

Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals


Abstract
:1. Introduction
2. Materials and Methods
2.1. K. pneumoniae Isolates and Cultural Conditions
2.2. Antibiotic Susceptibility and Heavy Metal Tolerance Assays
2.3. Growth Curve Assay
2.4. Polymerase Chain Reaction (PCR) Assay
2.5. Genome Sequencing, Assembly, and Annotation
2.6. Comparative Genome Analysis
2.7. Statistical Analysis
3. Results
3.1. Phenotypes and Genotypes of the K. pneumoniae Isolates
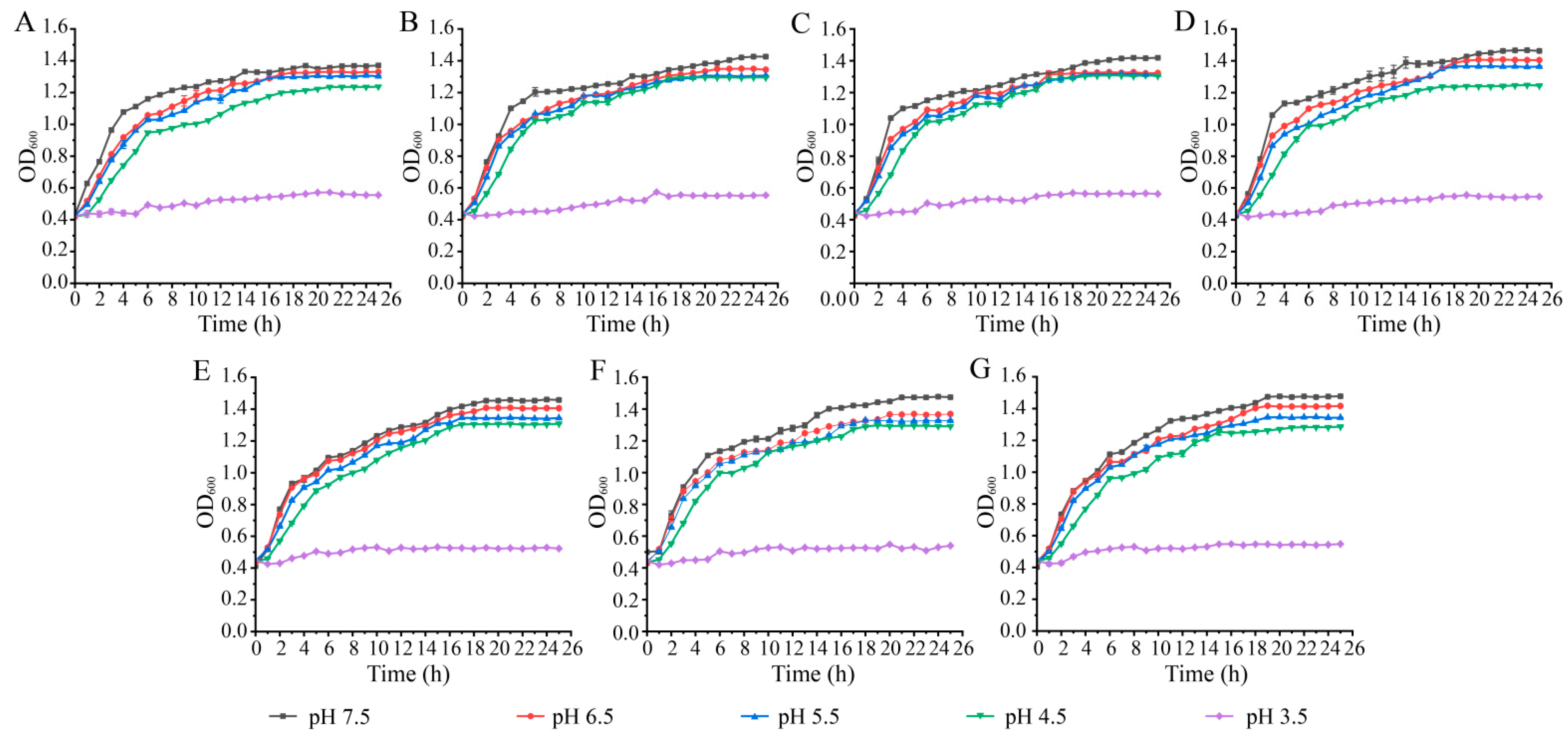
3.2. Survival of the K. pneumoniae Isolates at Different pH and NaCl Conditions
3.3. Genome Features of the K. pneumoniae Isolates of Aquatic Animal Origins
3.4. MGEs in the K. pneumoniae Genomes of Aquatic Animal Origins
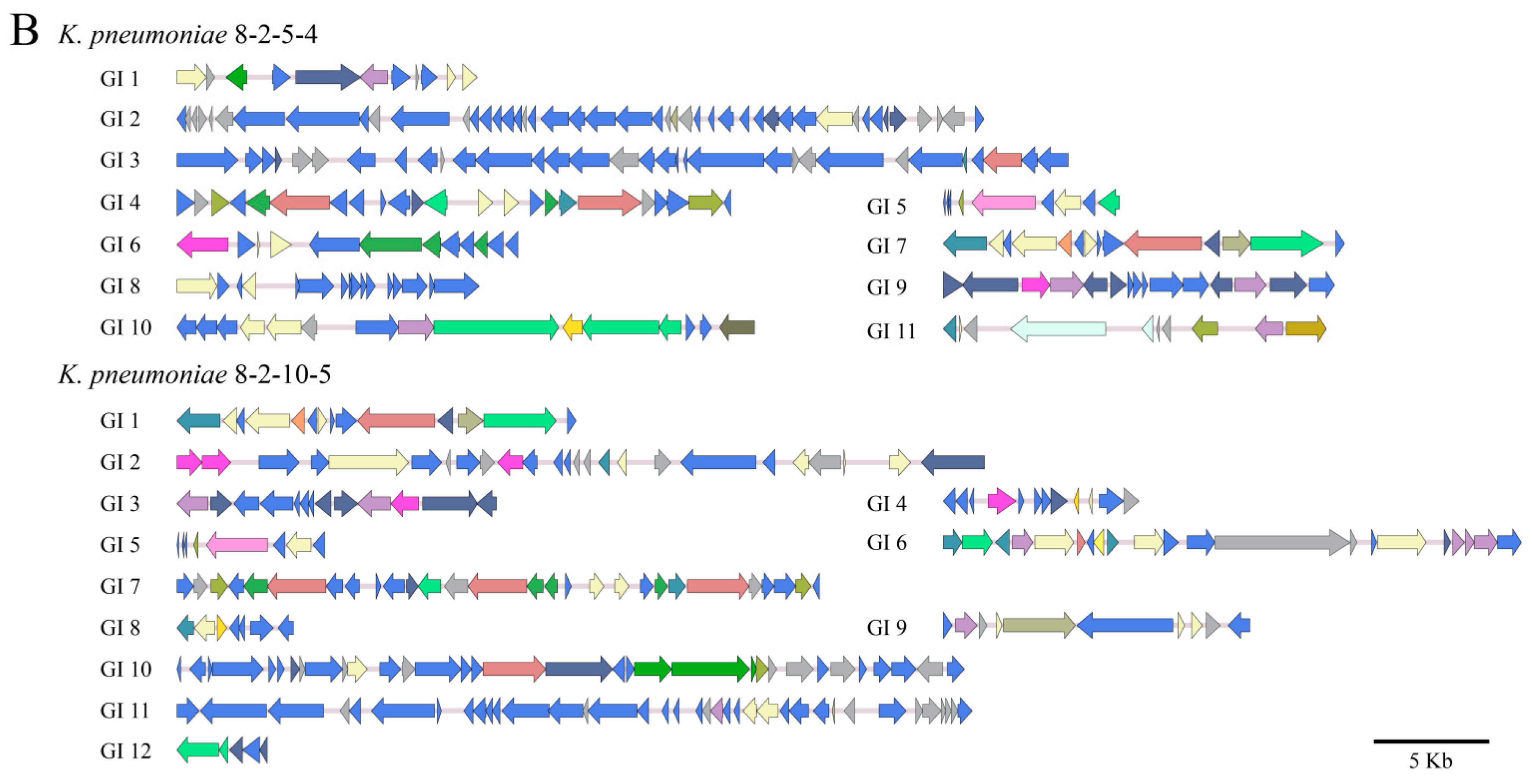
3.4.1. GIs
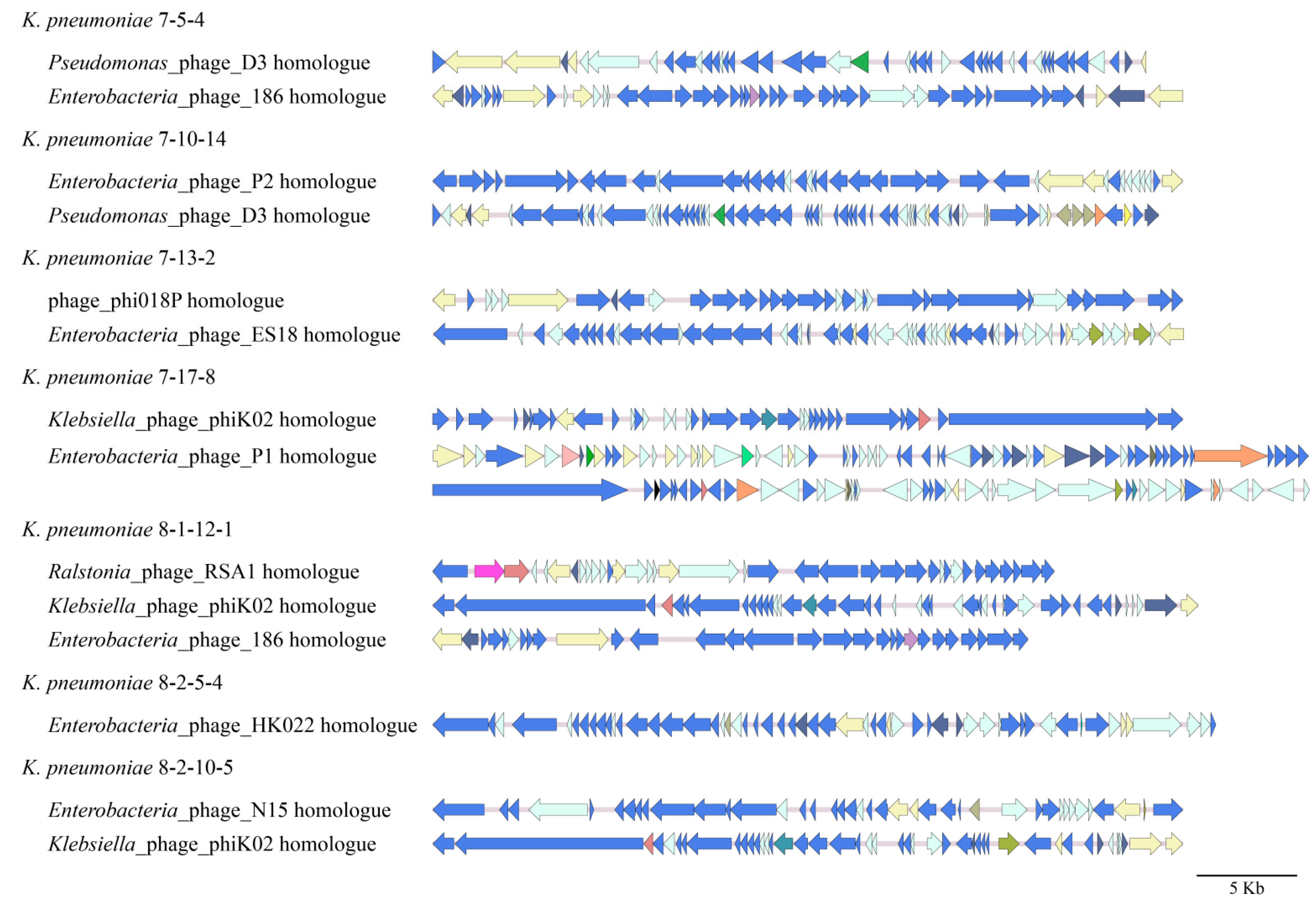
3.4.2. Prophages
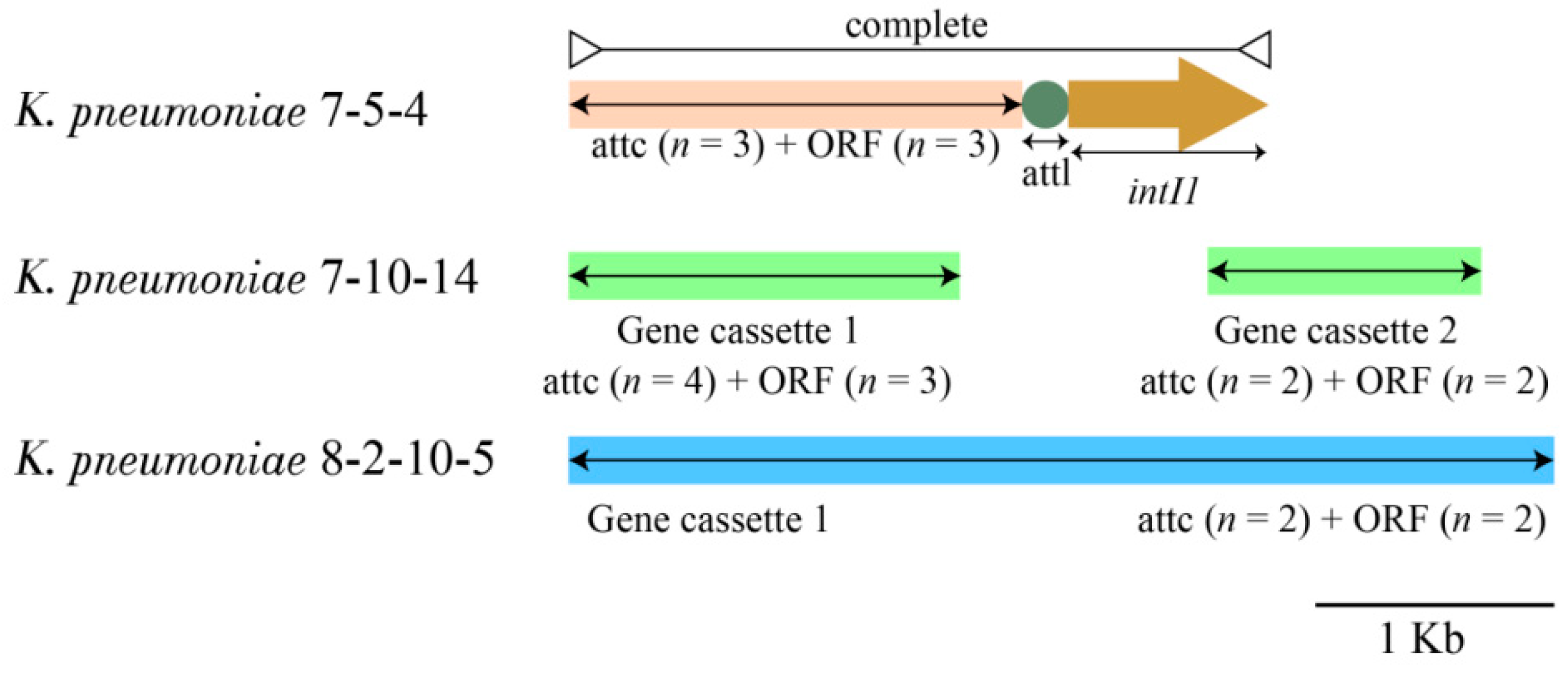
3.4.3. INs
3.4.4. ISs
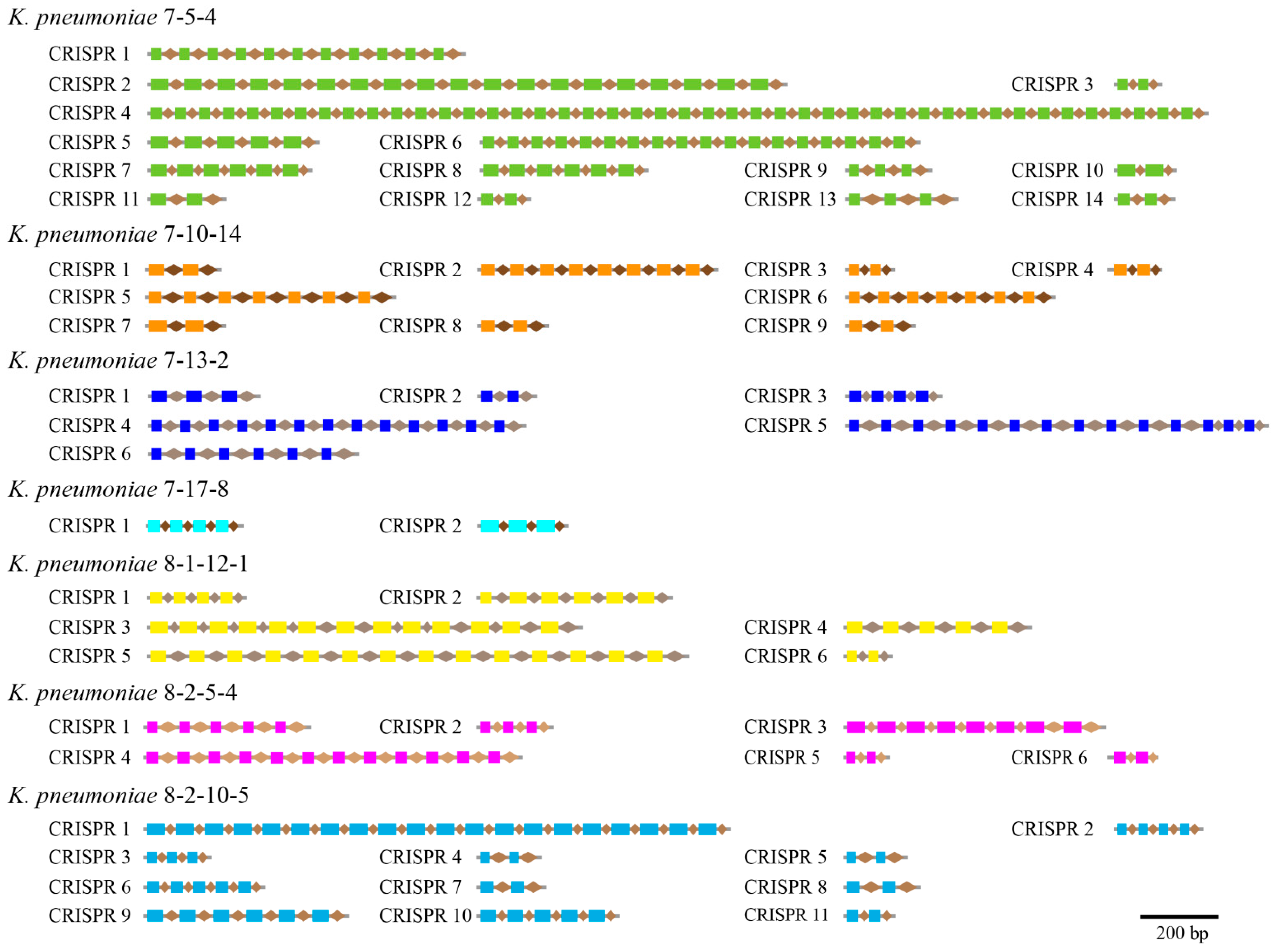
3.5. CRISPR-Cas Repeats
3.6. Putative Virulence-Associated Genes in the K. pneumoniae Genomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virulence-Related Gene | K. pneumoniae Genome | Reference |
|---|---|---|
| ecpABCDER | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [44] |
| entABCDEFS | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [45] |
| fepABCDG | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [46] |
| fes | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [46] |
| gnd | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [47] |
| kdsA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [48] |
| rcsAB | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [45] |
| tuf | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [49] |
| wza | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [50] |
| galU | 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [51] |
| hcp | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4 | [52] |
| impBCGHJKL | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4 | [52,53] |
| vasDG | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4 | [53] |
| yhjH | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [54] |
| fimABCDEFGI | 7-5-4, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [55] |
| fimH | 7-5-4, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [43] |
| manBC | 7-5-4, 7-10-14, 7-17-8, 8-1-12-1, 8-2-5-4 | [56] |
| vgrG | 7-5-4, 7-10-14, 7-13-2, 8-1-12-1, 8-2-5-4 | [57] |
| iroE | 7-13-2, 7-17-8, 8-2-5-4, 8-2-10-5 | [46] |
| glf | 7-10-14, 7-13-2, 8-2-5-4 | [58] |
| vasJ | 7-10-14, 8-2-5-4 | [59] |
| fyuA | 7-17-8, 8-1-12-1 | [60] |
| irp12345 | 7-17-8, 8-1-12-1 | [55,61] |
| mbtI | 7-17-8, 8-1-12-1 | [62] |
| ybtAX | 7-17-8, 8-1-12-1 | [63,64] |
| allABCDRS | 8-2-10-5 | [65] |
| iucABCD | 8-2-5-4 | [45] |
3.7. Antibiotic and Heavy Metal Resistance-Associated Genes in the K. pneumoniae Genomes
| Antibiotic/Heavy Metal | Gene | K. pneumoniae Genome | Reference |
|---|---|---|---|
| Cephalosporin | acrB | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [69] |
| Fluoroquinolone | hns | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [70] |
| emrR | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [71] | |
| marA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [72] | |
| ramA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [72] | |
| crp | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [73] | |
| qnrS1 | 7-5-4, 8-2-10-5 | [74] | |
| qnrB2 | 7-10-14, 8-2-10-5 | [75] | |
| Tetracycline | oqxAB | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [44] |
