Abstract
Neanthes japonica has high commercial value. The gut microbes in N. japonica can maintain the normal biological functions of the host. However, information on the gut bacterial community of N. japonica and its relationship with the surrounding environment is unclear. In this study, we used high-throughput sequencing technology to investigate the bacterial communities in the gut of N. japonica and soil. The results showed that the bacterial community diversity and structure differed obviously between the gut and soil samples. Bacterial richness and diversity in the gut samples decreased considerably compared to soil samples. In addition, dominant bacterial taxa varied significantly between the gut and soil samples. The dominant phyla in the gut and soil samples were Proteobacteria, Bacteroidota and Planctomycetota. The dominant genus in the gut was Burkholderia-Caballeronia-Paraburkholderia, while the dominant genera in the soil were Woeseia and Subgroup_23. In conclusion, the similarity between the bacterial communities in soil and the gut of N. japonica was small, indicating that soil had little effect on the establishment of the gut bacterial community. This study provides a better understanding of the gut bacterial community in N. japonica and the influence of the external environment on the colonization of the gut bacterial community.
1. Introduction
Gut microbes have attracted much attention due to their important role in maintaining the homeostasis of their hosts. It is widely accepted that gut microbes and their metabolites play an essential role in a range of biological functions of the host, such as nutrition and energy harvesting, immunity and inflammation regulation and neural transmission [1,2]. Several factors influence the colonization of microbial communities in the gut. Host phylogeny, growth stage, diet and health status have been shown to affect the composition and diversity of gut bacterial communities [3,4]. In addition, microorganisms in the external environment may contribute to the establishment of microbial communities in the guts of animals. It is documented that the gut microbes of soil invertebrates are gradually assembled from the soil micro-ecological zones [5]. Traditionally, the isolation and culture method is usually used to analyze the microbial community, but it cannot completely isolate the microorganisms from samples, which makes it difficult to accurately reflect the number of microorganisms [6]. PCR-DGGE and next-generation high-throughput sequencing based on 16S ribosomal RNA (rRNA) analysis are widely used for better identification of poorly described, rarely isolated bacterial strains [7]. The PCR-KGGE method has a length limitation, generally analyzing sequences less than 500 bp in length [6]. Conversely, high-throughput sequencing is an effective tool to comprehensively reflect microbial information in samples.
Neanthes japonica, a polychaete, is a euryhaline species native to China and Japan. It is widely present in estuarine systems, living as a deposit-feeder near the surface of sedimentary deposits rich in bio-detritus [8,9]. Due to its high nutritional value, it is considered as high-quality food for many aquatic organisms, such as fish and crustaceans, and is used as bait in sea fishing [9]. In addition, it has been widely recognized as an ideal feed for shrimp aquaculture. N. japonica is shrimp bait and provides a suitable living environment for the growth of shrimp through bioturbation [10]. For N. japonica, some papers have focused on the purification and characterization of fibrinolytic enzymes from N. japonica in recent years [11,12]. However, studies on the gut bacterial community diversity and composition structure of N. japonica, as well as their relationship with external environmental factors, are not well-known. In this study, we used high-throughput sequencing to analyze bacterial community diversity and composition in the gut of N. japonica and in soil, and further elucidated the relationship between bacterial communities in these two environments.
2. Materials and Methods
2.1. Samples Collection
Sampling was carried out at the Yellow River delta (37°16′ N–38°16′ N, 118°20′ E–119°20′ E). Four 1 m × 1 m plots were set up and the distance between the adjacent plots was 100 m. Visible plant litter and stones were removed, and soil samples were collected in 0–10 cm layers from each plot with a soil auger. After collection, soil samples were brought back in boxes filled with dry ice. N. japonica were removed from the soil and placed in the soil of the original habitat and then taken back to the laboratory alive. The N. japonica with a weight of 2.2 ± 0.6 g were randomly picked, placed in ice and washed with 75% alcohol and sterile distilled water. Afterwards, the samples were dissected aseptically, and the gut tissues were collected. The collected gut tissues (the Gut group) and soil samples (the Soil group) were stored at −80 °C until DNA extraction.
2.2. DNA Extraction and Polymerase Chain Reaction (PCR) Amplification
According to the manufacturer’s instruction, total genomic DNA from soil samples was extracted using E.Z.N.A. Soil DNA Kit (D4015, Omega, Inc., Norcross, GA, USA) and total genomic DNA in gut samples was extracted using E.Z.N.A. Stool DNA Kit. The extracted DNA purity and quality were assessed using an ultraviolet spectrophotometer and agarose gel electrophoresis. PCR was conducted to amplify the V3-V4 region of the bacterial 16S rRNA gene using the primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) under the following conditions: an initial denaturation at 98 °C for 30 s, 32 cycles of denaturation at 98 °C for 10 s, annealing at 54 °C for 30 s, extension at 72 °C for 45 s and final extension at 72 °C for 10 min [13]. The reaction mixture contained 25 ng of template DNA, 12.5 µL PCR Premix, 2.5 µL of each primer and PCR-grade water to adjust the volume. PCR products were confirmed with 2% agarose gel electrophoresis, purified using AMPure XT beads (Beckman Coulter Genomics, Danvers, MA, USA) and quantified using Qubit (Invitrogen, Carlsbad, CA, USA). Then, the amplicons pools were established and pair-end 2 × 250 bp sequencing was conducted using the NovaSeq 6000 sequencer at Lc-Bio Technologies Co., Ltd (Hangzhou, Zhejiang Province, China). Raw sequence data were deposited in the Sequence Read Archive (SRA) database of NCBI under accession number (SRR18906812-18906819), BioProject number (PRJNA830633) and Biosample number (SAMN27734950-27734957).
