8.12 (1 H, dd, J 7.8, 1.8, 6-H), 7.59 (1 H, ddd, J 8.4, 7.4, 1.8, 4-H), 7.22 (1 H, ddd, J 7.8, 7.4, 1.0, 5-H), 7.04 (1 H, dd, J 8.4, 1.0, 3-H) and 4.53 (2 H, q, JHF 8.0, CH2); 13C NMR (125 MHz): δ 169.2 (C, C=O), 157.3 (C, C–O), 134.9 (CH, Ar), 133.3 (CH, Ar), 123.3 (CH, Ar), 123.0 (q, JCF 277, CF3), 120.0 (C, Ar), 115.5 (CH, Ar) and 67.6 (q, JCF 35.8, CH2); 19F NMR (376 MHz): δF −73.8.
3.4. N-(1-Hydroxy-2-methylpropan-2-yl)-2-(2,2,2-trifluoroethoxy)benzamide 3
A solution of thionyl chloride (3.7 mL, 6.03 g, 50.7 mmol) and 2-(2,2,2-trifluoroethoxy)benzoic acid 2 (5.50 g, 25.0 mmol) in toluene (50 mL) was heated under reflux for 3 h and then evaporated to give 2-(2,2,2-trifluoroethoxy)benzoyl chloride as a pale yellow oil which was used without further purification. A solution of 2-(2,2,2-trifluoroethoxy)benzoyl chloride (25.0 mmol) in CH2Cl2 (50 mL) was added dropwise to a solution of 2-amino-2-methylpropan-1-ol (4.46 g, 50.0 mmol) in CH2Cl2 (50 mL) stirred at 0 °C. After the addition, the mixture was stirred at rt for 18 h and then poured into water. The organic layer was separated and the aqueous layer extracted with CH2Cl2 (50 mL). The combined organic extracts were washed with 2M HCl (50 mL), 2M NaOH (50 mL) and water (50 mL). Drying and evaporation gave 3 (3.95 g, 54%) as a colourless oil which crystallised on standing and was used without further purification; mp 49–52 °C; IR (ATR): νmax/cm−1 3416, 3354, 1638, 1541, 1487, 1452, 1285, 1229, 1157, 1107, 1059, 1040, 978, 864, 750, 689 and 579; 1H NMR (400 MHz): δ 8.17 (1 H, dd, J 8.0, 2.0, 6-H), 7.60 (1 H, br s, NH), 7.48 (1 H, ddd, J 8.4, 7.4, 2.0, 4-H), 7.19 (1 H, ddd, J 8.0, 7.4, 1.0, 5-H), 6.88 (1 H, dd, J8.4, 1.0, 3-H), 5.20 (1 H, t, J6.4, OH), 4.49 (2 H, q, JHF 8.0, CH2CF3), 3.69 (2 H, d, J 6.4, CH2OH) and 1.38 (6 H, s, CH3); 13C NMR (125 MHz): δ 165.1 (C, C=O), 154.4 (C, C–O), 133.0 (CH, Ar), 132.7 (CH, Ar), 123.2 (CH, Ar), 122.9 (4ry, q, JCF 276, CF3), 122.6 (C, Ar), 111.9 (CH, Ar), 70.9 (CH2OH), 66.0 (q, JCF 35.9, CH2CF3), 56.6 (C, C(CH3)2) and 24.7 (2 × CH3); 19F NMR (376 MHz): δ −73.5; HRMS (NSI): found, 292.1158. C13H17F3NO3 (M + H) requires 292.1155.
3.5. 4,4-Dimethyl-2-(2-(2,2,2-trifluoroethoxy)phenyl)-4,5-dihydrooxazole 4
Thionyl chloride (1.2 mL, 1.96 g, 16.5 mmol) was added to a solution of N-(1-hydroxy-2-methylpropan-2-yl)-2-(2,2,2-trifluoroethoxy)benzamide 3 (3.70 g, 12.7 mmol) in CH2Cl2 (120 mL), and the mixture was stirred at rt for 18 h. The mixture was washed with 2M NaOH (50 mL) and water (50 mL) and then dried and evaporated to give 4 (3.31 g, 95%) as a colourless oil; IR (ATR): νmax/cm−1 2970, 2901, 1649, 1493, 1452, 1285, 1242, 1157, 1065, 1038, 964, 862, 752 and 691; 1H NMR (500 MHz): δ 7.74 (1 H, dd, J 7.5, 1.8, 6-H), 7.42 (1 H, ddd, J 8.3, 7.5, 1.8, 4-H), 7.12 (1 H, td, J 7.5, 1.0, 5-H), 7.00 (1 H, dd, J 8.3, 1.0, 3-H), 4.40 (2 H, q, JHF 8.0, CH2CF3), 4.10 (2 H, s, oxazoline CH2) and 1.39 (6 H, s, CH3); 13C NMR (125 MHz): δ 160.3 (C, C=N), 156.4 (C, C–O), 132.1 (CH, Ar), 131.3 (CH, Ar), 123.3 (C, q, JCF 277, CF3), 123.1 (CH, Ar), 120.1 (C, Ar), 116.4 (CH, Ar), 79.1 (oxazoline CH2), 68.1 (q, JCF 35.3, CH2CF3), 67.7 (C, C(CH3)2) and 28.3 (2 × CH3); 19F NMR (470 MHz): δ −74.1; HRMS (NSI): found, 274.1047. C13H15F3NO2 (M + H) requires 274.1049.