| tet(A) | 7-5-4, 7-10-14, 8-2-10-5, 8-2-5-4 | [44] | |
| Aminoglycoside | KpnEFGH | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [76,77] |
| ompK37 | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [78] | |
| aph(3′)-Ia | 7-5-4, 7-10-14, 8-2-10-5 | [79] | |
| aph(3″)-Ib | 7-5-4, 7-10-14, 8-2-10-5 | [79] | |
| aph(6)-Id | 7-5-4, 7-10-14, 8-2-10-5 | [79] | |
| aac(3)-IId | 7-10-14, 8-2-10-5 | [79] | |
| aac(6′)-Ib-cr | 7-10-14, 8-2-10-5 | [80] | |
| aadA16 | 7-10-14, 8-2-10-5 | [79] | |
| aadA8 | 7-5-4 | [81] | |
| Diaminopyrimidine | dfrA27 | 7-10-14, 8-2-10-5 | [82] |
| Beta-lactam | SHV-11 | 7-5-4, 7-17-8 | [83] |
| SHV-1 | 7-10-14, 7-13-2 | [84] | |
| SHV-38 | 8-1-12-1, 8-2-5-4 | [85] | |
| TEM-1 | 7-5-4 | [85] | |
| OKP-B-7 | 8-2-10-5 | [86] | |
| TEM-116 | 8-2-5-4 | [85] | |
| Macrolide | mphA | 7-5-4, 7-10-14, 8-2-10-5 | [44] |
| ermB | 7-5-4 | [44] | |
| mphE | 7-5-4 | [87] | |
| msrE | 7-5-4 | [87] | |
| Nitroimidazole | msbA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [88] |
| Phenicol | floR | 7-5-4, 7-10-14, 8-2-10-5 | [89] |
| Peptide | pmrF | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [90] |
| ugd | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [91] | |
| yojI | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [92] | |
| Sulfonamide | sul1 | 7-5-4, 7-10-14, 8-2-10-5 | [44] |
| sul2 | 7-5-4, 8-2-10-5 | [44] | |
| Rifamycin | arr-2 | 7-5-4 | [80] |
| arr-3 | 7-10-14 | [46] | |
| Fosfomycin | FosA5 | 7-5-4, 7-10-14 | [80] |
| FosA6 | 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [80] | |
| mdtG | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [93] | |
| Heavy metal | cusARS | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [66,67] |
| Heavy metal | copA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [66] |
| Heavy metal | zntA | 7-10-14 | [94] |
| Heavy metal | arsABCR | 7-13-2 | [68] |
3.8. Strain-Specific Genes of the K. pneumoniae Isolates of Aquatic Animal Origins
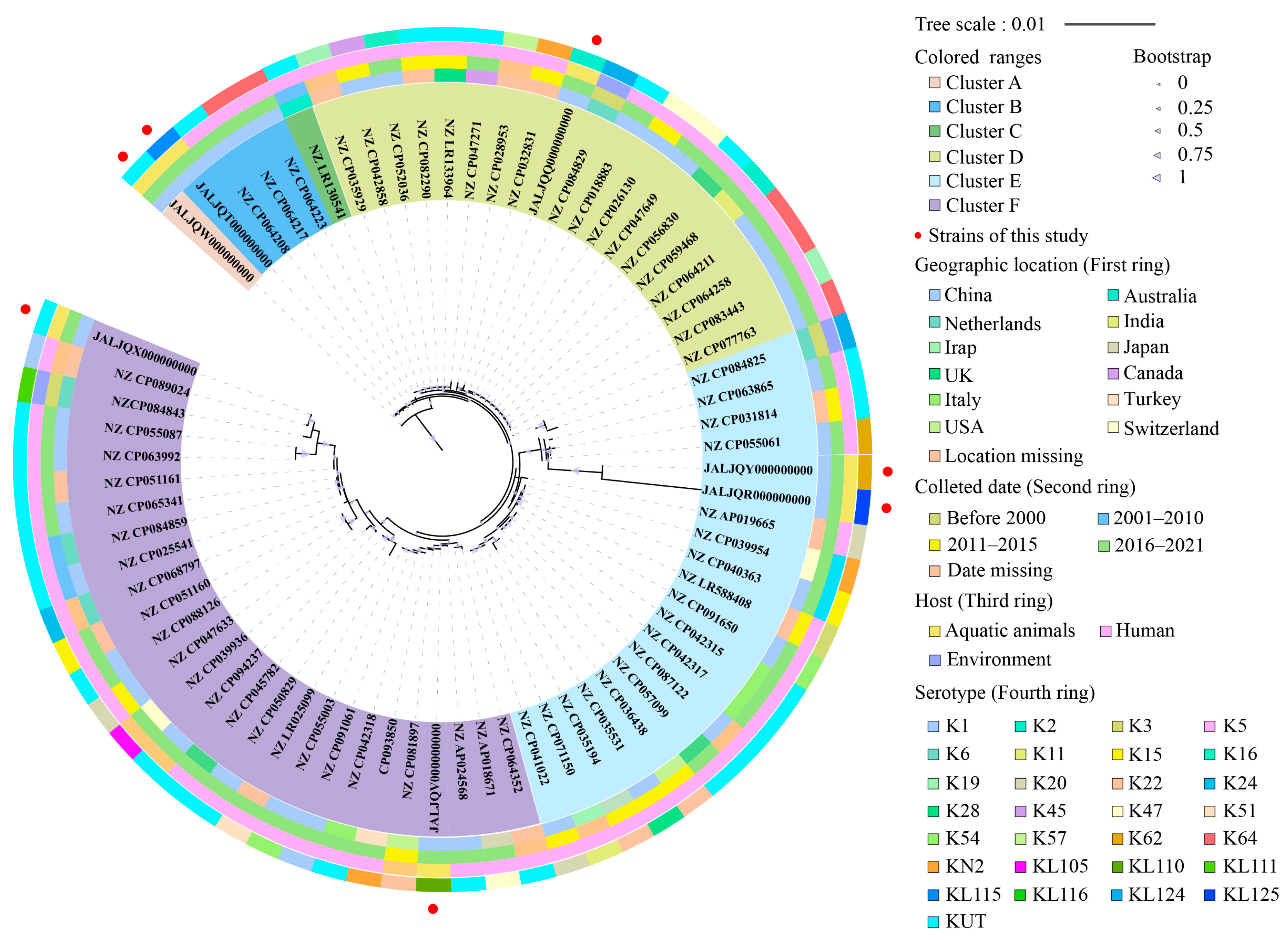
3.9. Phylogenetic Relatedness of the K. pneumoniae Isolates of Aquatic Animal Origins
3.10. Sequence Types (ST) of the K. pneumoniae Isolates of Aquatic Animal Origins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, R.M.; Bachman, M.A. Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Front. Cell Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girometti, N.; Lewis, R.E.; Giannella, M.; Ambretti, S.; Bartoletti, M.; Tedeschi, S.; Tumietto, F.; Cristini, F.; Trapani, F.; Gaibani, P.; et al. Klebsiella pneumoniae bloodstream infection: Epidemiology and impact of inappropriate empirical therapy. Medicine 2014, 93, 298–309. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arato, V.; Raso, M.M.; Gasperini, G.; Berlanda Scorza, F.; Micoli, F. Prophylaxis and treatment against Klebsiella pneumoniae: Current insights on this emerging anti-microbial resistant global threat. Int. J. Mol. Sci. 2021, 22, 4042. [Google Scholar] [CrossRef]
- Turton, J.F.; Perry, C.; Elgohari, S.; Hampton, C.V. PCR characterization and typing of Klebsiella pneumoniae using capsular type-specific, variable number tandem repeat and virulence gene targets. J. Med. Microbiol. 2010, 59, 541–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhao, Y.; Liu, C.; Chen, Z.; Zhou, D. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol. 2014, 9, 1071–1081. [Google Scholar] [CrossRef]
- Qin, X.; Wu, S.; Hao, M.; Zhu, J.; Ding, B.; Yang, Y. The colonization of carbapenem-resistant Klebsiella pneumoniae: Epidemiology, resistance mechanisms, and risk factors in patients admitted to intensive care units in China. J. Infect. Dis. 2020, 221, S206–S214. [Google Scholar] [CrossRef]