2.3. Sequencing Process and Statistical Analysis
Pair-end reads were assembled into samples based on barcode, and then barcode and primer sequences were cut. Pair-end reads were merged using FLASH v1.2.8. High-quality clean tags were obtained via filtering chimeras and removing sequences less than 100 bp in length. After dereplication using Divisive Amplicon Denoising Algorithm (DADA2) plugin in QIIME2, we obtained amplicon sequence variants (ASVs) table and singletons ASVs were removed [14]. Alpha diversity indices (Chao1 and Shannon indices) were calculated using QIIME2 software (version 2019.1). Chao1 and Shannon indices were used to evaluate bacterial community richness and diversity of samples, respectively. We used the Kruskal–Wallis to test the differences in bacterial alpha diversity. The Venn diagram was generated using the R VennDiagram package [15]. Principal coordinate analysis (PCoA) with unweighted UniFrac distance was used to visualize the differences in bacterial community structure. The plot of PCoA was generated using the R vegan package [16]. Additionally, permutational multivariate analysis of variance (PERMANOVA) was performed to provide statistical insight into the PCoA results. The heatmap with hierarchal clustering was performed to visualize similarities and differences in the microbial community composition among samples.
3. Results
3.1. Bacterial Diversity
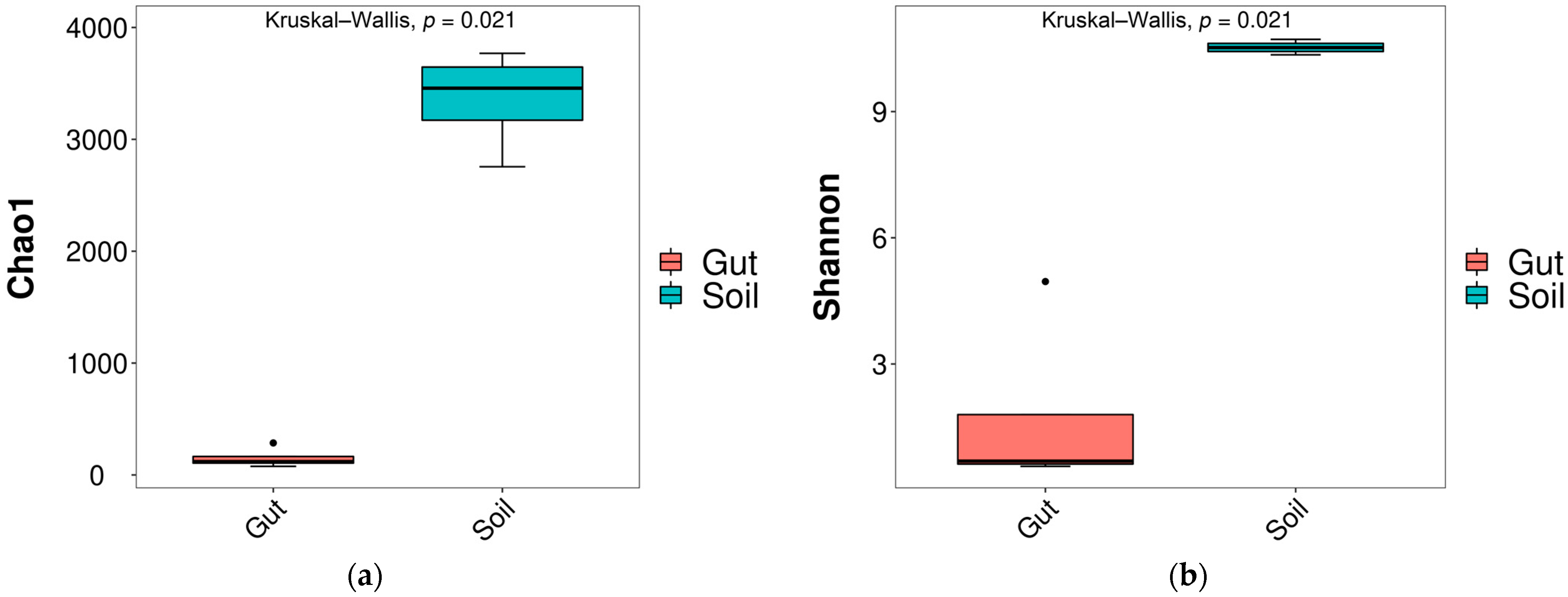
After sequencing, 565,680 raw reads were obtained. There were 565,680 effective reads after filtering raw reads. The richness and diversity of bacterial communities exhibited significant differences between the Gut and Soil groups. The diversity index (Shannon index) and richness index (Chao1 index) decreased significantly (p < 0.05) in the Gut group compared to the Soil group (Figure 1), indicating that the soil bacterial community was more diverse.
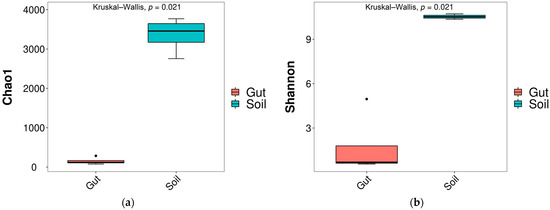
Figure 1.
Differences in alpha diversity indices of bacteria ((a) Chao1; (b) Shannon).
