
Yeast and Lactic Acid Bacteria Dominate the Core Microbiome of Fermented ‘Hairy’ Tofu (Mao Tofu)

 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Sampling
2.2. DNA Extraction, Amplification and NGS Library Preparation
2.3. Bioinformatics
2.4. Statistical Analyses
3. Results
3.1. Generating OTUs from MiSeq Data
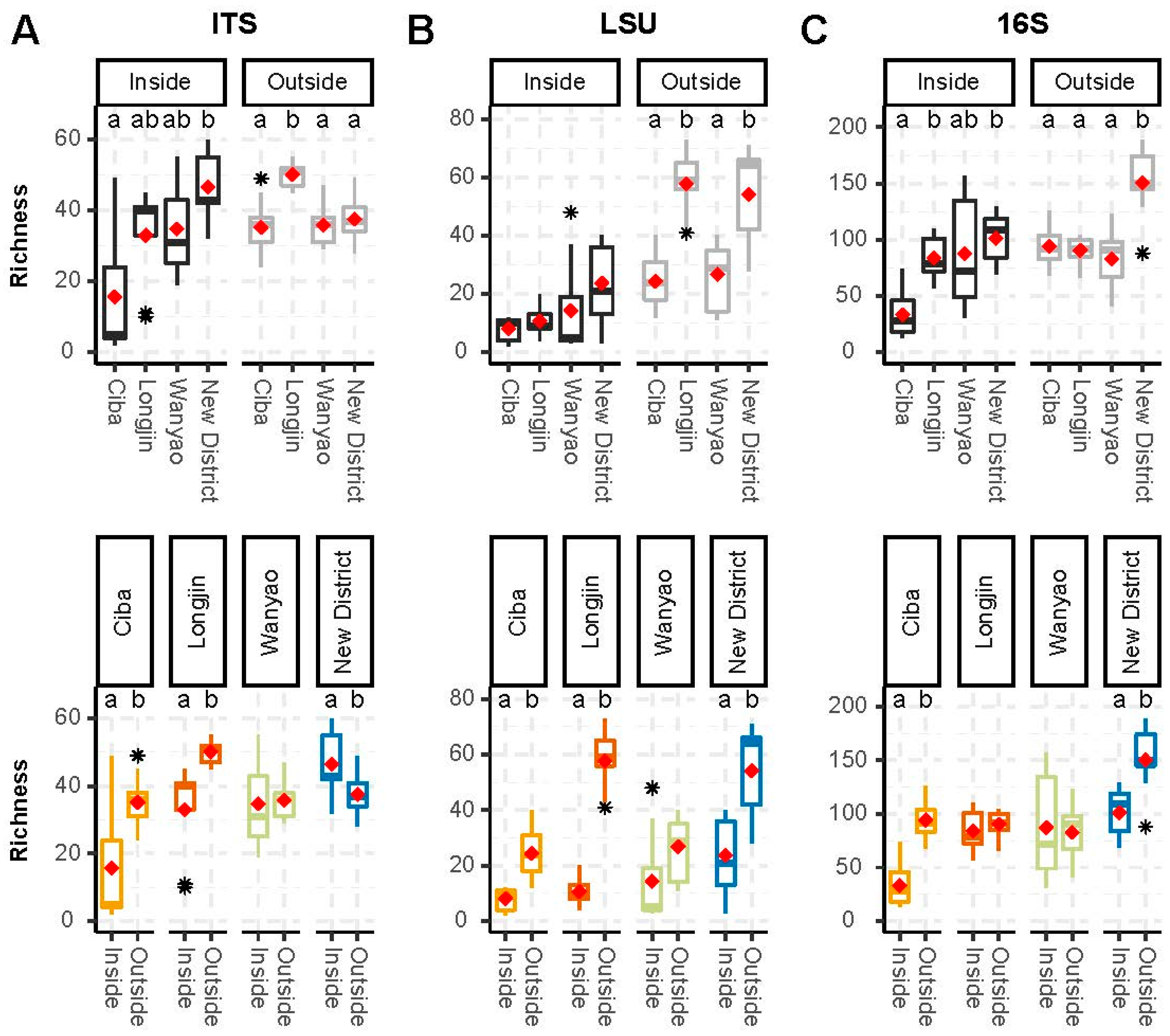
3.2. Alpha Diversity
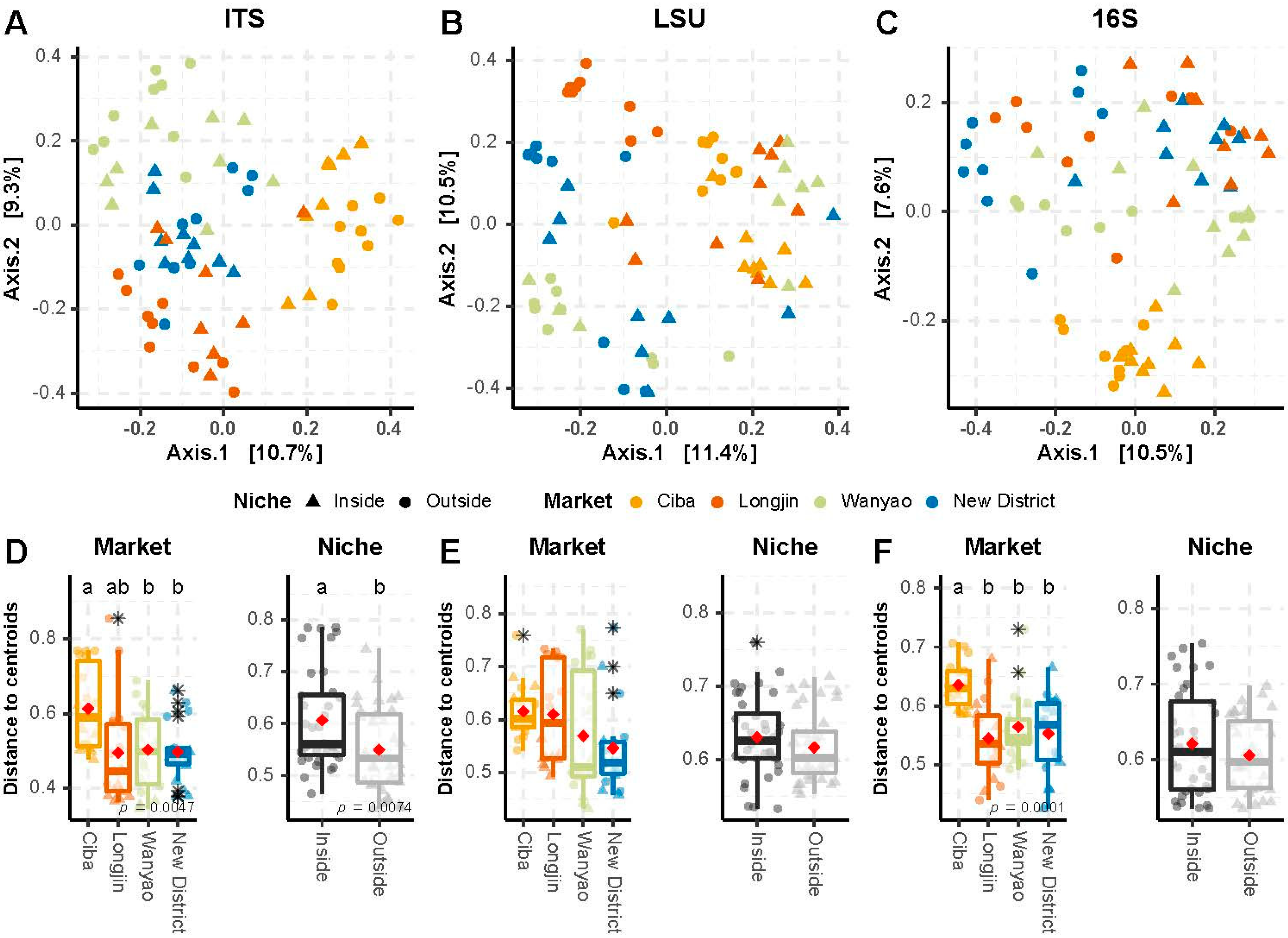
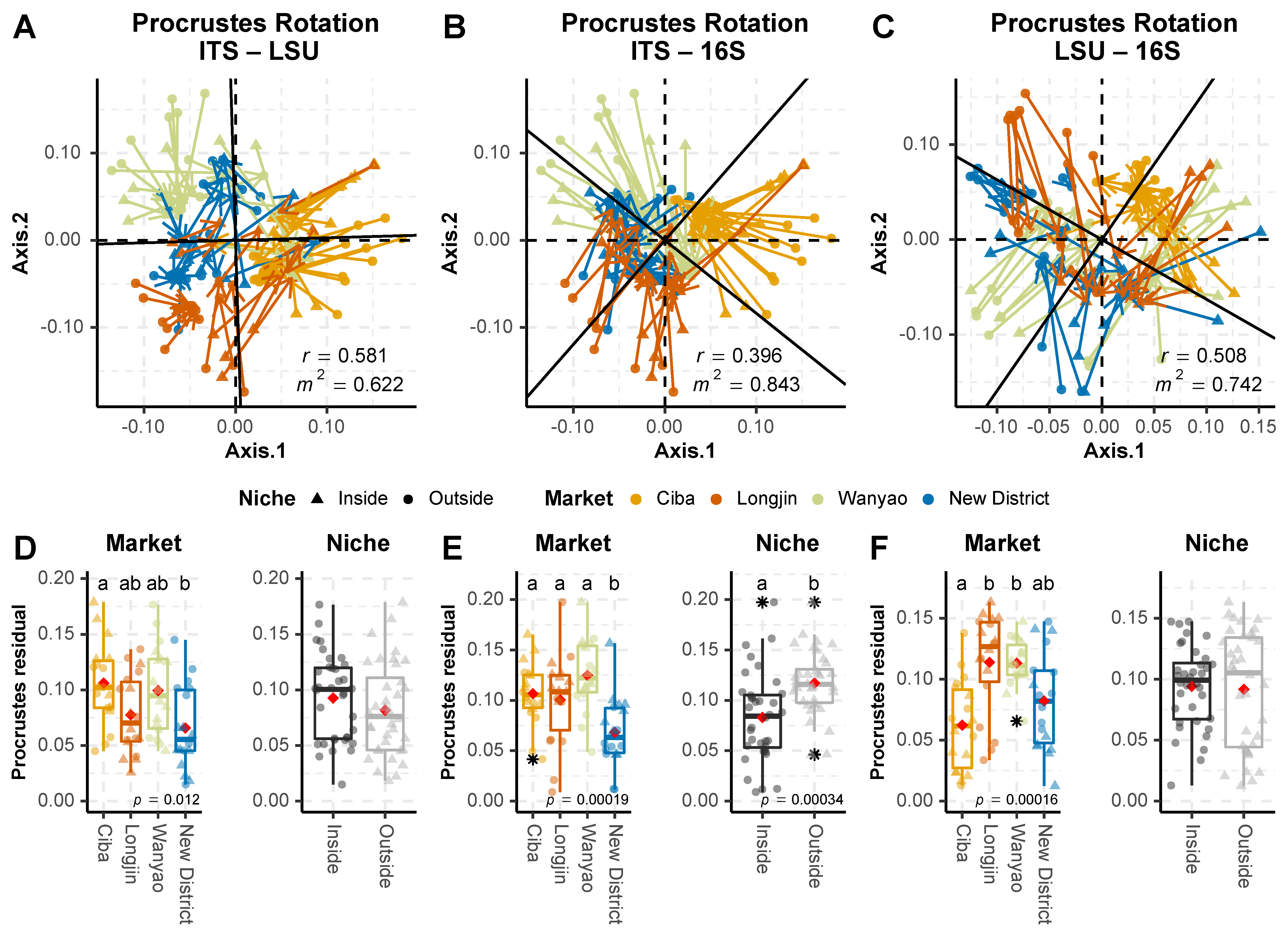
3.3. Beta Diversity
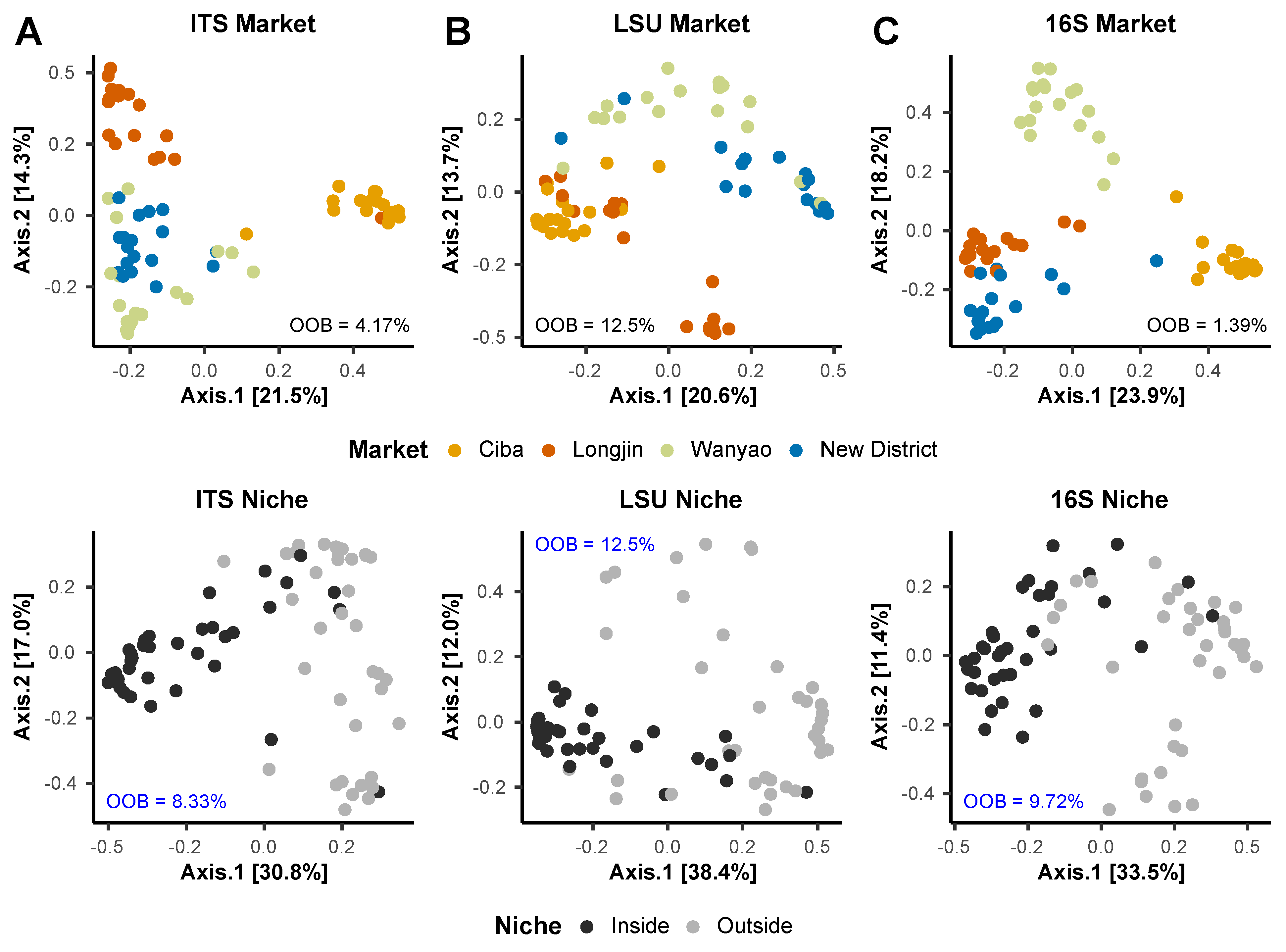
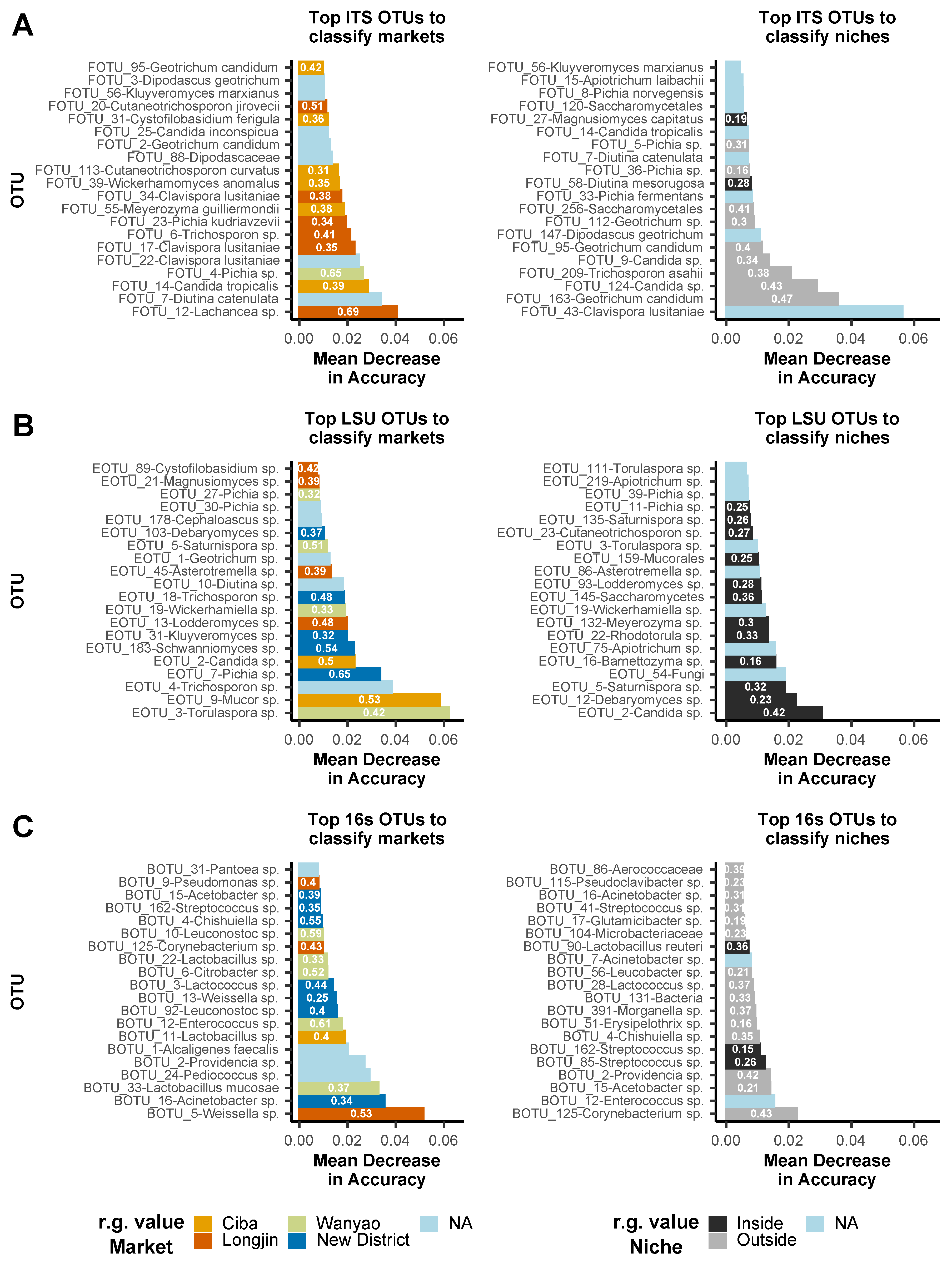
3.4. Random Forest Models and Indicator Taxa
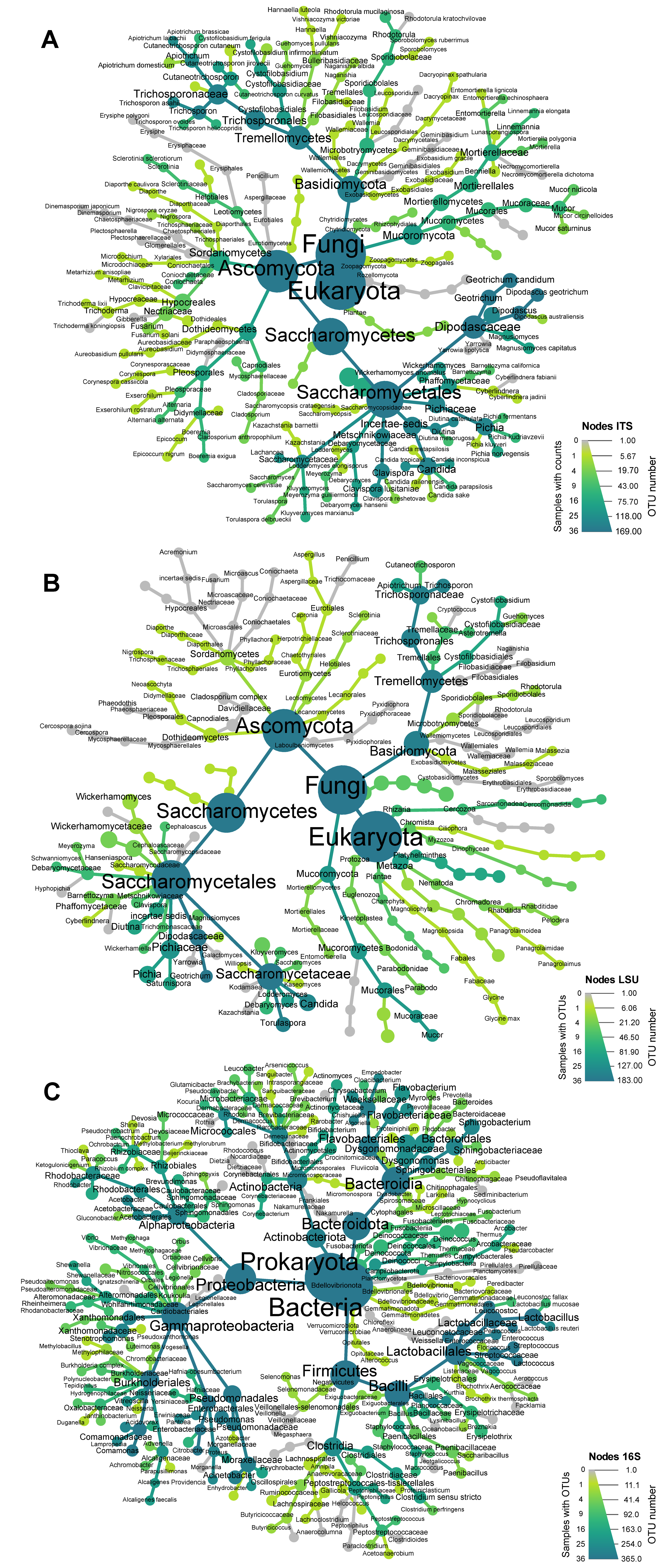
3.5. Microbial Diversity and Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jayachandran, M.; Xu, B. An Insight into the Health Benefits of Fermented Soy Products. Food Chem. 2019, 271, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.H.; Park, C.S.; Lim, J.K.; Kim, J.H.; Kwon, D.Y.; Kim, Y.S. Metabolism of Isoflavone Derivatives during Manufacturing of Traditional Meju and Doenjang. Food Sci. Biotechnol. 2008, 17, 442–445. [Google Scholar]
- Xu, L.; Cai, W.X.; Xu, B.J. A Systematic Assesment on Vitamins (B2, B12) and GABA Profiles in Fermented Soy Products Marketed in China. J. Food Processing Preserv. 2017, 41, e13126. [Google Scholar] [CrossRef]
- Kim, D.-J.; Jeong, Y.-J.; Kwon, J.-H.; Moon, K.-D.; Kim, H.-J.; Jeon, S.-M.; Lee, M.-K.; Park, Y.B.; Choi, M.-S. Beneficial Effect of Chungkukjang on Regulating Blood Glucose and Pancreatic Beta-Cell Functions in C75BL/KsJ-Db/db Mice. J. Med. Food 2008, 11, 215–223. [Google Scholar] [CrossRef]
- Yoon, G.-A.; Park, S. Antioxidant Action of Soy Isoflavones on Oxidative Stress and Antioxidant Enzyme Activities in Exercised Rats. Nutr. Res. Pract. 2014, 8, 618–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, P.; Parkin, D.M.; Bray, F.; Ferlay, J. Estimates of the Worldwide Mortality from 25 Cancers in 1990. Int. J. Cancer 1999, 83, 18–29. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, A.; Liu, M.C.; Zwart, A.; Lee, R.Y.; Gallagher, A.; Wang, Y.; Miller, W.R.; Dixon, J.M.; Clarke, R. Estrogen Receptor Alpha Positive Breast Tumors and Breast Cancer Cell Lines Share Similarities in Their Transcriptome Data Structures. Int. J. Oncol. 2006, 29, 1581–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-M.; Kim, Y.; Choi, H.J.; Choi, J.; Yi, Y.; Yoon, S. Soy Milk Suppresses Cholesterol-Induced Inflammatory Gene Expression and Improves the Fatty Acid Profile in the Skin of SD Rats. Biochem. Biophys. Res. Commun. 2013, 430, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Ren, N.N.; Chen, H.J.; Li, Y.; Mcgowan, G.W.; Lin, Y.G. A clinical study on the effect of nattokinase on carotid artery atherosclerosis and hyperlipidaemia. Zhonghua Yi Xue Za Zhi 2017, 97, 2038–2042. [Google Scholar] [PubMed]
- Tsai, J.S.; Lin, Y.S.; Pan, B.S.; Chen, T.J. Antihypertensive Peptides and γ-Aminobutyric Acid from Prozyme 6 Facilitated Lactic Acid Bacteria Fermentation of Soymilk. Process Biochem. 2006, 41, 1282–1288. [Google Scholar] [CrossRef]
- Kang, S.J.; Seo, J.Y.; Cho, K.M.; Lee, C.K.; Kim, J.H.; Kim, J.-S. Antioxidant and Neuroprotective Effects of Doenjang Prepared with Rhizopus, Pichia, and Bacillus. Prev. Nutr. Food Sci. 2016, 21, 221–226. [Google Scholar] [CrossRef]
- Gu, J.; Liu, T.; Sadiq, F.A.; Yang, H.; Yuan, L.; Zhang, G.; He, G. Biogenic Amines Content and Assessment of Bacterial and Fungal Diversity in Stinky Tofu—A Traditional Fermented Soy Curd. LWT 2018, 88, 26–34. [Google Scholar] [CrossRef]
- Zhao, X.; Zheng, X. A Primary Study on Texture Modification and Proteolysis of Mao-Tofu during Fermentation. Afr. J. Biotechnol. 2009, 8, 2294–2300. [Google Scholar]
- Liang, H.; Deng, L.; Lin, H. Distribution, Functions and Applications of Lactic Acid Bacteria in Traditional Fermented Soybean Foods. Food Sci. 2013, 34, 381–385. [Google Scholar]
- Chao, S.-H.; Tomii, Y.; Sasamoto, M.; Fujimoto, J.; Tsai, Y.-C.; Watanabe, K. Lactobacillus Capillatus Sp. Nov., a Motile Bacterium Isolated from Stinky Tofu Brine. Int. J. Syst. Evol. Microbiol. 2008, 58, 2555–2559. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Hu, H.; Li, L. Analysis of Microbial Flora in the Steep Juice of Zhejiang Shaoxing Stinky Tofu. Sci. Technol. Food Ind. 2012, 33, 183–187. [Google Scholar]
- Chao, S.-H.; Tomii, Y.; Watanabe, K.; Tsai, Y.-C. Diversity of Lactic Acid Bacteria in Fermented Brines Used to Make Stinky Tofu. Int. J. Food Microbiol. 2008, 123, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.P.; Zhang, X.J.; Wang, Y.; Wang, D.; Xie, J.L. The Investigation of Bacteria Diversity in Stinky Tofu Brine. Xiandai Shipin Keji 2010, 26, 1087–1091. [Google Scholar]
- Benucci, G.M.N.; Longley, R.; Zhang, P.; Zhao, Q.; Bonito, G.; Yu, F. Microbial Communities Associated with the Black Morel Cultivated in Greenhouses. PeerJ 2019, 7, e7744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal rna genes for phylogenetics. In PCR Protocols; Elsevier: Amsterdam, The Netherlands, 1990; pp. 315–322. [Google Scholar]
- Gardes, M.; Bruns, T.D. ITS Primers with Enhanced Specificity for Basidiomycetes--Application to the Identification of Mycorrhizae and Rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- Reynolds, N.K.; Benny, G.L.; Ho, H.-M.; Hou, Y.-H.; Crous, P.W.; Smith, M.E. Phylogenetic and Morphological Analyses of the Mycoparasitic Genus Piptocephalis. Mycologia 2019, 111, 54–68. [Google Scholar] [CrossRef]
- Hopple, J.S.; Vilgalys, R. Phylogenetic Relationships among Coprinoid Taxa and Allies Based on Data from Restriction Site Mapping of Nuclear rDNA. Mycologia 1994, 86, 96–107. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S rRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2010, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, R.; Benucci, G.M.N.; Mills, G.; Bonito, G. Fungal and Bacterial Community Dynamics in Substrates during the Cultivation of Morels (Morchella Rufobrunnea) Indoors. FEMS Microbiol. Lett. 2019, 366, fnz215. [Google Scholar] [CrossRef] [PubMed]
- Noel, Z.A.; Chang, H.-X.; Chilvers, M.I. Variation in Soybean Rhizosphere Oomycete Communities from Michigan Fields with Contrasting Disease Pressures. Appl. Soil Ecol. 2020, 150, 103435. [Google Scholar] [CrossRef]
- Caporaso, G.J.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Benucci, G.M.N.; Rennick, B.; Bonito, G. Patient Propagules: Do Soil Archives Preserve the Legacy of Fungal and Prokaryotic Communities? PLoS ONE 2020, 15, e0237368. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Flyvbjerg, H. Error Filtering, Pair Assembly and Error Correction for next-Generation Sequencing Reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. UCHIME2: Improved Chimera Prediction for Amplicon Sequencing. BioRxiv 2016, 074252. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gdanetz, K.; Benucci, G.M.N.; Vande Pol, N.; Bonito, G. CONSTAX: A Tool for Improved Taxonomic Resolution of Environmental Fungal ITS Sequences. BMC Bioinform. 2017, 18, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abarenkov, K.; Zirk, A.; Piirmann, T.; Pöhönen, R.; Ivanov, F.; Nilsson, R.H.; Kõljalg, U. UNITE General FASTA Release for Eukaryotes Version 04.02.2020; UNITE Community: London, UK, 2020. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 12 May 2020).
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.5-6. 2019. Available online: https://rdrr.io/cran/vegan/ (accessed on 12 May 2020).
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Murphy, M.A.; Evans, J.S.; Storfer, A. Quantifying Bufo Boreas Connectivity in Yellowstone National Park with Landscape Genetics. Ecology 2010, 91, 252–261. [Google Scholar] [CrossRef] [Green Version]
- De Cáceres, M.; Legendre, P.; Moretti, M. Improving Indicator Species Analysis by Combining Groups of Sites. Oikos 2010, 119, 1674–1684. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and Microbial Differential Abundance Strategies Depend upon Data Characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Leff, W.J. Mctoolsr: Microbial Community Data Analysis Tools. R package Version 0.1.1.2. Available online: https://github.com/leffj/mctoolsr (accessed on 26 May 2020). [CrossRef]
- Anderson, M.J.; Ellingsen, K.E.; McArdle, B.H. Multivariate Dispersion as a Measure of Beta Diversity. Ecol. Lett. 2006, 9, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Gower, J.C. Generalized Procrustes Analysis. Psychometrika 1975, 40, 33–51. [Google Scholar] [CrossRef]
- Jackson, D.A. PROTEST: A PROcrustean Randomization TEST of Community Environment Concordance. Écoscience 1995, 2, 297–303. [Google Scholar] [CrossRef]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R Package for Visualization and Manipulation of Community Taxonomic Diversity Data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 12 May 2020).
- Kassambara, A. Ggpubr: “Ggplot2” Based Publication Ready Plots. R Package Version 0.4.0. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 25 May 2020).
- Yan, S.; Liu, H.; Zhang, J.; Tong, Q. Lactobacillus Delbrueckii Is the Key Functional Microorganism of Natural Fermented Tofu Sour Water Involved in the Traditional Coagulation of Chinese Huizhou Mao-Tofu. LWT 2020, 131, 109706. [Google Scholar] [CrossRef]
- He, G.-Q.; Liu, T.-J.; Sadiq, F.A.; Gu, J.-S.; Zhang, G.-H. Insights into the Microbial Diversity and Community Dynamics of Chinese Traditional Fermented Foods from Using High-Throughput Sequencing Approaches. J. Zhejiang Univ. Sci. B 2017, 18, 289–302. [Google Scholar] [CrossRef]
- Tedersoo, L.; Lindahl, B. Fungal Identification Biases in Microbiome Projects. Environ. Microbiol. Rep. 2016, 8, 774–779. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, X.-H. Study of Mucor Spp. in Semi-Hard Cheese Ripening. J. Food Sci. Technol. 2010, 47, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Morel, G.; Sterck, L.; Swennen, D.; Marcet-Houben, M.; Onesime, D.; Levasseur, A.; Jacques, N.; Mallet, S.; Couloux, A.; Labadie, K.; et al. Differential Gene Retention as an Evolutionary Mechanism to Generate Biodiversity and Adaptation in Yeasts. Sci. Rep. 2015, 5, 11571. [Google Scholar] [CrossRef]
- Boutrou, R.; Guéguen, M. Interests in Geotrichum Candidum for Cheese Technology. Int. J. Food Microbiol. 2005, 102, 1–20. [Google Scholar] [CrossRef]
- Tamang, J.P.; Shin, D.-H.; Jung, S.-J.; Chae, S.-W. Functional Properties of Microorganisms in Fermented Foods. Front. Microbiol. 2016, 7, 578. [Google Scholar] [CrossRef] [Green Version]
- Büchl, N.R.; Seiler, H. Yeasts and Molds|Yeasts in Milk and Dairy Products; Encyclopedia of Dairy Sciences; Elsevier Ltd.: Amsterdam, The Netherlands, 2011; pp. 744–753. [Google Scholar]
- Dimidi, E.; Cox, S.R.; Rossi, M.; Whelan, K. Fermented Foods: Definitions and Characteristics, Impact on the Gut Microbiota and Effects on Gastrointestinal Health and Disease. Nutrients 2019, 11, 1806. [Google Scholar] [CrossRef] [Green Version]
- Padilla, B.; Gil, J.V.; Manzanares, P. Past and Future of Non-Saccharomyces Yeasts: From Spoilage Microorganisms to Biotechnological Tools for Improving Wine Aroma Complexity. Front. Microbiol. 2016, 7, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Breda, V.; Jolly, N.; van Wyk, J. Characterisation of Commercial and Natural Torulaspora Delbrueckii Wine Yeast Strains. Int. J. Food Microbiol. 2013, 163, 80–88. [Google Scholar] [CrossRef]
- Combina, M.; Elía, A.; Mercado, L.; Catania, C.; Ganga, A.; Martinez, C. Dynamics of Indigenous Yeast Populations during Spontaneous Fermentation of Wines from Mendoza, Argentina. Int. J. Food Microbiol. 2005, 99, 237–243. [Google Scholar] [CrossRef]
- Michel, M.; Kopecká, J.; Meier-Dörnberg, T.; Zarnkow, M.; Jacob, F.; Hutzler, M. Screening for New Brewing Yeasts in the Non-Saccharomyces Sector with Torulaspora Delbrueckii as Model. Yeast 2016, 33, 129–144. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.E.; McCarthy, C.G.P.; Walshe, A.E.; Shaw, D.R.; Sumski, D.A.; Krassowski, T.; Fitzpatrick, D.A.; Butler, G. Genome Analysis of the Yeast Diutina Catenulata, a Member of the Debaryomycetaceae/Metschnikowiaceae (CTG-Ser) Clade. PLoS ONE 2018, 13, e0198957. [Google Scholar]
- Delavenne, E.; Mounier, J.; Asmani, K.; Jany, J.-L.; Barbier, G.; Le Blay, G. Fungal Diversity in Cow, Goat and Ewe Milk. Int. J. Food Microbiol. 2011, 151, 247–251. [Google Scholar] [CrossRef]
- Fei, Y.; Li, L.; Chen, L.; Zheng, Y.; Yu, B. High-Throughput Sequencing and Culture-Based Approaches to Analyze Microbial Diversity Associated with Chemical Changes in Naturally Fermented Tofu Whey, a Traditional Chinese Tofu-Coagulant. Food Microbiol. 2018, 76, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Carini, P.; Marsden, P.J.; Leff, J.W.; Morgan, E.E.; Strickland, M.S.; Fierer, N. Relic DNA Is Abundant in Soil and Obscures Estimates of Soil Microbial Diversity. Nat. Microbiol. 2016, 2, 16242. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G. The Entomopathogenic fungusMetarhizium Anisopliaeand Its Potential as a Biocontrol Agent. Pestic. Sci. 1993, 37, 375–379. [Google Scholar] [CrossRef]
- Wrather, A.; Shannon, G.; Balardin, R.; Carregal, L.; Escobar, R.; Gupta, G.K.; Ma, Z.; Morel, W.; Ploper, D.; Tenuta, A. Effect of Diseases on Soybean Yield in the Top Eight Producing Countries in 2006. Plant Health Prog. 2010, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gray, L.E.; Kurle, J.E.; Grau, C.R. Specific Detection ofPhialophora gregataandPlectosporium Tabacinumin Infected Soybean Plants Using Polymerase Chain Reaction. Mol. Ecol. 1999, 8, 871–877. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Mills, D.A. Facility-Specific “House” Microbiome Drives Microbial Landscapes of Artisan Cheesemaking Plants. Appl. Environ. Microbiol. 2013, 79, 5214–5223. [Google Scholar] [CrossRef] [Green Version]
- Van Reckem, E.; Geeraerts, W.; Charmpi, C.; Van der Veken, D.; De Vuyst, L.; Leroy, F. Exploring the Link Between the Geographical Origin of European Fermented Foods and the Diversity of Their Bacterial Communities: The Case of Fermented Meats. Front. Microbiol. 2019, 10, 2302. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese Rind Communities Provide Tractable Systems for in Situ and in Vitro Studies of Microbial Diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.; Barbosa, R.; Zalar, P.; Imanishi, Y.; Shimizu, K.; Turchetti, B.; Legras, J.-L.; Serra, M.; Dequin, S.; Couloux, A.; et al. A Population Genomics Insight into the Mediterranean Origins of Wine Yeast Domestication. Mol. Ecol. 2015, 24, 5412–5427. [Google Scholar] [CrossRef]
- Dumas, E.; Feurtey, A.; Rodríguez de la Vega, R.C.; Le Prieur, S.; Snirc, A.; Coton, M.; Thierry, A.; Coton, E.; Le Piver, M.; Roueyre, D.; et al. Independent Domestication Events in the Blue-Cheese Fungus Penicillium Roqueforti. Mol. Ecol. 2020, 29, 2639–2660. [Google Scholar] [CrossRef] [Green Version]
- Mezzasalma, V.; Sandionigi, A.; Guzzetti, L.; Galimberti, A.; Grando, M.S.; Tardaguila, J.; Labra, M. Geographical and Cultivar Features Differentiate Grape Microbiota in Northern Italy and Spain Vineyards. Front. Microbiol. 2018, 9, 946. [Google Scholar] [CrossRef]
- Longley, R.; Noel, Z.A.; Benucci, G.M.N.; Chilvers, M.I.; Trail, F.; Bonito, G. Crop Management Impacts the Soybean (Glycine Max) Microbiome. Front. Microbiol. 2020, 11, 1116. [Google Scholar] [CrossRef] [PubMed]
- Statnikov, A.; Henaff, M.; Narendra, V.; Konganti, K.; Li, Z.; Yang, L.; Pei, Z.; Blaser, M.J.; Aliferis, C.F.; Alekseyenko, A.V. A Comprehensive Evaluation of Multicategory Classification Methods for Microbiomic Data. Microbiome 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | ITS | LSU | 16S | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Df | F-Value | R2 | p-Value | Df | F-Value | R2 | p-Value | Df | F-Value | R2 | p-Value | |
| Market | 3 | 6.070 | 0.193 | 0.0003 | 3 | 4.891 | 0.158 | 0.0003 | 3 | 5.423 | 0.170 | 0.0003 |
| Niche | 1 | 4.708 | 0.050 | 0.0003 | 1 | 5.244 | 0.057 | 0.0003 | 1 | 5.420 | 0.057 | 0.0003 |
| Market:Niche | 3 | 2.502 | 0.079 | 0.0003 | 3 | 2.934 | 0.095 | 0.0003 | 3 | 3.260 | 0.102 | 0.0003 |
| Residuals | 64 | 0.678 | 62 | 0.690 | 64 | 0.670 | ||||||
| Total | 71 | 1.000 | 69 | 1.000 | 71 | 1.000 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benucci, G.M.N.; Wang, X.; Zhang, L.; Bonito, G.; Yu, F. Yeast and Lactic Acid Bacteria Dominate the Core Microbiome of Fermented ‘Hairy’ Tofu (Mao Tofu). Diversity 2022, 14, 207. https://doi.org/10.3390/d14030207
Benucci GMN, Wang X, Zhang L, Bonito G, Yu F. Yeast and Lactic Acid Bacteria Dominate the Core Microbiome of Fermented ‘Hairy’ Tofu (Mao Tofu). Diversity. 2022; 14(3):207. https://doi.org/10.3390/d14030207
Chicago/Turabian StyleBenucci, Gian Maria Niccolò, Xinxin Wang, Li Zhang, Gregory Bonito, and Fuqiang Yu. 2022. "Yeast and Lactic Acid Bacteria Dominate the Core Microbiome of Fermented ‘Hairy’ Tofu (Mao Tofu)" Diversity 14, no. 3: 207. https://doi.org/10.3390/d14030207
APA StyleBenucci, G. M. N., Wang, X., Zhang, L., Bonito, G., & Yu, F. (2022). Yeast and Lactic Acid Bacteria Dominate the Core Microbiome of Fermented ‘Hairy’ Tofu (Mao Tofu). Diversity, 14(3), 207. https://doi.org/10.3390/d14030207