- Liu, P.; Li, X.; Luo, M.; Xu, X.; Su, K.; Chen, S. Risk factors for carbapenem-resistant Klebsiella pneumoniae infection: A meta-analysis. Microb. Drug Resist. 2018, 24, 190–198. [Google Scholar] [CrossRef]
- Galler, H.; Feierl, G.; Petternel, C.; Reinthaler, F.F.; Haas, D.; Grisold, A.J. KPC-2 and OXA-48 carbapenemase-harbouring Enterobacteriaceae detected in an Austrian wastewater treatment plant. Clin. Microbiol. Infect. 2014, 20, O132–O134. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.; Moura, R.A.; Silva, K.C.; Pavez, M.; McCulloch, J.A.; Dropa, M. Isolation of KPC-2-producing Klebsiella pneumoniae strains belonging to the high-risk multiresistant clonal complex 11 (ST437 and ST340) in urban rivers. J. Antimicrob. Chemother. 2014, 69, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Jelić, M.; Hrenović, J.; Dekić, S.; Goić-Barišić, I.; Tambić Andrašević, A. First evidence of KPC-producing ST258 Klebsiella pneumoniae in river water. J. Hosp. Infect. 2019, 103, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, S.; Rosado, D.; Moreno-Andrés, J.; Cartuche, L.; Cruz, D.; Acevedo-Merino, A. Inactivation of a wild isolated Klebsiella pneumoniae by photo-chemical processes: UV-C, UV-C/H2O2 and UV-C/H2O2/Fe3+. Catal. Today. 2018, 313, 94–99. [Google Scholar] [CrossRef]
- Chen, J.; Sun, R.; Pan, C.; Sun, Y.; Mai, B.; Li, Q.X. Antibiotics and food safety in aquaculture. J. Agric. Food Chem. 2020, 68, 11908–11919. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lu, S.; Huang, C.; Wang, F.; Ren, Y.; Cao, H. A survey of chloramphenicol residues in aquatic products of Shenzhen, South China. Food Addit. Contam. Part A 2021, 38, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ni, L.; Guan, H.; Chen, D.; Qin, S.; Chen, L. First report of potentially pathogenic Klebsiella pneumoniae from serotype K2 in Mollusk Tegillarca granosa and genetic diversity of Klebsiella pneumoniae in 14 species of edible aquatic animals. Foods 2022, 11, 4058. [Google Scholar] [CrossRef]
- Pal, A.; Bhattacharjee, S.; Saha, J.; Sarkar, M.; Mandal, P. Bacterial survival strategies and responses under heavy metal stress: A comprehensive overview. Crit. Rev. Microbiol. 2022, 48, 327–355. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, T.; Yang, Y.; Yu, S.; Wu, J.; Lin, R.; Li, Y.; Fang, J.; Zhu, C. Co-occurrence of antibiotic and heavy metal resistance and sequence type diversity of Vibrio parahaemolyticus isolated from Penaeus vannamei at freshwater farms, feawater farms, and markets in Zhejiang Province, China. Front. Microbiol. 2020, 11, 1294. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic mechanisms of five heavy metals: Mercury, lead, chromium, cadmium, and arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Dickinson, A.W.; Power, A.; Hansen, M.G.; Brandt, K.K.; Piliposian, G.; Appleby, P. Heavy metal pollution and co-selection for antibiotic resistance: A microbial palaeontology approach. Environ. Int. 2019, 132, 105117. [Google Scholar] [CrossRef]
- Kunhikannan, S.; Thomas, C.J.; Franks, A.E.; Mahadevaiah, S.; Kumar, S.; Petrovski, S. Environmental hotspots for antibiotic resistance genes. Microbiologyopen 2021, 10, e1197. [Google Scholar] [CrossRef]
- Xu, D.; Peng, X.; Xie, L.; Chen, L. Survival and genome diversity of Vibrio parahaemolyticus isolated from edible aquatic animals. Diversity 2022, 14, 350. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Ni, L.; Xu, D.; Xu, Y.; Ding, Y.; Xie, L.; Chen, L. First experimental evidence for the presence of potentially toxic Vibrio cholerae in snails, and virulence, cross-resistance and genetic diversity of the bacterium in 36 species of aquatic food animals. Antibiotics 2021, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, T.; Peng, X.; Chen, L. Insights into Vibrio parahaemolyticus CHN25 response to artificial gastric fluid stress by transcriptomic analysis. Int. J. Mol. Sci. 2014, 15, 22539–22562. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Yu, P.; Ren, S.; Zhu, Z.; Jin, Y.; Yan, J.; Peng, X.; Chen, L. Prophage-related gene VpaChn25_0724 contributes to cell membrane integrity and growth of Vibrio parahaemolyticus CHN25. Front. Cell. Infect. Microbiol. 2020, 10, 595709. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Cury, J.; Jové, T.; Touchon, M.; Néron, B.; Rocha, E.P. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016, 44, 4539–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.Y.; Liu, C.X.; Liu, P.; Wu, Z.Y.; Zhang, Y.D.; Xiong, X.S. Regulation of gene expression of hcp, a core gene of the type VI secretion system in Acinetobacter baumannii causing respiratory tract infection. J. Med. Microbiol. 2018, 67, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, Y.; Ji, X. Research progress of prophages. Yi Chuan 2021, 43, 240–248. [Google Scholar] [CrossRef]
- Li, Y.; Yang, L.; Fu, J.; Yan, M.; Chen, D.; Zhang, L. Genotyping and high flux sequencing of the bacterial pathogenic elements-integrons. Microb. Pathog. 2018, 116, 22–25. [Google Scholar] [CrossRef]
- Huyan, J.; Tian, Z.; Zhang, Y.; Zhang, H.; Shi, Y.; Gillings, M.R. Dynamics of class 1 integrons in aerobic biofilm reactors spiked with antibiotics. Environ. Int. 2020, 140, 105816. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Jiang, S.; Wang, Y.; Tian, X.; Zhao, P.; Xu, J. CRISPR-Cas adaptive immune systems in Sulfolobales: Genetic studies and molecular mechanisms. Sci. China Life Sci. 2021, 64, 678–696. [Google Scholar] [CrossRef]
- Wang, G.; Song, G.; Xu, Y. Association of CRISPR/Cas system with the drug resistance in Klebsiella pneumoniae. Infect. Drug Resist. 2020, 13, 1929–1935. [Google Scholar] [CrossRef]
- Kamruzzaman, M.; Iredell, J.R. CRISPR-Cas system in antibiotic resistance plasmids in Klebsiella pneumoniae. Front. Microbiol. 2020, 10, 2934. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, J.; Liu, P.; Li, X.; Su, K.; Yuan, L. Genome sequencing and comparative genome analysis of 6 hypervirulent Klebsiella pneumoniae strains isolated in China. Arch. Microbiol. 2021, 203, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Sarshar, M.; Behzadi, P.; Ambrosi, C.; Zagaglia, C.; Palamara, A.T.; Scribano, D. FimH and anti-adhesive therapeutics: A disarming strategy against uropathogens. Antibiotics 2020, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Kim, Y.; Han, D.; Hur, H.G. Emergence of high level carbapenem and extensively drug resistant Escherichia coli ST746 producing NDM-5 in influent of wastewater treatment plant, seoul, South Korea. Front. Microbiol. 2021, 12, 645411. [Google Scholar] [CrossRef]
- Yuan, L.; Li, X.; Du, L.; Su, K.; Zhang, J.; Liu, P. RcsAB and fur coregulate the iron-acquisition system via entC in Klebsiella pneumoniae NTUH-K2044 in response to iron availability. Front. Cell Infect. Microbiol. 2020, 10, 282. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L.; Hu, X.; Liang, X.; Gong, G.; Xie, C. Co-occurrence of Klebsiella variicola and Klebsiella pneumoniae both carrying bla (KPC) from a respiratory intensive care unit patient. Infect. Drug Resist. 2021, 14, 4503–4510. [Google Scholar] [CrossRef]
