

Testing the Effect of Sampling Effort on Inferring Phylogeographic History in Psolodesmus mandarinus (Calopterygidae, Odonata)


Abstract
:1. Introduction
2. Materials and Methods
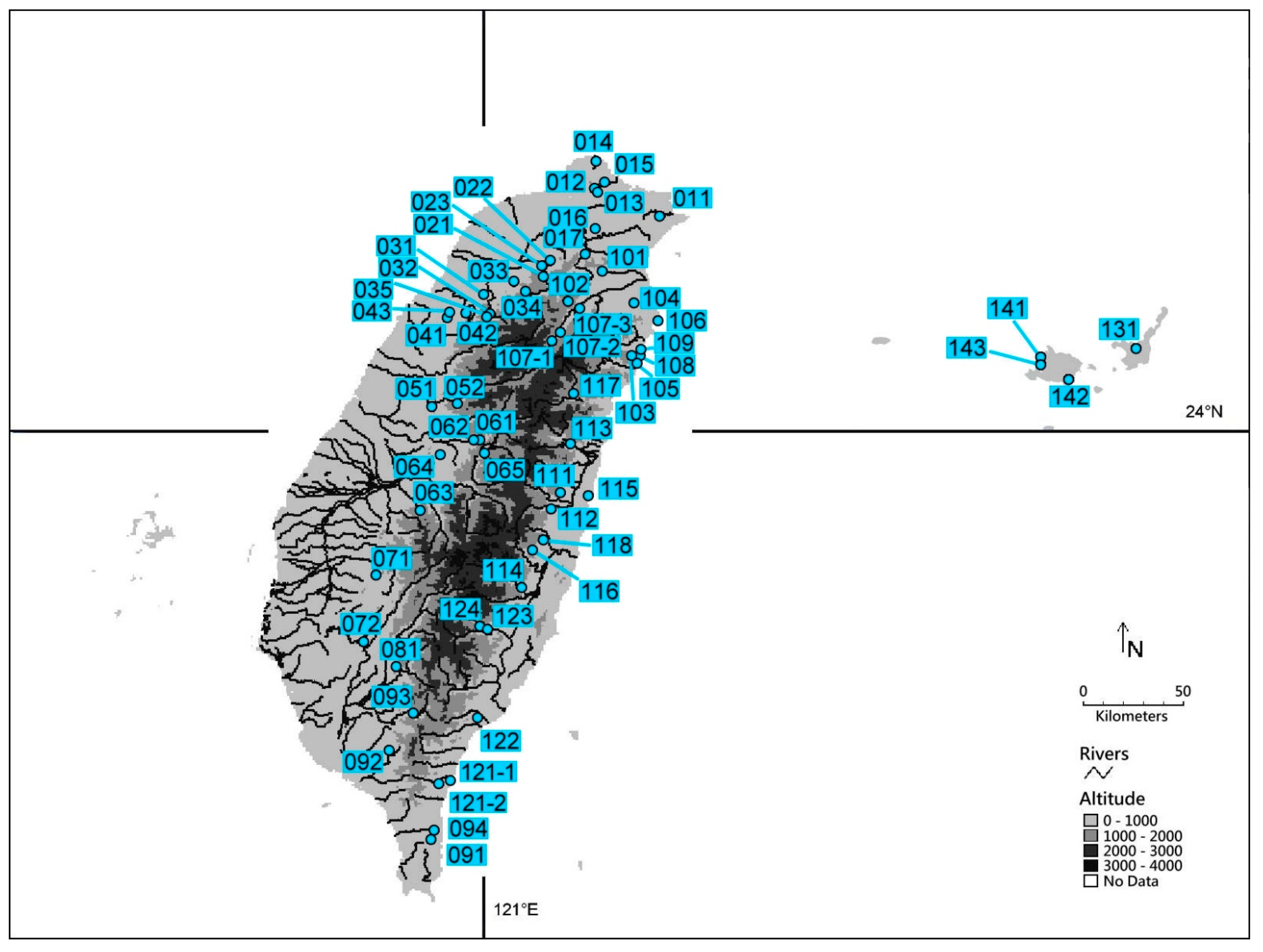
2.1. Sampling, DNA Extraction, Sequencing, and Alignment
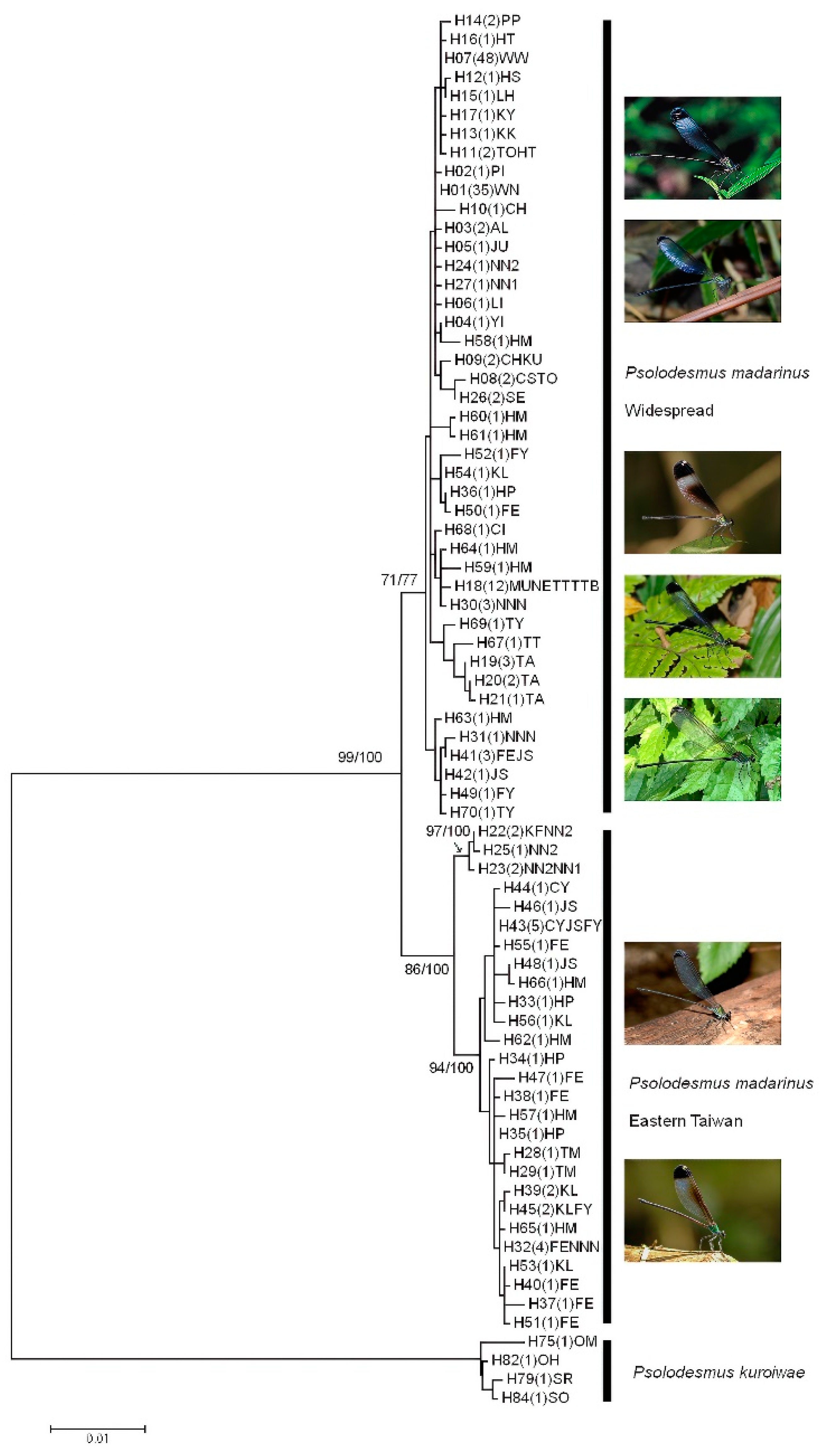
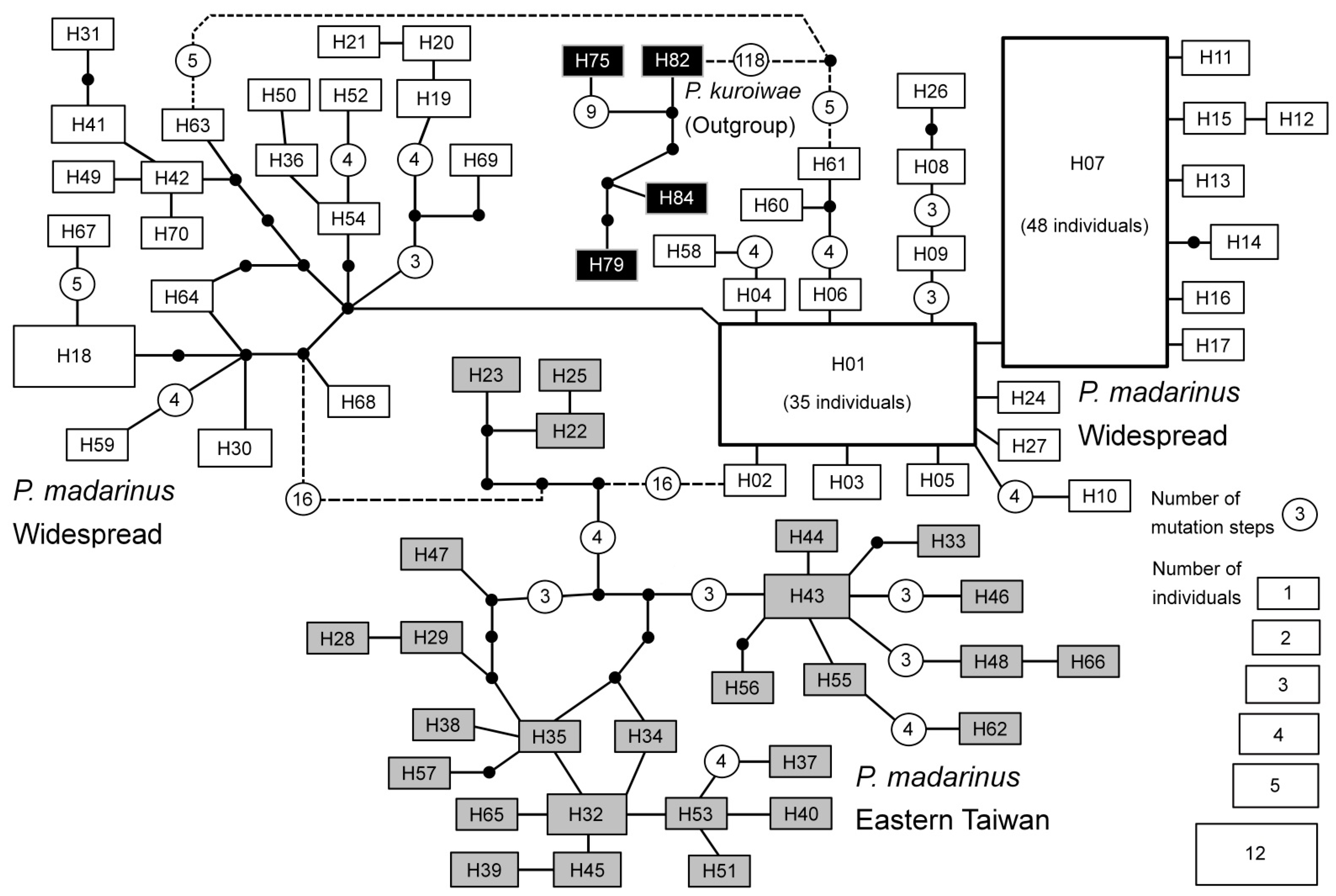
2.2. Phylogenetic and Network Analyses
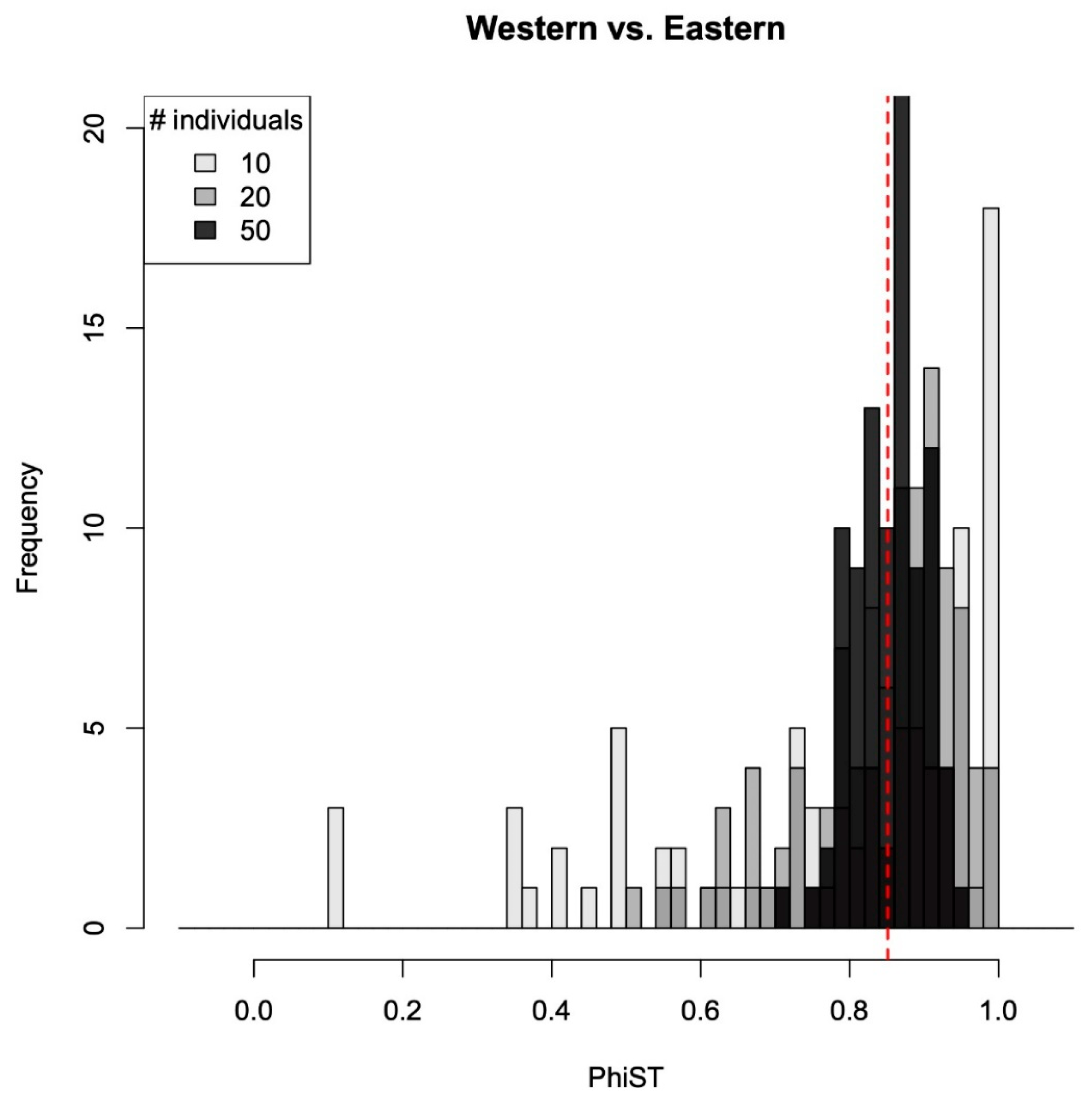
2.3. Population Genetic Analyses
2.4. Testing the Effect of Sampling Effort on Inferring Population Structure Phylogenetic Reconstruction
3. Results
3.1. Sequence Alignment, Phylogenetic, and Network Analyses
3.2. Population Genetic Analyses
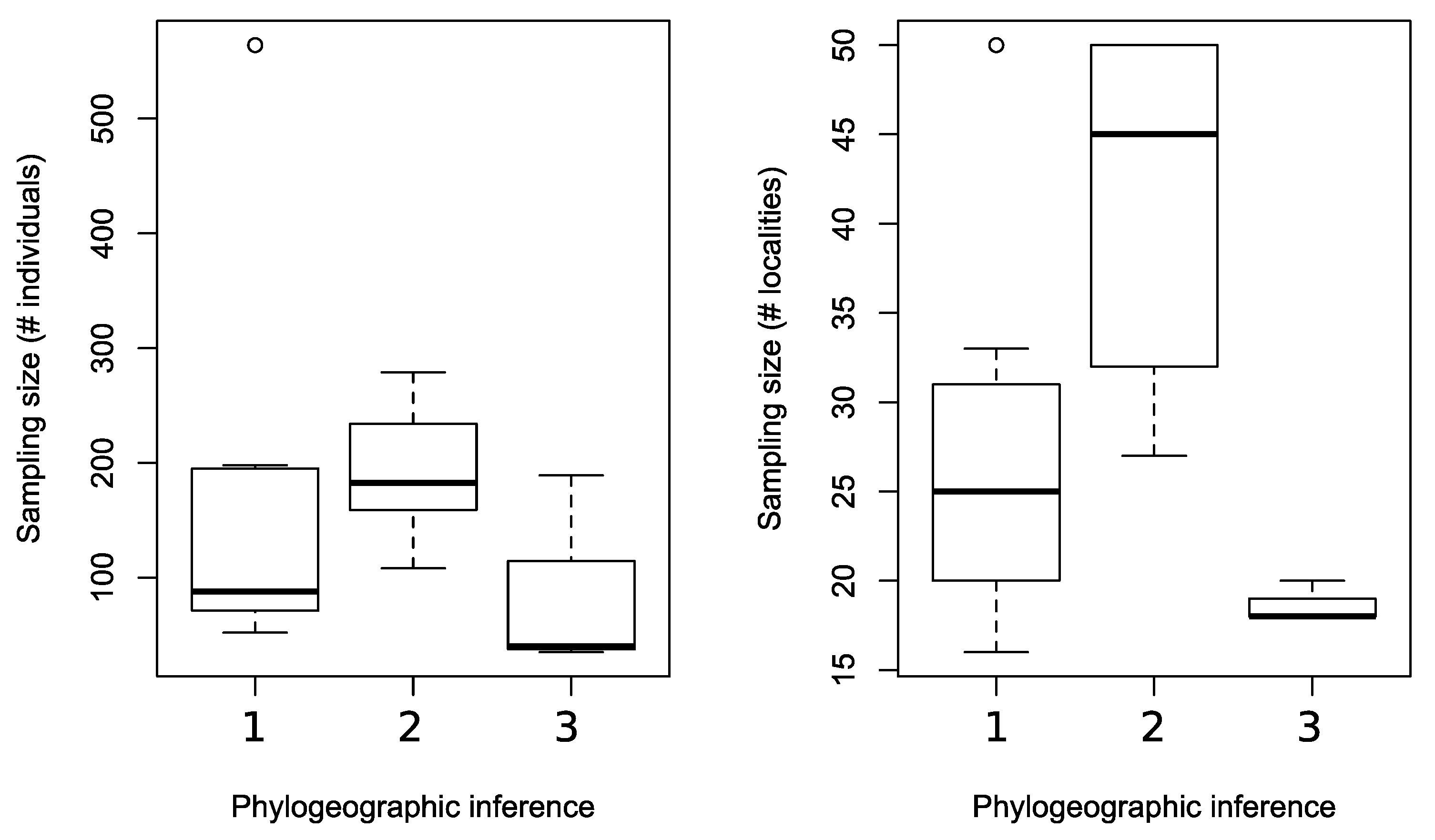
3.3. The Effect of Sampling Effort on Phylogenetic Reconstruction and Inferring Population Structure
4. Discussion
4.1. The Effect of Sampling Effort on the Reconstructed Phylogeographic History
4.2. The Phylogeographic History of Psolodesmus mandarinus
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Templeton, A.R. Statistical phylogeography: Methods of evaluating and minimizing inference errors. Mol. Ecol. 2004, 13, 789–809. [Google Scholar] [CrossRef] [PubMed]
- Knowles, L.L. Statistical phylogeography. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 593–612. [Google Scholar] [CrossRef]
- Gutiérrez-García, T.; Vázquez-Domínguez, E. Comparative phylogeography: Designing studies while surviving the process. BioScience 2011, 61, 857–868. [Google Scholar] [CrossRef]
- Meirmans, P.G. Seven common mistakes in population genetics and how to avoid them. Mol. Ecol. 2015, 24, 3223–3231. [Google Scholar] [CrossRef]
- Nielsen, R.; Beaumont, M.A. Statistical inferences in phylogeography. Mol. Ecol. 2009, 18, 1034–1047. [Google Scholar] [CrossRef]
- Heath, T.A.; Hedtke, S.M.; Hillis, D.M. Taxon sampling and the accuracy of phylogenetic analyses. J. Syst. Evol. 2008, 46, 239–257. [Google Scholar]
- Malaney, J.L.; Cook, J.A. Using biogeographical history to inform conservation: The case of Preble’s meadow jumping mouse. Mol. Ecol. 2013, 22, 6000–6017. [Google Scholar] [CrossRef]
- Yang, Y.J.; Lin, Y.S.; Wu, J.L.; Hui, C.F. Variation in mitochondrial DNA and population structure of the Taipei treefrog Rhacophorus taipeianus in Taiwan. Mol. Ecol. 1994, 3, 219–228. [Google Scholar] [CrossRef]
- Huang, J.P.; Lin, C.P. Diversification in subtropical mountains: Phylogeography, Pleistocene demographic expansion, and evolution of polyphonic mandibles in Taiwanese stag beetle, Lucanus formosanus. Mol. Phylogenet. Evol. 2010, 57, 1149–1161. [Google Scholar] [CrossRef]
- Wang, Y.H.; Hsiao, Y.W.; Lee, K.H.; Tseng, H.Y.; Lin, Y.P.; Komaki, S.; Lin, S.M. Acoustic differentiation and behavioral response reveals cryptic species within Buergeria treefrogs (Anura, Rhacophoridae) from Taiwan. PLoS ONE 2017, 12, e0184005. [Google Scholar] [CrossRef]
- Kuo, H.C.; Chen, S.F.; Fang, Y.P.; Flanders, J.; Rossiter, S.J. Comparative rangewide phylogeography of four endemic Taiwanese bat species. Mol. Ecol. 2014, 23, 3566–3586. [Google Scholar] [CrossRef] [PubMed]
- Creer, S.; Malhotra, A.; Thorpe, R.A.; Chou, W.H. Multiple causation of phylogeographical pattern as revealed by nested clade analysis of the bamboo viper (Trimeresurus stejnegeri) within Taiwan. Mol. Ecol. 2001, 10, 1967–1981. [Google Scholar] [CrossRef] [PubMed]
- Creer, S.; Thorpe, R.A.; Malhotra, A.; Chou, W.H.; Stenson, A.G. The utility of AFLP for supporting mitochondrial DNA phylogeographical analyses in the Taiwanese bamboo viper, Trimeresurus stejnegeri. J. Evol. Biol. 2004, 17, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P.; Lin, C.P. Lineage-specific late Pleistocene expansion of an endemic subtropical gossamer-wing damselfly, Euphaea formosa, in Taiwan. BMC Evol. Biol. 2011, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.L.; Lin, H.D.; Weng, C.F. A New Phylogeographic Pattern of Endemic Bufo bankorensis in Taiwan Island Is Attributed to the Genetic Variation of Populations. PLoS ONE 2014, 9, e98029. [Google Scholar] [CrossRef]
- Lin, S.C.; Chen, Y.F.; Shieh, S.H.; Yang, P.S. A revision of the status of Psolodesmus mandarinus based on molecular and morphological evidence (Odonata: Calopterygidae). Odonatologica 2014, 43, 51066. [Google Scholar]
- Oshida, T.; Lin, L.K.; Chang, S.W.; Chen, Y.J.; Lin, J.K. Phylogeography of two sympatric giant flying squirrel subspecies, Petaurista alborufus lena and P. philippensis grandis (Rodentia: Sciuridae), in Taiwan. Biol. J. Linn. Soc. 2011, 102, 404–419. [Google Scholar] [CrossRef]
- Shih, H.T.; Hung, H.C.; Schubart, C.D.; Chen, C.A.; Chang, H.W. Intraspecific genetic diversity of the endemic freshwater crab Candidopotamon rathbunae (Decapoda, Brachyura, Potamidae) reflects five million years of the geological history of Taiwan. J. Biogeogr. 2006, 33, 980–989. [Google Scholar] [CrossRef]
- Liu, M.Y.; Tzeng, C.S.; Lin, H.D. Phylogeography and the genetic structure of the land-locked freshwater prawn Macrobrachium asperulum (Crustacea: Decapoda: Palaemonidae) in Taiwan. Hydrobiologia 2011, 671, 1–12. [Google Scholar] [CrossRef]
- Jang-Liaw, N.H.; Chou, W.H. Phylogeography of the fanged dicroglossine frog, Limnonectes fujianensis (Anura, Ranidae), in Taiwan. Zoolog. Sci. 2011, 28, 254–263. [Google Scholar] [CrossRef]
- Lee, J.W.; Jiang, L.; Su, Y.C.; Tso, I.M. Is central mountain range a geographic barrier to the giant wood spider Nephila pilipes (Araneae: Tetragnathidae) in Taiwan? A population genetic approach. Zool. Stud. 2004, 43, 112–122. [Google Scholar]
- Oshida, T.; Lee, J.K.; Lin, L.K.; Chen, Y.J. Phylogeography of Pallas’s squirrel in Taiwan: Geographical isolation in an arboreal small mammal. J. Mammal. 2006, 87, 247–254. [Google Scholar] [CrossRef]
- Wang, T.Y.; Liao, T.Y.; Tzeng, C.S. Phylogeography of the Taiwanese Endemic Hillstream Loaches, Hemimyzon formosanus and H. taitungensis (Cypriniformes: Balitoridae). Zool. Stud. 2007, 46, 547–560. [Google Scholar]
- Ožana, S.; Dolný, A.; Pánek, T. Nuclear copies of mitochondrial DNA as a potential problem for phylogenetic and population genetic studies of Odonata. Syst. Enotmol. 2022, 47, 591–602. [Google Scholar] [CrossRef]
- Ballard, J.M.O.; Whitloch, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef]
- Zink, R.M.; Barrowclough, G.F. Mitochondrial DNA under siege in avian phylogeography. Mol. Ecol. 2008, 17, 2107–2121. [Google Scholar] [CrossRef]
- Fujisawa, T.; Barraclough, T.G. Delimiting species using single-locus data and the general mixed Yule coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef]
- Lieftinck, M.A.; Lien, J.C.; Maa, T.C. Catalogue of Taiwanese Dragonflies (Insecta: Odonata); Asian Ecological Society: Taichung, China, 1984. [Google Scholar]
- Wang, L.J. Dragonflies of Taiwan; JemJen Publishing: New Taipei, China, 2000. [Google Scholar]
- Karjalainen, S.; Hämäläinen, M. Demoiselle Damselflies. Winged Jewels of Silvery Streams; Caloptera Publishing: Helsinki, Finland, 2013. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Tamura, K.G.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1660. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.D.; Chen, Y.R.; Lin, S.M. Strict consistency between genetic and topographic landscapes of the brown tree frog (Buergeria robusta) in Taiwan. Mol. Phylogenet. Evol. 2012, 62, 251–262. [Google Scholar] [CrossRef]
- Paradis, E. Pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef]
- Winter, D.J. MMOD: An R library for the calculation of population differentiation statistics. Mol. Ecol. Resour. 2012, 12, 1158–1160. [Google Scholar] [CrossRef]
- Paradis, E. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Dijkstra, K.D.B.; Kalkman, V.J.; Dow, R.A.; Stokvis, F.R.; Van, T.J. Redefining the damselfly families: A comprehensive molecular phylogeny of Zygoptera (Odonata). Syst. Entomol. 2014, 39, 68–96. [Google Scholar] [CrossRef]
- Liao, C.C.; Chen, C.H. Investigation of floristic similarities between Taiwan and terrestrial ecoregions in Asia using GBIF data. Bot. Stud. 2017, 58, 15. [Google Scholar] [CrossRef]
- He, J.; Gao, Z.; Su, Y.; Lin, S.; Jiang, H. Geographical and temporal origins of terrestrial vertebrates endemic to Taiwan. J. Biogeogr. 2018, 45, 2458–2470. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Sample Size | # Localities | Inferred Pattern * | Reference |
|---|---|---|---|---|
| Bamboo viper | 201 | 40 | 2 | [12] |
| Bat1 | 108 | 50 | 2 | [11] |
| Bat2 | 146 | 50 | 1 | [11] |
| Bat3 | 234 | 50 | 2 | [11] |
| Bat4 | 164 | 50 | 2 | [11] |
| Toad | 279 | 27 | 2 | [15] |
| Damselfly1 | 159 | 32 | 2 | [14] |
| Damselfly2 $ | 60 | 20 | 1 | [16] |
| Flying squirrel1 | 40 | 20 | 3 | [17] |
| Flying Squirrel2 | 35 | 18 | 3 | [17] |
| Freshwater Crab | 88 | 18 | 1 | [18] |
| Freshwater Prawn | 195 | 20 | 1 | [19] |
| Frog | 198 | 31 | 1 | [20] |
| Spider | 189 | 18 | 3 | [21] |
| Small mammal | 71 | 29 | 1 | [22] |
| Stag beetle | 52 | 25 | 1 | [9] |
| Freshwater fish | 71 | 16 | 1 | [23] |
| Tree frog | 564 | 33 | 1 | [18] |
| No. | Acronym | Locality | GPS Coordinates | Altitude | Haplotype ● | Accession Numbers |
|---|---|---|---|---|---|---|
| Taiwan | ||||||
| 11 | TI | Tinglanku, Shuanghsi District, New Taipei city | 25°01′04.4″ N 121°52′32.0″ E | 42 m | H01 (2) | KM360534 |
| 12 | PI | Pingtenli, Shihlin District, Taipei city | 25°08′24.6″ N 121°34′43.1″ E | 500 m | H02 (1) | KM360535 |
| 13 | PA | Pamierh Park, Shihlin District, Taipei city | 25°07′20.6″ N 121°35′35.5″ E | 330 m | H01 (2) | KM360534 |
| 14 | AL | Alipang, Shihmen District, New Taipei city | 25°15′50.5″ N 121°35′05.2″ E | 140 m | H01 (1), H03 (2) | KM360534, KM360536 |
| 15 | LU | Lukuping, Wanli District, New Taipei city | 25°10′07.9″ N 121°37′18.3″ E | 419 m | H01 (4) | KM360534 |
| 16 | YI | Yinhotung, Hsintien District, New Taipei city | 24°57′30.5″ N 121°34′55.9″ E | 212 m | H01 (1), H04 (1) | KM360534, KM360537 |
| 17 | WU | Wulai, Wulai District, New Taipei city | 24°50′20.6″ N 121°32′08.4″ E | 219 m | H01 (4) | KM360534 |
| 21 | JU | Junghua, Fuhsing Township, Taoyuan County | 24°44′05.5″ N 121°21′02.1″ E | 505 m | H01 (1), H05 (1) | KM360534, KM360538 |
| 22 | LI | Liuhsia, Fuhsing Township, Taoyuan County | 24°48′34.3″ N 121°22′25.5″ E | 364 m | H01 (2), H06 (1) | KM360534, KM360539 |
| 23 | FU | Fuhsing, Fuhsing Township, Taoyuan County | 24°47′20.9″ N 121°20′22.8″ E | 369 m | H07 (3) | KM360540 |
| 31 | PE | Peipu, Peipu Township, Hsinchu County | 24°39′27.8″ N 121°04′45.5″ E | 264 m | H07 (4) | KM360540 |
| 32 | SH | Shihlu, Chienshih Township, Hsinchu County | 24°33′58.6″ N 121°06′23.7″ E | 1110 m | H07 (2) | KM360540 |
| 33 | CS | Chienshihhsienho, Chienshih Township, Hsinchu County | 24°42′48.7″ N 121°12′32.4″ E | 300 m | H07 (1), H08 (1), H09 (1) | KM360540-KM360542 |
| 34 | CH | Chienshih, Chienshih Township, Hsinchu County | 24°40′11.1″ N 121°15′57.7″ E | 851 m | H01 (1), H07 (1), H10 (1) | KM360534, KM360540, KM360543 |
| 35 | KU | Kuanwu, Wufeng Township, Hsinchu County | 24°33′48.3″ N 121°05′35.8″ E | 812 m | H07 (2), H09 (1) | KM360540, KM360542 |
| 41 | ST | Shihtanpeitawo, Shihtan Township, Miaoli County | 24°33′09.5″ N 120°54′58.5″ E | 272 m | H07 (1) | KM360540 |
| 42 | NA | Nanchuang, Nanchuang Township, Miaoli County | 24°34′16.9″ N 121°00′00.1″ E | 332 m | H07 (4) | KM360540 |
| 43 | TO | Touwu, Touwu Township, Miaoli County | 24°34′40.8″ N 120°55′34.3″ E | 179 m | H07 (1), H08 (1), H11 (1) | KM360540, KM360541, KM360544 |
| 51 | HS | Hsinshe, Hsinshe District, Taichung city | 24°08′54.9″ N 120°50′38.7″ E | 605 m | H07 (3), H12 (1) | KM360540, KM360545 |
| 52 | KK | Kukuan, Hoping District, Taichung city | 24°09′28.3″ N 120°57′38.9″ E | 687 m | H07 (2), H13 (1) | KM360540, KM360546 |
| 61 | PP | Penpusi, Puli Township, Nantou County | 23°59′41.4″ N 121°03′41.1″ E | 735 m | H14 (2) | KM360547 |
| 62 | KY | Kuanyinpupu, Puli Township, Nantou County | 23°59′32.9″ N 121°02′06.0″ E | 646 m | H07 (1), H17 (1) | KM360540, KM360550 |
| 63 | HT | Hsitou, Luku Township, Nantou County | 23°40′27.8″ N 120°47′26.9″ E | 1082 m | H07 (7), H11 (1), H16 (1) | KM360540, KM360544, KM360549 |
| 64 | LH | Lienhuachih, Yuchih Township, Nantou County | 23°55′26.1″ N 120°53′03.5″ E | 735 m | H07 (2), H15 (1) | KM360540, KM360548 |
| 65 | JE | Jenai, Jenai Township, Nantou County | 23°55′42.4″ N 121°04′56.1″ E | 1120 m | H07 (2) | KM360540 |
| 71 | CP | Chungpu, Chungpu Township, Chiayi County | 23°23′13.2″ N 120°35′34.1″ E | 816 m | H07 (3) | KM360540 |
| 72 | NH | Nanhua Dam, Nanhua District, Tainan city | 23°04′38.3″ N 120°32′03.5″ E | 198 m | H07 (3) | KM360540 |
| 81 | SP | Shanping, Liukuei District, Kaohsiung city | 22°58′00.1″ N 120°41′02.5″ E | 660 m | H07 (4) | KM360540 |
| 91 | MU | Mutan, Mutan Township, Pintung County | 22°10′45.5″ N 120°50′26.5″ E | 280 m | H18 (3) | KM360551 |
| 92 | TA | Taiwu, Taiwu Township, Pintung County | 22°35′11.8″ N 120°38′55.3″ E | 395 m | H19 (3), H20 (2), H21 (1) | KM360552-KM360554 |
| 93 | WT | Wutai, Wutai Township, Pintung County | 22°45′22.8″ N 120°45′34.2″ E | 438 m | H07 (2) | KM360540 |
| 94 | NE | Neiwen, Neiwen Township, Pintung County | 22°13′24.4″ N 120°51′22.1″ E | 321 m | H18 (3) | KM360551 |
| 101 | TP | Tsaopi, Yuanshan Township, Yilan County | 24°45′41.4″ N 121°36′42.6″ E | 603 m | H01 (1) | KM360534 |
| 102 | MI | Mingchih, Tatung Township, Yilan County | 24°37′54.6″ N 121°27′11.7″ E | 1047 m | H01 (2) | KM360534 |
| 103 | SM | Shenmihu, Nanao Township, Yilan County | 24°22′41.3″ N 121°44′48.8″ E | 1100 m | H01 (1) | KM360534 |
| 104 | TU | Sanfu, Tungshan Township, Yilan County | 24°37′03.1″ N 121°45′23.9″ E | 140 m | H01 (2) | KM360534 |
| 105 | KF | Kufeng, Nanao Township, Yilan County | 24°20′41.0″ N 121°46′15.7″ E | 18 m | H01 (3), H22 (1) | KM360534, KM360555 |
| 106 | SU | Suao, Nanao Township, Yilan County | 24°32′18.6″ N 121°51′55.4″ E | 314 m | H01 (3) | KM360534 |
| 107-1 | SE | Province Highway 7A, Nanao Township, Yilan County | 24°26′41.3″ N 121°23′02.5″ E | 1088 m | H26 (1) | KM360559 |
| 107-2 | SE | Province Highway 7A, Nanao Township, Yilan County | 24°29′09.7″ N 121°25′30.5″ E | 781 m | H01 (1), H26 (1) | KM360534, KM360559 |
| 107-3 | SE | Province Highway 7A, Nanao Township, Yilan County | 24°35′37.7″ N 121°30′32.5″ E | 355 m | H01 (1) | KM360534 |
| 108 | NN2 | Nanao II, Nanao Township, Yilan County | 24°22′57.3″ N 121°47′02.0″ E | 220 m | H01 (2), H22 (1), H23 (1), H24 (1), H25 (1) | KM360534, KM360555-KM360558 |
| 109 | NN1 | Nanao I, Nanao Township, Yilan County | 24°24′03.2″ N 121°47′09.7″ E | 190 m | H01 (1), H23 (1), H27 (1) | KM360534, KM360556, KM360560 |
| 111 | FE | Fenglin, Fenglin Township, Hualien County | 23°45′29.9″ N 121°25′23.6″ E | 249 m | H32 (3), H37 (1), H38 (1), H40 (1), H41 (1), H47 (1), H50 (1), H51 (1), H55 (1) | KM360565, KM360570, KM360571, KM360573, KM360574, KM360580, KM360583, KM360584, KM360588 |
| 112 | KL | Kuangfulintao, Wanjung Township, Hualien County | 23°40′57.0″ N 121°22′58.1″ E | 229 m | H39 (1), H45 (1), H53 (1), H54 (1), H56 (1) | KM360572, KM360578, KM360586, KM360587, KM360589 |
| 113 | TM | Tungmen, Hsiulin Township, Hualien County | 23°58′39.4″ N 121°28′22.0″ E | 198 m | H28 (1), H29 (1) | KM360561, KM360562 |
| 114 | NNN | Nanan, Chohsi Township, Hualien County | 23°19′35.7″ N 121°14′26.3″ E | 445 m | H30 (3), H31 (1), H32 (1) | KM360563-KM360565 |
| 115 | CY | Chienying, Fenglin Township, Hualien County | 23°44′52.6″ N 121°32′53.9″ E | 160 m | H43 (1), H44(1) | KM360576, KM360577 |
| 116 | JS | Juisui, JuiSui Township, Hualien County | 23°29′45.0″ N 121°17′43.8″ E | 1141 m | H41 (2), H42 (1), H43 (3), H46 (1), H48 (1) | KM360574-KM360576, KM360579, KM360581 |
| 117 | HP | Hsipao, Hsiulin Township, Hualien County | 24°12′26.3″ N 121°28′54.6″ E | 939 m | H33 (1), H34 (1), H35 (1), H36 (1) | KM360566-KM360569 |
| 118 | FY | Fuyuan, JuiSui Township, Hualien County | 23°32′40.7″ N 121°20′37.1″ E | 898 m | H43 (1), H45 (1), H49 (1), H52 (1) | KM360576, KM360578, KM360582, KM360585 |
| 121-1 | TT | Tachu Main Stream, Tawu Township, Taitung County | 22°25′59.2″ N 120°52′45.4″ E | 288 m | H18 (3), H67 (1) | KM360551, KM360600 |
| 121-2 | TTB | Tachu Tributary, Tawu Township, Taitung County | 22°26′59.0″ N 120°55′46.1″ E | 123 m | H18 (3) | KM360551 |
| 122 | CI | Chihpen, Peinan Township, Taitung County | 22°44′09.2″ N 121°03′00.8″ E | 137 m | H68 (1) | KM360601 |
| 123 | TY | Tsiayunchiao, Haituan Township, Taitung County | 23°08′21.1″ N 121°05′59.8″ E | 475 m | H69 (1), H70 (1) | KM360602, KM360603 |
| 124 | HM | Hsiama, Haituan Township, Taitung County | 23°09′09.3″ N 121°03′53.7″ E | 680 m | H57 (1), H58 (1), H59 (1), H60 (1), H61 (1), H62 (1), H63 (1), H64 (1), H65 (1), H66 (1) | KM360590-KM360599 |
| Japan | ||||||
| 131 | OM | Ishigaki, Mt.Omoto | 24°25′15″ N 124°11′02″ E | 300 m | H72 (3), H73 (1), H74 (1), H75 (1), H76 (1), H77 (1) | H75: KM360604 |
| 141 | SO | Iriomote, Sonai | 24°23′17″ N 123°44′59″ E | 50 m | H78 (1), H82 (1), H85 (1) | H82: KM360606 |
| 142 | OH | Iriomotea, Otomi | 24°17′09″ N 123°52′55″ E | 80 m | H79 (1), H81 (2), H83 (1), H84 (1) | H79: KM360605, H84: KM360607 |
| 143 | SR | Iriomotea, Sirahama | 24°21′35″ N 123°45′06″ E | 60 m | H80 (1), H86 (1), H87 (1), H88 (1), H89 (1) | |
| Set Name | Primer Name | Primer Sequence (5′-3′) | Direction | Length | Amplification Region (Mt Gene) |
|---|---|---|---|---|---|
| Pmk-005 | Pmk-F001 (Pmk-COI-1684F) | CCCACGACTAAACAACATAAG | forward | 663 bp | COI |
| Pmk-R005 (Pmk-COI-2346R) | GGAACAGCAATTACTATTGTGG | reverse | |||
| Pmk-006 | Pmk-F006 (Pmk-COI-2178F) | CCCAAGAAAGAGGAAAGAAG | forward | 740 bp | COI |
| Pmk-R006 (Pmk-COI-2917R) | GAATCTATGTTCTGTTGGTGG | reverse | |||
| Pmk-007 | Pmk-F007 (Pmk-COI2895F) | CACCACCAACAGAACATAG | forward | 814 bp | COI-tRNA-Leu-COII |
| Pmk-R007 (Pmk-COI3708) | GTCATCTAGTGAGGCTTCAC | reverse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.-J.; Chou, Y.-W.; Huang, J.-P. Testing the Effect of Sampling Effort on Inferring Phylogeographic History in Psolodesmus mandarinus (Calopterygidae, Odonata). Diversity 2022, 14, 809. https://doi.org/10.3390/d14100809
Wang L-J, Chou Y-W, Huang J-P. Testing the Effect of Sampling Effort on Inferring Phylogeographic History in Psolodesmus mandarinus (Calopterygidae, Odonata). Diversity. 2022; 14(10):809. https://doi.org/10.3390/d14100809
Chicago/Turabian StyleWang, Liang-Jong, Yen-Wei Chou, and Jen-Pan Huang. 2022. "Testing the Effect of Sampling Effort on Inferring Phylogeographic History in Psolodesmus mandarinus (Calopterygidae, Odonata)" Diversity 14, no. 10: 809. https://doi.org/10.3390/d14100809
APA StyleWang, L.-J., Chou, Y.-W., & Huang, J.-P. (2022). Testing the Effect of Sampling Effort on Inferring Phylogeographic History in Psolodesmus mandarinus (Calopterygidae, Odonata). Diversity, 14(10), 809. https://doi.org/10.3390/d14100809