Supplementary Materials
The following are available online; 1H, 13C and 19F NMR spectra for 1–4.
Author Contributions
A.D.H. prepared the compounds and recorded the spectroscopic data; R.A.A. designed the study, analysed the data and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
We thank EPSRC (UK) for a DTA studentship to ADH (Grant EP/L505079/1) and the EPSRC UK National Mass Spectrometry Facility at Swansea University.
Data Availability Statement
The research data supporting this publication (original NMR files) can be accessed at https://doi.org/10.17630/a2165975-3542-44ed-aa69-72f767b1877c.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Meyers, A.I.; Mihelich, E.D. The synthetic utility of 2-oxazolines. Angew. Chem. Int. Ed. Engl. 1976, 15, 270–281. [Google Scholar] [CrossRef]
- Meyers, A.I.; Reumann, M. The synthetic utility of oxazolines in aromatic substitution. Tetrahedron 1985, 41, 837–860. [Google Scholar] [CrossRef]
- Gant, T.G.; Meyers, A.I. The chemistry of 2-oxazolines (1985-present). Tetrahedron 1994, 50, 2297–2360. [Google Scholar] [CrossRef]
- Aitken, R.A.; Harper, A.D.; Slawin, A.M.Z. Base-induced cyclisation of ortho-substituted 2-phenyloxazolines to give 3-aminobenzofurans and related heterocycles. Synlett 2017, 28, 1738–1742. [Google Scholar] [CrossRef]
- Aitken, R.A.; Harper, A.D.; Slawin, A.M.Z. Rationalisation of patterns of competing reactivity by X-ray structure determination: Reaction of isomeric (benzyloxythienyl)oxazolines with a base. Molecules 2021, 26, 7690. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Harper, A.D.; Inwood, R.A.; Slawin, A.M.Z. Access to diarylmethanols by Wittig rearrangement of ortho-, meta-, and para-benzyloxy-N-butylbenzamides. J. Org. Chem. 2022, 87, 4692–4701. [Google Scholar] [CrossRef] [PubMed]
- Uneyama, K.; Katagiri, T.; Amii, H. α-Trifluoromethylated carbanion synthons. Acc. Chem. Res. 2008, 41, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Murray, L. New gas-phase cascade reactions of stabilized phosphorus ylides leading to ring-fused indoles and to quinolines. J. Org. Chem. 2008, 73, 9781–9783. [Google Scholar] [CrossRef] [PubMed]
- Idoux, J.P.; Madenwald, M.L.; Garcia, B.S.; Chu, D.-L. Aromatic fluoroalkylation vis direct displacement of a nitro or fluoro group. J. Org. Chem. 1985, 50, 1876–1878. [Google Scholar] [CrossRef]
- Priepke, H.; Doods, H.; Kuelzer, R.; Pfau, R.; Stenkamp, D.; Pelcman, B.; Roenn, R. New compounds. PCT Int. Appl. WO 2012/022793 A1, 23 February 2012. [Google Scholar]
- Mendel, A. Polyfluoroalkoxy-substituted aromatic carboxylic acids and esters and salts thereof. US Patent 3766247, 16 October 1973. [Google Scholar]
- Huang, S.-T.; Hsei, I.-J.; Chen, C. Synthesis and anticancer evaluation of bis(benzimidazoles), bis(benzoxazoles), and benzothiazoles. Bioorg. Med. Chem. 2006, 14, 6106–6119. [Google Scholar] [CrossRef] [PubMed]
- Liverton, N.; Kuduk, S.D.; Luo, Y.; Meng, N.; Yu, T. Piperidine, oxadiazole and thiadiazole orexin receptor antagonists. PCT Int. Appl. WO 2016/069510 A1, 6 May 2016. [Google Scholar]
- Pourkaveh, R.; Svatunek, D.; Schnürch, M. Palladium-catalyzed ortho alkoxylation of oxazoline derivatives: An avenue to reach meta-substituted electron-rich arenes exploiting oxazoline as a removeable directing group. ACS Omega 2024, 9, 44224–44232. [Google Scholar] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).