- Cookson, A.L.; Biggs, P.J.; Marshall, J.C.; Reynolds, A.; Collis, R.M.; French, N.P. Culture independent analysis using gnd as a target gene to assess Escherichia coli diversity and community structure. Sci. Rep. 2017, 7, 841. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.B.; Wu, S.L.; Zhu, D.P.; Wu, H.; Jiang, T.; Qian, Y.H. Expression of the 2-dehydro-3-deoxyphosphooc-tonate aldolase (KdsA) gene in mulberry leaves (Morus alba L.) is down-regulated under high salt and drought stress. Genet. Mol. Res. 2015, 14, 11955–11964. [Google Scholar] [CrossRef]
- Harvey, K.L.; Jarocki, V.M.; Charles, I.G.; Djordjevic, S.P. The diverse functional roles of elongation factor Tu (EF-Tu) in microbial pathogenesis. Front. Microbiol. 2019, 10, 2351. [Google Scholar] [CrossRef]
- Niu, T.; Guo, L.; Luo, Q.; Zhou, K.; Yu, W.; Chen, Y. Wza gene knockout decreases Acinetobacter baumannii virulence and affects wzy-dependent capsular polysaccharide synthesis. Virulence 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- De Luca, E.; Alvarez-Narvaez, S.; Maboni, G.; Baptista, R.P.; Nemeth, N.M.; Niedringhaus, K.D. Comparative genomics analyses support the reclassification of bisgaard taxon 40 as Mergibacter gen. nov., with Mergibacter septicus sp. nov. as type species: Novel insights into the phylogeny and virulence factors of a pasteurellaceae family member associated with mortality events in seabirds. Front. Microbiol. 2021, 12, 667356. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, L.; Huang, L.; Qin, Y.; Zhang, J.; Zhang, J. Integration of RNA-seq and RNAi provides a novel insight into the immune responses of Epinephelus coioides to the impB gene of Pseudomonas plecoglossicida. Fish Shellfish. Immunol. 2020, 105, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Pira, H.; Risdian, C.; Musken, M.; Schupp, P.J.; Wink, J. Photobacterium arenosum WH24, isolated from the gill of pacific oyster Crassostrea gigas from the north sea of germany: Co-cultivation and prediction of virulence. Curr. Microbiol. 2022, 79, 219. [Google Scholar] [CrossRef] [PubMed]
- Claret, L.; Miquel, S.; Vieille, N.; Ryjenkov, D.A.; Gomelsky, M.; Darfeuille-Michaud, A. The flagellar sigma factor fliA regulates adhesion and invasion of crohn disease-associated Escherichia coli via a cyclic dimeric GMP-dependent pathway. J. Biol. Chem. 2007, 282, 33275–33283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, B.F.; Ferreira, M.L.; Campos, P.A.; Royer, S.; Gonçalves, I.R.; da Fonseca Batistão, D.W. Hypervirulence and biofilm production in KPC-2-producing Klebsiella pneumoniae CG258 isolated in Brazil. J. Med. Microbiol. 2018, 67, 523–528. [Google Scholar] [CrossRef]
- Jensen, S.O.; Reeves, P.R. Molecular evolution of the GDP-mannose pathway genes (manB and manC) in Salmonella enterica. Microbiology-SGM 2001, 147 Pt 3, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, Z.; He, F.; Ruan, Z.; Jiang, Y.; Hua, X. The role of the type VI secretion system vgrG gene in the virulence and antimicrobial resistance of Acinetobacter baumannii ATCC 19606. PLoS ONE 2018, 13, e0192288. [Google Scholar] [CrossRef] [Green Version]
- Barnell, W.O.; Liu, J.; Hesman, T.L.; O’Neill, M.C.; Conway, T. The zymomonas mobilis glf, zwf, edd, and glk genes form an operon: Localization of the promoter and identification of a conserved sequence in the regulatory region. J. Bacteriol. 1992, 174, 2816–2823. [Google Scholar] [CrossRef] [Green Version]
- Planamente, S.; Salih, O.; Manoli, E.; Albesa-Jové, D.; Freemont, P.S.; Filloux, A. TssA forms a gp6-like ring attached to the type VI secretion sheath. EMBO J. 2016, 35, 1613–1627. [Google Scholar] [CrossRef]
- Chen, Y.; Song, K.; Chen, X.; Li, Y.; Lv, R.; Zhang, Q. Attenuation of Yersinia pestis fyuA mutants caused by iron uptake inhibition and decreased survivability in macrophages. Front. Cell. Infect. Microbiol. 2022, 12, 874773. [Google Scholar] [CrossRef]
- Magistro, G.; Magistro, C.; Stief, C.G.; Schubert, S. The high-pathogenicity island (HPI) promotes flagellum-mediated motility in extraintestinal pathogenic. Escherichia coli. PLoS ONE 2017, 12, e0183950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneghetti, F.; Villa, S.; Gelain, A.; Barlocco, D.; Chiarelli, L.R.; Pasca, M.R. Iron acquisition pathways as targets for antitubercular drugs. Curr. Med. Chem. 2016, 23, 4009–4026. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Wang, Q.; Perlaza-Jimenez, L.; Zheng, X.; Zhao, Y.; Dhanasekaran, V. First description of antimicrobial resistance in carbapenem-susceptible Klebsiella pneumoniae after imipenem treatment, driven by outer membrane remodeling. BMC Microbiol. 2020, 20, 218. [Google Scholar] [CrossRef] [PubMed]
- Bobrov, A.G.; Kirillina, O.; Fosso, M.Y.; Fetherston, J.D.; Miller, M.C.; VanCleave, T.T. Zinc transporters YbtX and ZnuABC are required for the virulence of Yersinia pestis in bubonic and pneumonic plague in mice. Metallomics. 2017, 9, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Arena, F.; Henrici De Angelis, L.; Pieralli, F.; Di Pilato, V.; Giani, T.; Torricelli, F. Draft genome sequence of the first hypermucoviscous Klebsiella quasipneumoniae subsp. quasipneumoniae isolate from a bloodstream infection. Genome Announc. 2015, 3, e00952-15. [Google Scholar] [CrossRef] [Green Version]
- Besaury, L.; Bodilis, J.; Delgas, F.; Andrade, S.; De la Iglesia, R.; Ouddane, B. Abundance and diversity of copper resistance genes cusA and copA in microbial communities in relation to the impact of copper on Chilean marine sediments. Mar. Pollut. Bull. 2013, 67, 16–25. [Google Scholar] [CrossRef]
- Shafer, C.M.; Tseng, A.; Allard, P.; McEvoy, M.M. Strength of Cu-efflux response in Escherichia coli coordinates metal resistance in Caenorhabditis elegans and contributes to the severity of environmental toxicity. J. Biol. Chem. 2021, 297, 101060. [Google Scholar] [CrossRef]
- Zhen, Z.; Yan, C.; Zhao, Y. Influence of epiphytic bacteria on arsenic metabolism in Hydrilla verticillata. Environ. Pollut. 2020, 261, 114232. [Google Scholar] [CrossRef]
- Bratu, S.; Landman, D.; George, A.; Salvani, J.; Quale, J. Correlation of the expression of acrB and the regulatory genes marA, soxS and ramA with antimicrobial resistance in clinical isolates of Klebsiella pneumoniae endemic to New York City. J. Antimicrob. Chemother. 2009, 64, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Escobar, G.A.; Grepinet, O.; Raymond, P.; Abed, N.; Velge, P.; Virlogeux-Payant, I. H-NS is the major repressor of Salmonella Typhimurium Pef fimbriae expression. Virulence 2019, 10, 849–867. [Google Scholar] [CrossRef] [Green Version]
- Barroso, K.C.M.; Previato-Mello, M.; Batista, B.B.; Batista, J.H.; da Silva Neto, J.F. EmrR-dependent upregulation of the efflux pump emrCAB contributes to antibiotic resistance in Chromobacterium violaceum. Front. Microbiol. 2018, 9, 2756. [Google Scholar] [CrossRef] [PubMed]
- Holden, E.R.; Webber, M.A. MarA, ramA, and soxS as mediators of the stress response: Survival at a cost. Front. Microbiol. 2020, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Aiba, H. Autoregulation of the Escherichia coli crp gene: CRP is a transcriptional repressor for its own gene. Cell 1983, 32, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Monárrez, R.; Wang, Y.; Fu, Y.; Liao, C.H.; Okumura, R.; Braun, M.R. Genes and proteins involved in qnrS1 induction. Antimicrob. Agents Chemother. 2018, 62, e00806-18. [Google Scholar] [CrossRef] [Green Version]
- Furlan, J.P.R.; Lopes, R.; Gonzalez, I.H.L.; Ramos, P.L.; von Zeska Kress, M.R.; Stehling, E.G. Hypermucoviscous/hypervirulent and extensively drug-resistant QnrB2-, QnrS1-, and CTX-M-3-coproducing Klebsiella pneumoniae ST2121 isolated from an infected elephant (Loxodonta africana). Vet. Microbiol. 2020, 251, 108909. [Google Scholar] [CrossRef]
- Srinivasan, V.B.; Rajamohan, G. KpnEF, a new member of the Klebsiella pneumoniae cell envelope stress response regulon, is an SMR-type efflux pump involved in broad-spectrum antimicrobial resistance. Antimicrob. Agents Chemother. 2013, 57, 4449–4462. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V.B.; Singh, B.B.; Priyadarshi, N.; Chauhan, N.K.; Rajamohan, G. Role of novel multidrug efflux pump involved in drug resistance in Klebsiella pneumoniae. PLoS ONE 2014, 9, e96288. [Google Scholar] [CrossRef] [Green Version]
- Doménech-Sánchez, A.; Hernández-Allés, S.; Martínez-Martínez, L.; Benedí, V.J.; Albertí, S. Identification and characterization of a new porin gene of Klebsiella pneumoniae: Its role in beta-lactam antibiotic resistance. J. Bacteriol. 1999, 181, 2726–2732. [Google Scholar] [CrossRef] [Green Version]
- Galetti, R.; Filho, R.; Ferreira, J.C.; Varani, A.M.; Sazinas, P.; Jelsbak, L. The plasmidome of multidrug-resistant emergent Salmonella serovars isolated from poultry. Infect. Genet. Evol. 2021, 89, 104716. [Google Scholar] [CrossRef]
- Naha, S.; Sands, K.; Mukherjee, S.; Roy, C.; Rameez, M.J.; Saha, B. KPC-2-producing Klebsiella pneumoniae ST147 in a neonatal unit: Clonal isolates with differences in colistin susceptibility attributed to acrAB-TolC pump. Int. J. Antimicrob. Agents. 2020, 55, 105903. [Google Scholar] [CrossRef]
- Ceccarelli, D.; Salvia, A.M.; Sami, J.; Cappuccinelli, P.; Colombo, M.M. New cluster of plasmid-located class 1 integrons in Vibrio cholerae O1 and a dfrA15 cassette-containing integron in Vibrio parahaemolyticus isolated in Angola. Antimicrob. Agents. Chemother. 2006, 50, 2493–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.F.; Chen, G.S.; Kong, Q.X.; Gao, L.P.; Chen, X.; Ye, Y. Increase in the prevalence of resistance determinants to trimethoprim/sulfamethoxazole in clinical Stenotrophomonas maltophilia isolates in China. PLoS ONE 2016, 11, e0157693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miryala, S.K.; Anbarasu, A.; Ramaiah, S. Role of SHV-11, a class A beta-lactamase, gene in multidrug resistance among Klebsiella pneumoniae strains and understanding Its mechanism by gene Network analysis. Microb. Drug. Resist. 2020, 26, 900–908. [Google Scholar] [CrossRef]
- Dadashpour, R.; Moghaddam, M.J.M.; Salehi, Z. Prevalence of non-extended spectrum beta-lactamases SHV-1 and TEM-1 or -2 types in multidrug-resistant Enterobacteriaceae in northern Iran. Biol. Futur. 2020, 71, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.H.; Siddiqui, M.T.; Sultan, I.; Haq, Q.M.R. Prevalence and diversity of blaTEM, blaSHV and blaCTX-M variants among multidrug resistant Klebsiella spp. from an urban riverine environment in India. Int. J. Environ. Health Res. 2019, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Fevre, C.; Passet, V.; Weill, F.X.; Grimont, P.A.; Brisse, S. Variants of the Klebsiella pneumoniae OKP chromosomal beta-lactamase are divided into two main groups, OKP-A and OKP-B. Antimicrob. Agents Chemother. 2005, 49, 5149–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Lu, W.; Zhou, D.; Zheng, G.; Liu, H.; Qian, C. Characterization of two macrolide resistance-related genes in multidrug-resistant Pseudomonas aeruginosa isolates. Pol. J. Microbiol. 2020, 69, 349–356. [Google Scholar] [CrossRef]
- Chiu, H.C.; Lin, T.L.; Yang, J.C.; Wang, J.T. Synergistic effect of imp/ostA and msbA in hydrophobic drug resistance of Helicobacter pylori. BMC Microbiol. 2009, 9, 136. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhang, J.; Xu, L.; Liu, Y.; Li, P.; Zhu, T. Spread of the florfenicol resistance floR gene among clinical Klebsiella pneumoniae isolates in China. Antimicrob. Resist. Infect. Control. 2018, 7, 127. [Google Scholar] [CrossRef] [Green Version]
- Breazeale, S.D.; Ribeiro, A.A.; Raetz, C.R. Oxidative decarboxylation of UDP-glucuronic acid in extracts of polymyxin-resistant Escherichia coli. origin of lipid a species modified with 4-amino-4-deoxy-L-arabinose. J. Biol. Chem. 2002, 277, 2886–2896. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.S.; Lin, T.Y.; Wang, W.B.; Liu, M.C.; Hsueh, P.R.; Liaw, S.J. Characterization of UDP-glucose dehydrogenase and UDP-glucose pyrophosphorylase mutants of Proteus mirabilis: Defectiveness in polymyxin B resistance, swarming, and virulence. Antimicrob. Agents Chemother. 2010, 54, 2000–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, G.R.; Maurya, A.; Yadav, D.K.; Khan, F.; Gupta, M.K.; Gupta, P.; Darokar, M.P.; Srivastava, S.K. Comparative drug resistance reversal potential of natural glycosides: Potential of synergy niaziridin & niazirin. Curr. Top. Med. Chem. 2019, 19, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Fàbrega, A.; Martin, R.G.; Rosner, J.L.; Tavio, M.M.; Vila, J. Constitutive soxS expression in a fluoroquinolone-resistant strain with a truncated soxR protein and identification of a new member of the marA-soxS-rob regulon, mdtG. Antimicrob. Agents Chemother. 2010, 54, 1218–1225. [Google Scholar] [CrossRef] [Green Version]
- Maunders, E.A.; Ganio, K.; Hayes, A.J.; Neville, S.L.; Davies, M.R.; Strugnell, R.A. The role of zntA in Klebsiella pneumoniae zinc homeostasis. Microbiol. Spectr. 2022, 10, e0177321. [Google Scholar] [CrossRef]
- Chen, J.; Huang, L.; Wang, Q.; Zeng, H.; Xu, J.; Chen, Z. Antibiotics in aquaculture ponds from Guilin, south of China: Occurrence, distribution, and health risk assessment. Environ. Res. 2022, 204, 112084. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Liaqat, F.; Akbar, A.; Sahfee, M.; Samad, A.; Anwar, M. Virulent and multidrug-resistant Klebsiella pneumoniae from clinical samples in Balochistan. Int. Wound J. 2021, 18, 510–518. [Google Scholar] [CrossRef]
- Marques, C.; Menezes, J.; Belas, A.; Aboim, C.; Cavaco-Silva, P.; Trigueiro, G. Klebsiella pneumoniae causing urinary tract infections in companion animals and humans: Population structure, antimicrobial resistance and virulence genes. J. Antimicrob. Chemother. 2019, 74, 594–602. [Google Scholar] [CrossRef]
- Varol, M.; Sunbul, M.R. Organochlorine pesticide, antibiotic and heavy metal residues in mussel, crayfish and fish species from a reservoir on the Euphrates River, Turkey. Environ. Pollut. 2017, 230, 311–319. [Google Scholar] [CrossRef]
- Ni, L.; Chen, D.; Fu, H.; Xie, