
Spatial Genetic Structure of Prunus mongolica in Arid Northwestern China Based on RAD Sequencing Data



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Collection and DNA Extraction
2.2. RAD Sequencing and SNP Calling
2.3. Genetic Structure Analyses
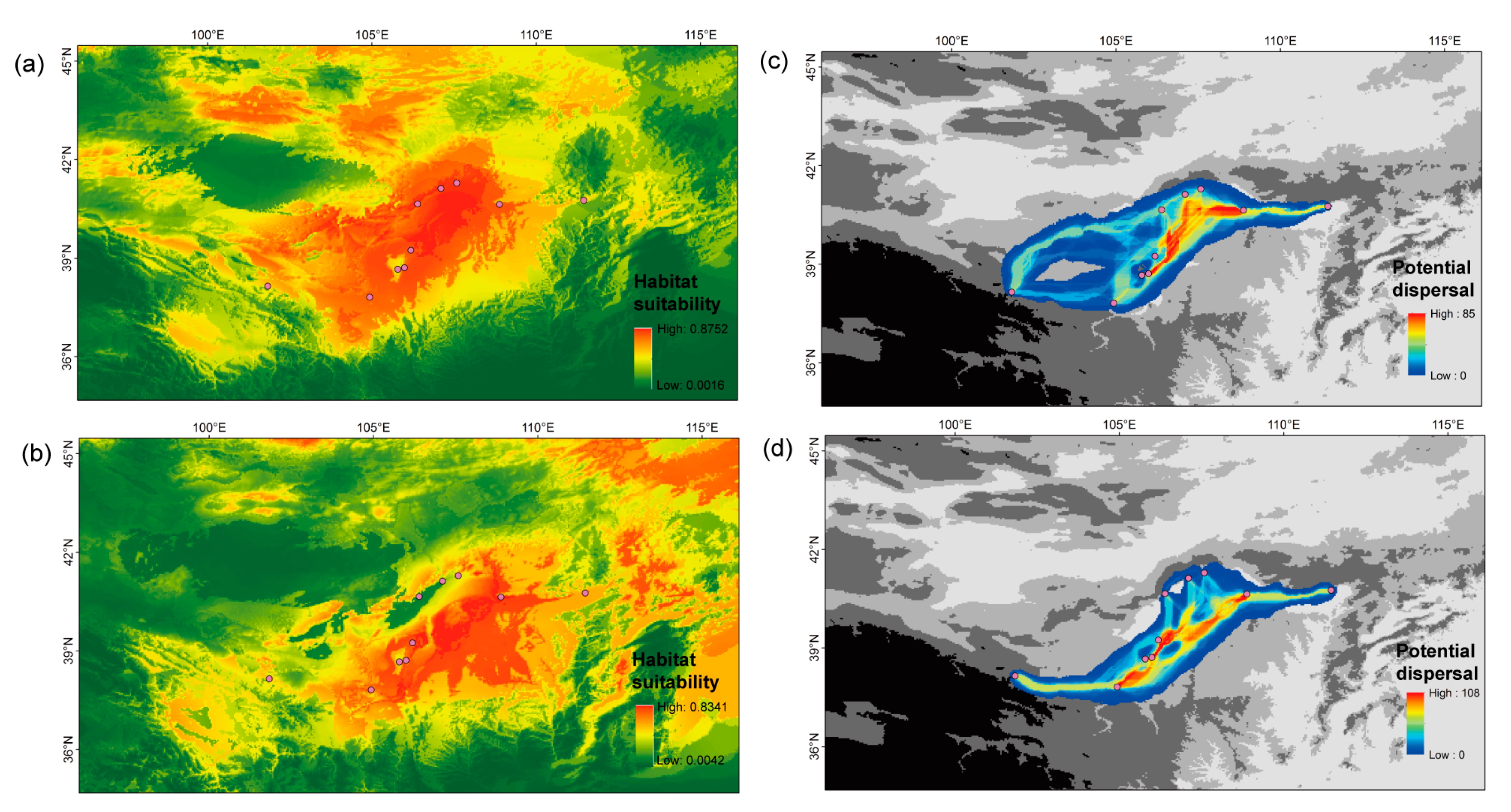
2.4. Species Distribution Model and Potential Migration Corridors
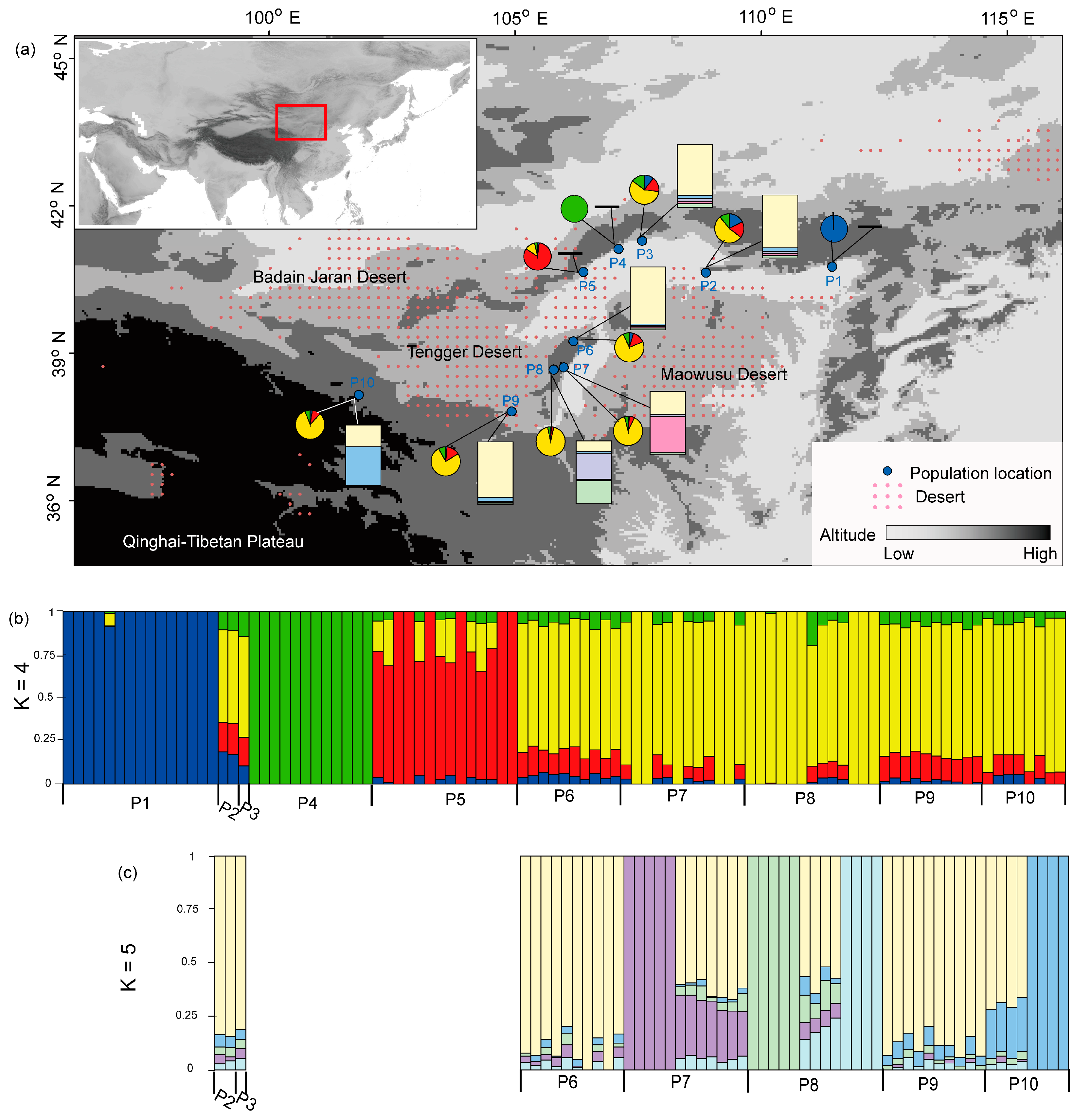
3. Results
4. Discussion
4.1. Effect of Habitat Fragmentation on Population Structure of P. mongolica
4.2. Conservation Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.-X.; Li, X.-S.; Wang, J.-C.; Zhang, D.-Y. Insights into the aridification history of Central Asian Mountains and international conservation strategy from the endangered wild apple tree. J. Biogeogr. 2021, 48, 332–344. [Google Scholar] [CrossRef]
- Su, Z.; Pan, B.; Zhang, M.; Shi, W. Conservation genetics and geographic patterns of genetic variation of endangered shrub Ammopiptanthus (Fabaceae) in northwestern China. Conserv. Genet. 2016, 17, 485–496. [Google Scholar] [CrossRef]
- Cheng, J.; Kao, H.; Dong, S. Population genetic structure and gene flow of rare and endangered Tetraena mongolica Maxim. revealed by reduced representation sequencing. BMC Plant Biol. 2020, 20, 391. [Google Scholar] [CrossRef]
- Su, Z.; Richardson, B.A.; Zhuo, L.; Jiang, X. Divergent population genetic structure of the endangered Helianthemum (Cistaceae) and its implication to conservation in Northwestern China. Front. Plant Sci. 2017, 7, 2010. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-X.; Wang, Q.; Jia, S.-W. Genomic phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into habitat fragmentation in arid Northwestern China. Diversity 2020, 12, 335. [Google Scholar] [CrossRef]
- Yang, X.; Scuderi, L.; Paillou, P.; Liu, Z.; Li, H.; Ren, X. Quaternary environmental changes in the drylands of China—A critical review. Quat. Sci. Rev. 2011, 30, 3219–3233. [Google Scholar] [CrossRef]
- Guan, Q.; Pan, B.; Li, N.; Zhang, J.; Xue, L. Timing and significance of the initiation of present day deserts in the northeastern Hexi Corridor, China. Palaeogeogr. Palaeocl. 2011, 306, 70–74. [Google Scholar]