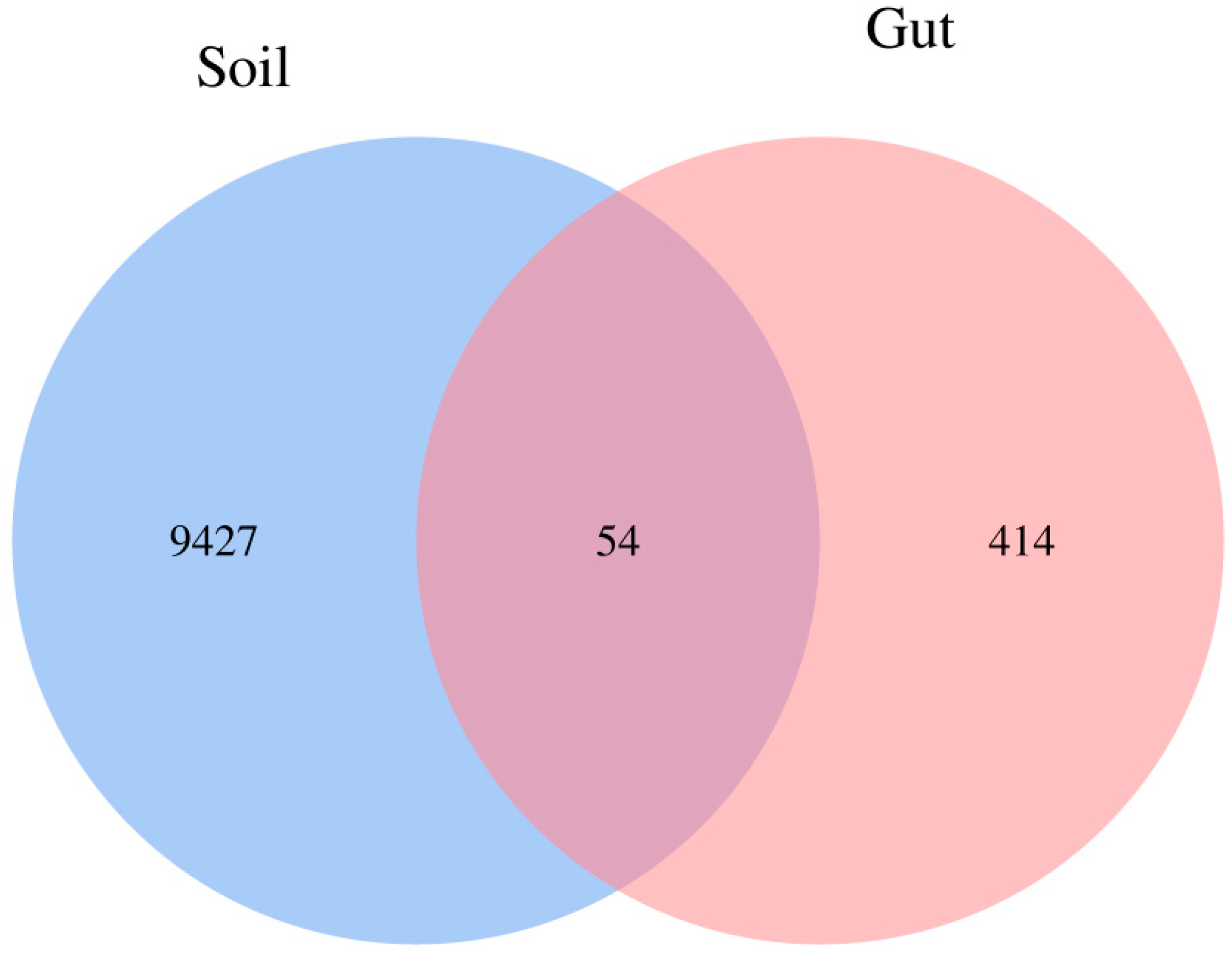
The Venn diagram showed the number of shared and unique ASVs between Soil and Gut groups. The number of unique ASVs in the Gut group and Soil group was 414 and 9427, respectively. The number of shared ASVs was 54 (Figure 2). The small proportion of shared ASVs in the total ASVs of the two groups represented the notable difference in bacterial communities.
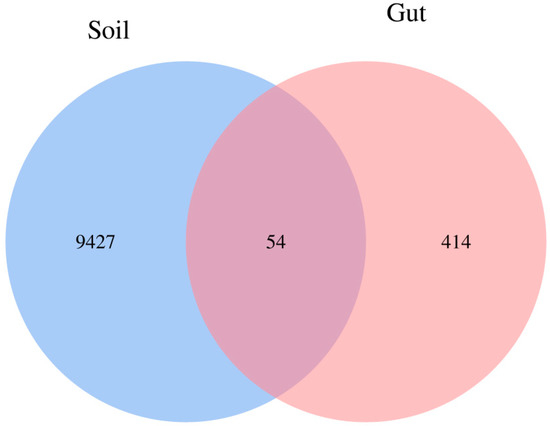
Figure 2.
Venn diagram depicting the number of shared and unique bacterial amplicon sequence variants (ASVs) between groups. Each circle represents sampled compartments. Values within intersections represent shared ASVs, values outside intersections represent unique ASVs.