- Ma, S.M.; Nie, Y.B.; Duan, X.; Yu, C.S.; Wang, R.X. The potential distribution and population protection priority of Amygdalus mongolica. Acta Ecol. Sinica 2015, 35, 2960–2966. [Google Scholar]
- Zheng, Y. The Analysis about the Meteorological Elements in the Surrounding Area of Tengger Desert during the Recent 49a; Lanzhou University: Lanzhou, China, 2014. [Google Scholar]
- Ma, J. Reproductive Biology of Amygalus monogolica; Northwest University: Xi’an, China, 2010. [Google Scholar]
- Wang, W.; Yang, T.; Wang, H.-L.; Li, Z.-J.; Ni, J.-W.; Su, S.; Xu, X.-Q. Comparative and phylogenetic analyses of the complete chloroplast genomes of six Almond species (Prunus spp. L.). Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Fu, L.G. China Plant. Red Data Book; Science Press: Beijing, China, 1992. [Google Scholar]
- Ma, S.M.; Nie, Y.B.; Jiang, X.L.; Xu, Z.; Ji, W.Q. Genetic structure of the endangered, relict shrub Amygdalus mongolica (Rosaceae) in arid northwest China. Aust. J. Bot. 2019, 67, 128–139. [Google Scholar] [CrossRef]
- Duan, Y.; Bai, C.; Duan, C.; Shen, Y. Molecular phylogeography of Amygdalus mongolica (Rosaceae) based on chloroplast DNA non-coding sequences. Acta Bot. Boreali-Occident. Sin. 2018, 38, 1625–1633. [Google Scholar]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-generation population genetics. Brief. Funct. Genom. 2010, 9, 416–423. [Google Scholar] [CrossRef]
- Emerson, K.J.; Merz, C.R.; Catchen, J.M.; Hohenlohe, P.A.; Cresko, W.A.; Bradshaw, W.E.; Holzapfel, C.M. Resolving postglacial phylogeography using high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 16196–16200. [Google Scholar] [CrossRef] [Green Version]
- Uckele, K.A.; Adams, R.P.; Schwarzbach, A.E.; Parchman, T.L. Genome-wide RAD sequencing resolves the evolutionary history of serrate leaf Juniperus and reveals discordance with chloroplast phylogeny. Mol. Phylogenet. Evol. 2021, 156, 107022. [Google Scholar] [CrossRef]
- Cutter, A.D. Integrating phylogenetics, phylogeography and population genetics through genomes and evolutionary theory. Mol. Phylogenet. Evol. 2013, 69, 1172–1185. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Vandergast, A.G.; Perry, W.M.; Lugo, R.V.; Hathaway, S.A. Genetic landscapes GIS Toolbox: Tools to map patterns of genetic divergence and diversity. Mol. Ecol. Resour. 2011, 11, 158–161. [Google Scholar] [CrossRef]
- Brown, J.L.; Hill, D.J.; Dolan, A.M.; Carnaval, A.C.; Haywood, A.M. PaleoClim, high spatial resolution paleoclimate surfaces for global land areas. Sci. Data 2018, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.-L.; Xu, G.; Deng, M. Spatial genetic patterns and distribution dynamics of the rare oak Quercus chungii: Implications for biodiversity conservation in Southeast China. Forests 2019, 10, 821. [Google Scholar] [CrossRef] [Green Version]
- Loera, I.; Ickert-Bond, S.M.; Sosa, V. Pleistocene refugia in the Chihuahuan Desert: The phylogeographic and demographic history of the gymnosperm Ephedra compacta. J. Biogeogr. 2017, 44, 2706–2716. [Google Scholar] [CrossRef]
- Salinas-Ivanenko, S.; Múrria, C. Macroecological trend of increasing values of intraspecific genetic diversity and population structure from temperate to tropical streams. Global Ecol. Biogeogr. 2021, 30, 1685–1697. [Google Scholar] [CrossRef]
- Peter, B.M.; Slatkin, M. The effective founder effect in a spatially expanding population. Evolution 2015, 69, 721–734. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Wu, H.; Zhu, S.; Li, Q.; Zhang, C.; Yu, Y.; Sun, A. Cenozoic aridification in Northwest China evidenced by paleovegetation evolution. Palaeogeogr. Palaeocl. 2020, 557, 109907. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, H.; Pan, B.; Zhang, M. Intraspecific divergences and phylogeography of Panzerina lanata (Lamiaceae) in northwest China. PeerJ 2019, 7, e6264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Liu, Y.; Que, P.; Zhang, Z. Quaternary climate and environmental changes have shaped genetic differentiation in a Chinese pheasant endemic to the eastern margin of the Qinghai-Tibetan Plateau. Mol. Phylogenet. Evol. 2013, 67, 129–139. [Google Scholar] [CrossRef]
- Liu, Y.; Dietrich, C.H.; Wei, C. Genetic divergence, population differentiation and phylogeography of the cicada Subpsaltria yangi based on molecular and acoustic data: An example of the early stage of speciation? BMC Evol. Biol. 2019, 19, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Yang, H.; Lu, Q.; Zhang, X. Endemic shrubs in temperate arid and semiarid regions of northern China and their potentials for rangeland restoration. AOB Plants 2015, 7, plv063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.H. The western Ordos plateau as a biodiversity center of relic shrubs in arid areas of China. Biodivers. Conserv. 2005, 14, 3187–3200. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Code | Longitude/Latitude | Nind | Voucher Specimen | π (SD) | Ho (SD) | He (SD) |
|---|---|---|---|---|---|---|
| Overall | — | 97 | 0.339 (0.160) | 0.378 (0.187) | 0.350 (0.128) | |
| P1 | 111.44/40.76 | 15 | — | 0.216 (0.105) | 0.363 (0.415) | 0.213 (0.221) |
| P2 | 108.87/40.64 | 2 | — | — | — | |
| P3 | 107.58/41.29 | 1 | AM-WZQ-001 | — | — | |
| P4 | 107.09/41.12 | 12 | AM-WHQ-001 | 0.249 (0.122) | 0.388 (0.382) | 0.241 (0.211) |
| P5 | 106.38/40.65 | 14 | AM-DK-001 | 0.299 (0.146) | 0.398 (0.295) | 0.287 (0.177) |
| P6 | 106.18/39.24 | 10 | AM-ZQZ-001 | 0.335 (0.166) | 0.385 (0.254) | 0.321 (0.164) |
| P7 | 105.98/38.71 | 12 | AM-YC-001 | 0.321 (0.158) | 0.394 (0.293) | 0.305 (0.176) |
| P8 | 105.78/38.66 | 13 | AM-ZQG-001 | 0.322 (0.158) | 0.393 (0.288) | 0.313 (0.172) |
| P9 | 104.93/37.81 | 10 | — | 0.336 (0.167) | 0.397 (0.252) | 0.323 (0.162) |
| P10 | 101.83/38.15 | 8 | AM-YCX-001 | 0.301 (0.151) | 0.370 (0.324) | 0.281 (0.192) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-X.; Wang, Q.; Wen, Z.-B. Spatial Genetic Structure of Prunus mongolica in Arid Northwestern China Based on RAD Sequencing Data. Diversity 2021, 13, 397. https://doi.org/10.3390/d13080397
Zhang H-X, Wang Q, Wen Z-B. Spatial Genetic Structure of Prunus mongolica in Arid Northwestern China Based on RAD Sequencing Data. Diversity. 2021; 13(8):397. https://doi.org/10.3390/d13080397
Chicago/Turabian StyleZhang, Hong-Xiang, Qian Wang, and Zhi-Bin Wen. 2021. "Spatial Genetic Structure of Prunus mongolica in Arid Northwestern China Based on RAD Sequencing Data" Diversity 13, no. 8: 397. https://doi.org/10.3390/d13080397
APA StyleZhang, H.-X., Wang, Q., & Wen, Z.-B. (2021). Spatial Genetic Structure of Prunus mongolica in Arid Northwestern China Based on RAD Sequencing Data. Diversity, 13(8), 397. https://doi.org/10.3390/d13080397