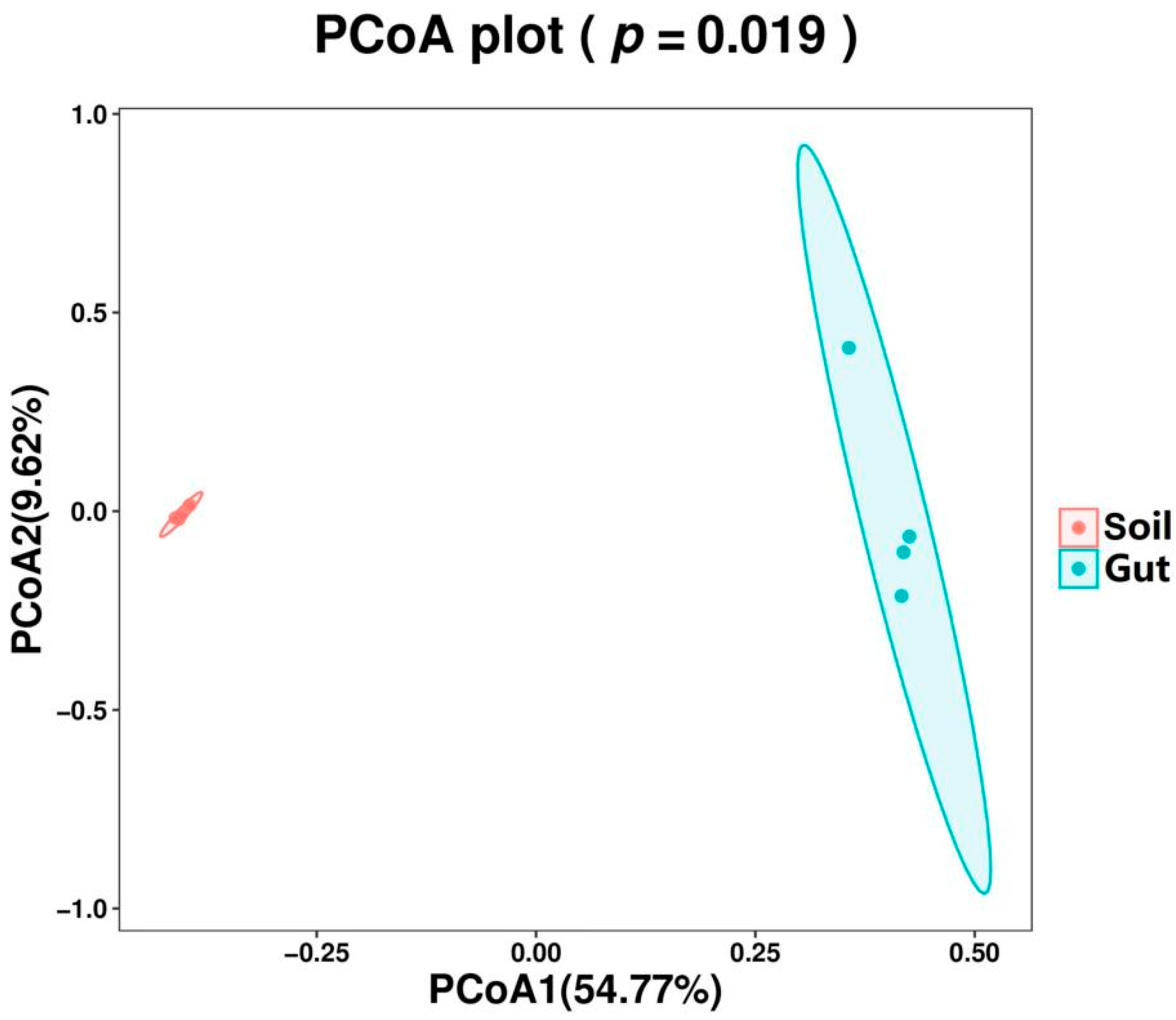
PCoA based on unweighted UniFrac distance was performed to determine the difference in the composition structure between the groups. The PCoA1 and the PCoA2 explained 54.77% and 9.62% of variations, respectively (Figure 3). The result of PCoA showed clear separations into the two groups, representing the difference in bacterial communities. In addition, samples from the same group were clustered together and samples in the Soil group showed higher similarity than those in the Gut group. To statistically support the clustering of the bacterial communities between the two groups, p-value was evaluated via PERMANOVA method. The bacterial community showed significant dissimilarity (p < 0.05) between the two groups.
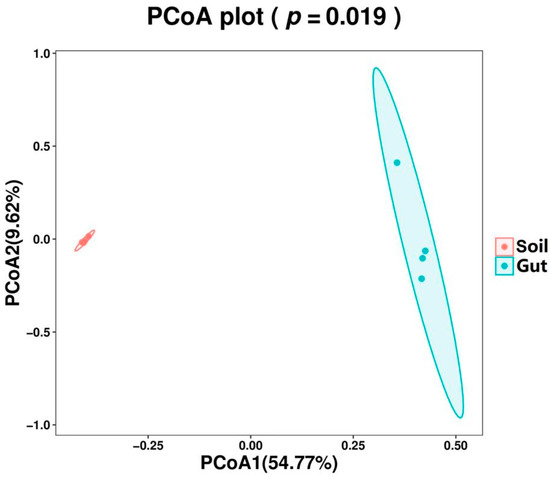
Figure 3.
Principal coordinates analysis (PCoA) with unweighted UniFrac distance of bacterial community composition.
3.2. Bacterial Community Composition
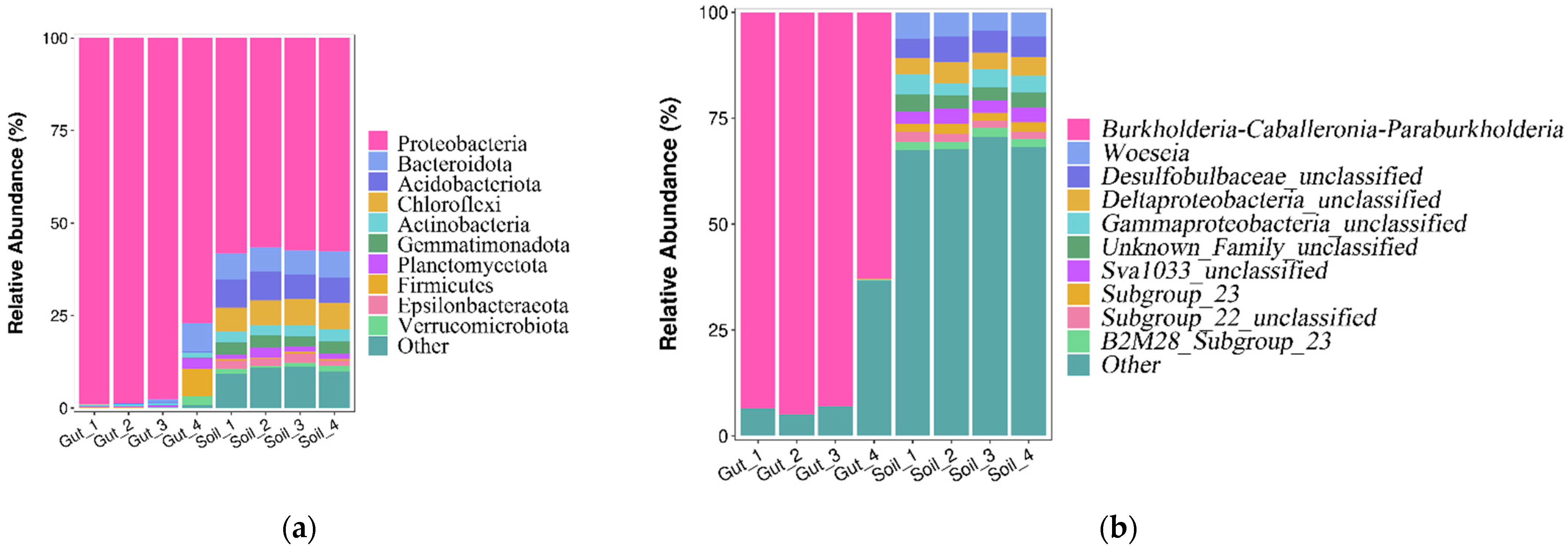
The relative abundance of the bacterial communities at the phylum level is shown in Figure 4a. The dominant phyla (relative abundance more than 1%) in the Gut group were Proteobacteria (93.06%), Bacteroidota (2.11%), Firmicutes (1.96%) and Planctomycetota (1.02%). The dominant phyla (relative abundance more than 1%) in the Soil group were Proteobacteria (57.41%), Acidobacteriota (7.25%), Chloroflexi (6.83%), Bacteroidota (6.78%), Gemmatimonadota (3.12%), Actinobacteria (3.01%), Epsilonbacteraeota (2.01%), Planctomycetota (1.72%) and Verrucomicrobiota (1.16%). Proteobacteria were the most abundant phylum in the two groups.
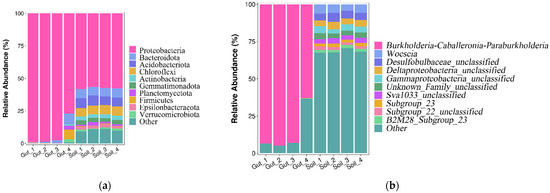
Figure 4.
Bacterial community composition at the phylum (a) and genus (b) level.
There were significant differences (p < 0.05) in the relative abundance of bacterial communities, including Proteobacteria, Acidobacteriota, Chloroflexi, Gemmatimonadota and Actinobacteria (Figure S1).
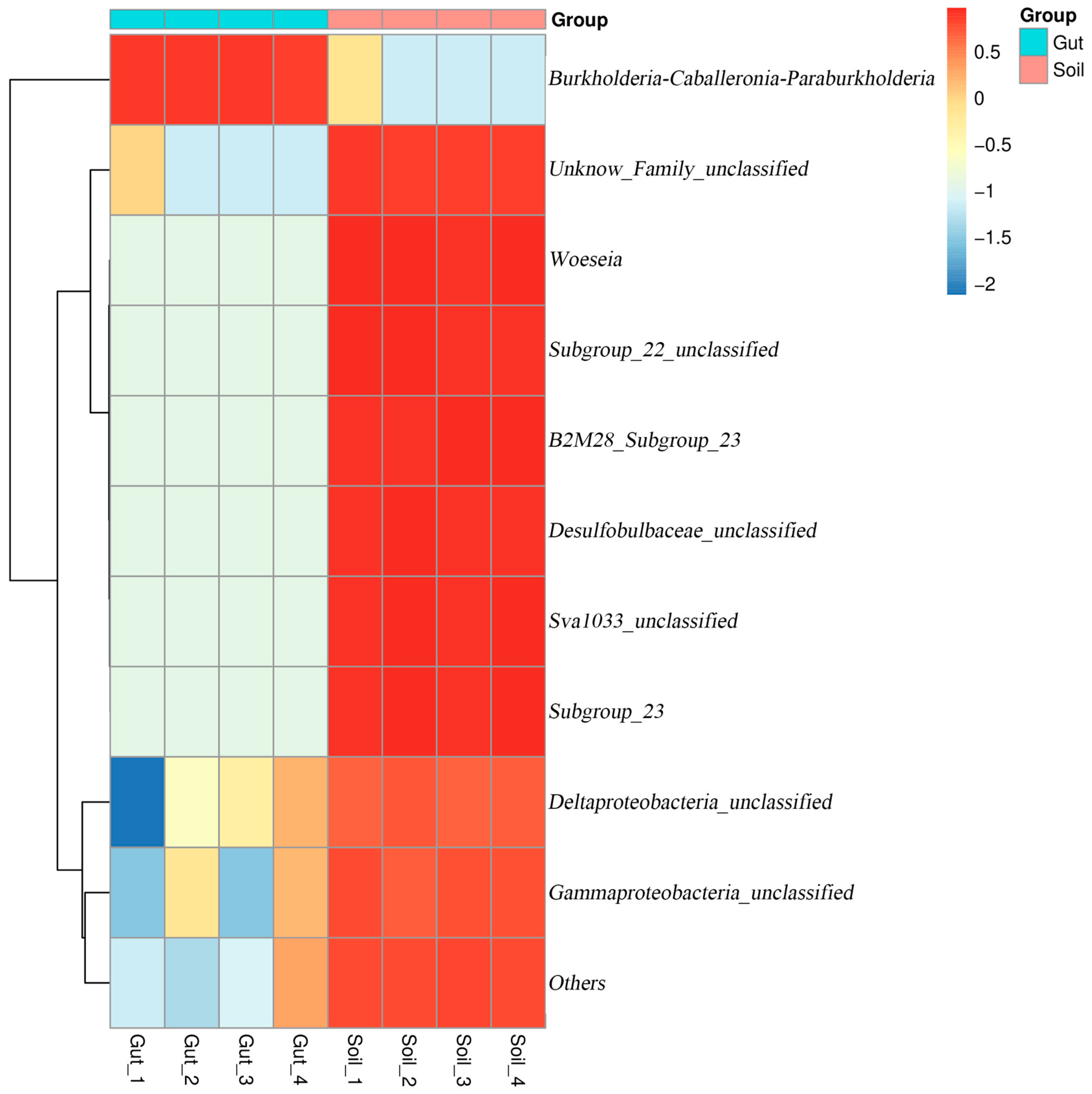
The relative abundance of the bacterial communities at the genus level are shown in Figure 4b. Except for the unclassified genera, the dominant genus (relative abundance more than 1%) in the Gut group was Burkholderia-Caballeronia-Paraburkholderia (86.04%). The dominant genera in the Soil group were Woeseia (5.45%) and Subgroup_23 (2.06%). The heatmap also reflected the distribution of bacteria genera in all samples (Figure 5). These results indicated that there were considerable differences in the bacterial community composition between the Gut group and the Soil group.
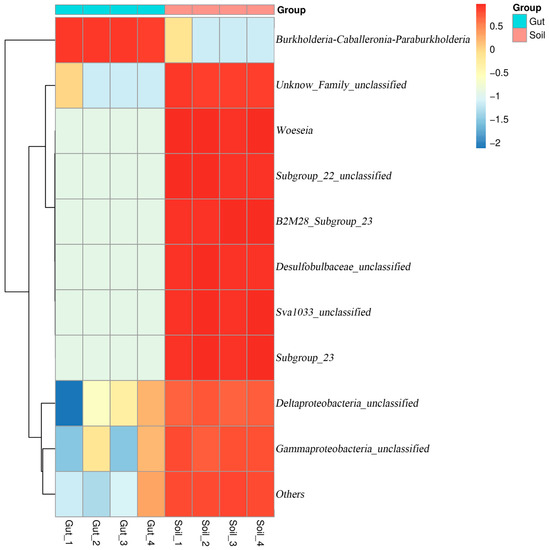
Figure 5.
Heatmap with hierarchal clustering showing differences in the relative abundance of bacterial genera among different groups.
4. Discussion
Gut microbes contribute to the health and metabolism of hosts. The development of next-generation sequencing has enabled a comprehensive identification of taxa in the microbial community. The variation in the gut microbes is induced by a host (i.e., host diet, age and behavior) and external environmental factors. In this study, we used high-throughput sequencing to explore the bacterial community composition and diversity in the gut of N. japonica, and compared it with the bacterial community in soil, subsequently discussing the relationship between them.
In the gut of N. japonica, we found that the dominant phyla including Proteobacteria, Bacteroidota and Firmicutes also constituted the dominant bacterial phyla in the gut of some crustaceans, for example, the black tiger shrimp [17], the Chinese mitten crab [18] and Penaeus vannamei Boone [19]. However, the proportion of the dominant phyla in the gut of N. japonica differed from that in other animals, manifested as the decreased proportions of Bacteroidota and Firmicutes. The three dominant phyla can promote the health and development of the host. Proteobacteria were the most abundant phylum identified in the gut of N. japonica, with an absolutely high relative abundance. This result was in line with previous studies that Proteobacteria were the most abundant phylum in the gut of earthworms, the domesticated silkworm and the black tiger shrimp [20,21,22]. The dominance of Proteobacteria may be explained by their stronger nutrition acquisition capacity compared to other phyla [8]. Proteobacteria are often facultatively or obligately anaerobic and thus can adapt to a range of oxic conditions. Therefore, Proteobacteria contribute towards maintaining the homeostasis of the anaerobic environment of the gut [23]. Firmicutes have been shown to be beneficial to carbon metabolism [7]. Bacteroidota have been found to promote digestion. They can help the host degrade high molecular weight organic matter and plant cell wall compounds [24]. Members of the phylum Bacteroidota were not identified in all gut samples and their proportion in the bacterial community was low. According to the previous study, a low relative abundance of Bacteroidota was due to diet or biopolymer degradation functions of other gut microbes [24]. Therefore, Bacteroidota are not likely commensal inhabitants to N. japonica and their presence in the gut of N. japonica may be due to dietary reasons.
In previous studies, in the gut of some animals, such as the black tiger shrimp, Chinese shrimp and sea cucumbers, Vibrio were the most commonly found bacterial genus [20,25,26]. Although Vibrio are commonly considered a pathogen for aquatic animals, some avirulent strains have been used as probiotics, increasing disease resistance in the host [17,26]. In addition, Zhang et al. used PCR-KGGE technology to analyze the gut bacterial in Perinereis aibuhitensis, finding that the most abundant genera were Propionigenium, followed by Pseudoalteromonas, which can maintain the normal physiological functions of the host [27]. However, in the present study, except for the unclassified genera, the bacterial community in the gut of N. japonica was dominated by Burkholderia-Caballeronia-Paraburkholderia, which was different from previous studies. Burkholderia-Caballeronia-Paraburkholderia belong to Proteobacteria, and have been found in the gut of triatomines [28]. They may be a kind of biological probiotic [29].
Numerous studies have been carried out to understand the correlation of microorganisms in the gut of animals with those from the associated environment. It has been proposed that some of the bacteria identified in the gut of fish came from water or sediment [30]. In addition, the gut microbial community structure in shrimp also changes in response to the variation in the microorganisms in the external environment, showing a close link between bacterial communities in the gut of shrimp and those in its living environment [19,31]. However, it has also been shown that the bacterial community diversity and composition in the half-smooth tongue sole gut were distinct from that in the surrounding environment [32]. In the present study, our results showed that the bacterial community diversity and structure in the gut of N. japonica differed distinctly from those in soil, which reflected that bacterial communities in the N. japonica gut might be independent. Changes in environments caused a considerable variation in bacterial community diversity. The diversity of bacterial communities in the gut of N. japonica was significantly lower than that in soil, supported by the Shannon and Chao1 indices. The decrease in bacterial diversity from the external environment to the gut was consistent with previous studies, which showed that the diversity of bacterial communities in the gut of mud crab [33], Penaeus vannamei Boone [19] and half-smooth tongue sole [32] was lower than that in the environment they inhabit. The dominant bacterial taxa differed and the relative abundance of the same taxa also varied in the two environments. The dominant phyla identified in both the gut of N. japonica and soil were only Proteobacteria, Bacteroidota and Planctomycetota. The relative abundance of Proteobacteria was considerably higher in the gut of N. japonica than that of soil, whereas there were no significant differences in the relative abundance of Bacteroidota and Planctomycetota in the two environments. At the genus level, the relative abundance of Desulfobulbus increased considerably in soil (Figure S2). Desulfobulbus are sulfate-reducing bacteria, which can drive the reduction reactions in the sulfur cycling in soil ecosystems [34]. The relative abundance of Delftia increased considerably in the gut of N. japonica (Figure S2). They are strictly aerobic and chemo-organotrophic with the characteristic of degrading organic matter. It was reported that they might increase the chance of disease in a host with a weakened resistance [35]. We did not obtain the reasons for the differences between bacterial communities in the gut and soil, thus it can be investigated in the future.
5. Conclusions
In this study, we identified the gut bacterial community in N. japonica and found a small similarity between the bacterial communities in the gut of N. japonica and soil. The bacterial diversity and richness in soil were significantly higher, which was consistent with previous studies. Bacterial taxa also differed significantly in the two environments. In the gut of N. japonica, we found that Proteobacteria absolutely dominated the bacterial community. At the genus level, the potential pathogen Delftia was identified in the gut. The reasons for the differences in bacterial communities in soil and the gut of N. japonica need to be further explored.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/d14070514/s1, Figure S1: The boxplot of bacterial taxa with significant difference at the phylum level; Figure S2: The boxplot of bacterial taxa with significant difference at the genus level; Table S1: Alpha diversity indices.
Author Contributions
Writing—original draft preparation, S.S.; writing—review and editing, L.L., X.L. and J.W.; funding acquisition, X.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of Shandong Province (ZR2021QDO82), the NSFC-Shandong Joint Found (No. U1806213), and the National Innovation and Entrepreneurship Training Program for College Students (202110449141).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw sequence data were deposited in the Sequence Read Archive (SRA) database of NCBI under accession number (SRR18906812-18906819), BioProject number (PRJNA830633), and Biosample number (SAMN27734950-27734957).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lee, W.-J.; Hase, K. Gut microbiota–generated metabolites in animal health and disease. Nat. Chem. Biol. 2014, 10, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Ajuwon, K.M.; Fang, R. Mechanistic insight into the gut microbiome and its interaction with host immunity and inflammation. Anim. Nutr. 2020, 6, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Wang, K.; Wu, J.; Qiuqian, L.; Yang, K.; Qian, Y.; Zhang, D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 2015, 99, 6911–6919. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Liu, B.; Wu, H.; Feng, J.; Jiang, T. Seasonal dietary shifts alter the gut microbiota of avivorous bats: Implication for adaptation to energy harvest and nutritional utilization. mSphere 2021, 6, e0046721. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Lu, T.; Yu, Y.; Penuelas, J.; Zhu, Y.-G.; Qian, H. Gammaproteobacteria, a core taxon in the guts of soil fauna, are potential responders to environmental concentrations of soil pollutants. Microbiome 2021, 9, 196. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Zhou, Z.; He, S.; Cao, Y.; Shi, P.; Yao, B.; Ringø, E. Analysis of bacterial diversity in the intestine of grass carp (Ctenopharyngodon idellus) based on 16S rDNA gene sequences. Aquac. Res. 2010, 42, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Gong, L.; Khan, T.A.; Pan, L.; Yan, L.; Li, D.; Cao, L.; Li, Y.; Ding, X.; Yi, G.; et al. Comparative analysis and gut bacterial community assemblages of grass carp and crucian carp in new lineages from the Dongting Lake area. MicrobiologyOpen 2020, 9, e996. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, W.; Jiang, Y.; Dai, W.; Li, P.; Yao, D.; Wang, J.; Shi, Y.; Cui, Z.; Cao, H.; et al. Variations in bacterial taxonomic profiles and potential functions in response to the gut transit of earthworms (Eisenia fetida) feeding on cow manure. Sci. Total Environ. 2021, 787, 147392. [Google Scholar] [CrossRef]
- Liu, Y.; Xian, W. The effect of temperature on growth and energy budget of the polychaete, Neanthes japonica Izuka. J. Ocean Univ. China 2009, 8, 177–183. [Google Scholar] [CrossRef]
- Ye, J. Biological Characteristics of Neanthes japonica and their application in shrimp aquaculture. J. Anhui Agric. Sci. 2010, 48, 7883–7938. [Google Scholar]
- Wang, S.; Deng, Z.; Li, Q.; Ge, X.; Bo, Q.; Liu, J.; Cui, J.; Jiang, X.; Liu, J.; Zhang, L.; et al. A novel alkaline serine protease with fibrinolytic activity from the polychaete, Neanthes japonica. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2011, 159, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, S.; Li, Q.; Ji, X.; Zhang, L.; Hong, M. Purification and characterization of a novel fibrinolytic enzyme from the polychaete, Neanthes japonica (Iznka). Bioresour. Technol. 2010, 101, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.B.; Stedmon, C.; Kellerman, A.M.; Nielsen, N.J.; Andersson, A.F.; Laudon, H.; Lindström, E.; Kritzberg, E.S. Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. ISME J. 2016, 10, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler dia-grams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Wagner, H.H. Vegan Community Ecology Package Version 2.5-7 November; R. Foundation: Vienna, Austria, 2020. [Google Scholar]
- Rungrassamee, W.; Klanchui, A.; Chaiyapechara, S.; Maibunkaew, S.; Tangphatsornruang, S.; Jiravanichpaisal, P.; Karoonuthaisiri, N. Bacterial Population in Intestines of the Black Tiger Shrimp (Penaeus monodon) under Different Growth Stages. PLoS ONE 2013, 8, e60802. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Guan, W.; Wei, G.; Liu, B.; Xu, J.; Zhao, L.; Zhang, Y. Phylogenetic analysis of intestinal bacteria in the Chinese mitten crab (Eriocheir sinensis). J. Appl. Microbiol. 2007, 103, 675–682. [Google Scholar] [CrossRef]
- Sun, Z.; Xuan, Y.; Zhang, H.; Jiang, M.; Pan, Y.; Zhang, Y.; Gong, Y.; Lu, X.; Yu, D.; Xue, R.; et al. Bacterial diversity in the Penaeus vannamei Boone intestine and aquaculture environment. J. Fish. Sci. China 2016, 23, 594–605. [Google Scholar]
- Chaiyapechara, S.; Rungrassamee, W.; Suriyachay, I.; Kuncharin, Y.; Klanchui, A.; Karoonuthaisiri, N.; Jiravanichpaisal, P. Bacterial Community Associated with the Intestinal Tract of P. monodon in Commercial Farms. Microb. Ecol. 2012, 63, 938–953. [Google Scholar] [CrossRef]
- Liu, D.; Lian, B.; Wu, C.; Guo, P. A comparative study of gut microbiota profiles of earthworms fed in three different substrates. Symbiosis 2017, 74, 21–29. [Google Scholar] [CrossRef]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, C.D.; Young, W.; Maclean, P.H.; Cookson, A.L.; Bermingham, E.N. Metagenomic insights into the roles of Proteobacteria in the gastrointestinal microbiomes of healthy dogs and cats. Microbiologyopen 2018, 7, e00677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xu, D.; Zhu, J.; Wang, S.; Liu, M.; Sun, M.; Wang, G.; Song, L.; Liu, X.; Xie, T. Habitat environmental factors influence intestinal microbial diversity of the short-faced moles (Scaptochirus moschata). AMB Express 2021, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, L.; Liu, M.; Wang, B.; Jiang, K.; Ma, S.; Li, Q. The intestinal microbial diversity in Chinese shrimp (Fenneropenaeus chinensis) as determined by PCR–DGGE and clone library analyses. Aquaculture 2011, 317, 32–36. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Y.; Wu, P.; Chen, M.; He, L.; Xu, Q.; Wang, A. Bacterial community composition in gut content and ambient sediment of two tropical wild sea cucumbers (Holothuria atra and H. leucospilota). J. Oceanol. Limnol. 2021, 40, 360–372. [Google Scholar] [CrossRef]
- Zhang, B.; Lian, B.; Wang, B.; Zhou, Y.; He, J. PCR-DGGE analysis of immobilized microbial diversity in digestive tract of sand worm Perinereis aibuhitensis. J. Dalian Ocean. Univ. 2013, 28, 413–417. [Google Scholar]
- Hu, Y.; Xie, H.; Gao, M.; Huang, P.; Zhou, H.; Ma, Y.; Zhou, M.; Liang, J.; Yang, J.; Lv, Z. Dynamic of Composition and Diversity of Gut Microbiota in Triatoma rubrofasciata in Different Developmental Stages and Environmental Conditions. Front. Cell. Infect. Microbiol. 2020, 10, 587708. [Google Scholar] [CrossRef]
- Zla, B.; Sp, A.; Zhe, Z.A.; Xw, A.; Jla, B.; Yha, B.; Wza, B.; Qla, B.; Pba, B.; Msa, B. Novel pathway of acephate degra-dation by the microbial consortium ZQ01 and its potential for environmental bioremediation. J. Hazard. Mater. 2021, 426, 127841. [Google Scholar]
- Ge, L. Advances in the study of bacteria in intestines of fishes. Reserv. Fish. 2006, 26, 17–20. [Google Scholar]
- Jin, R.; Jiang, M.; Sun, S.; Dai, X.; Wu, H.; Zhou, J.; Yu, Z.; Zhang, F. Microbial community in Litopenaeus vannamei intestine and its aquaculture environment. J. Fish. China 2020, 44, 2037–2054. [Google Scholar]
- Zhang, Z.; Li, B.; Wang, Y.; Liao, M.; Wang, L.; Rong, X. The microflora structure in digestive tract of half-smooth tongue sole (Cynoglossus semilaevis Gunther) cultured in outdoor pond basing on high-through sequencing technique. Acta Hydrobiol. Sin. 2015, 39, 38–45. [Google Scholar]
- Wang, X.; Zhao, Y.; Song, Z.; Zhong, S.; Huang, G.; Tong, T.; Nie, Z.; Su, Q.; Yang, J. Application of high-throughput sequencing techniques for analyzing bacterial communities in pond-raised mud crab (Scylla paramamosain) intestine and its aquaculture environment. J. Fish. Sci. China 2017, 24, 1245. [Google Scholar] [CrossRef]
- Fang, A.Q.; He, Z.L.; Wang, C.; Yang, C.; Yan, Q.Y. Progress in studying microbially-driven sulfur cycling in man-grove sediments. Acta Microbiol. Sin. 2020, 60, 13–25. [Google Scholar]
- Li, Z.; Liu, L. Advance of study in Delftia. China Trop. Med. 2008, 18, 2254–2255. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).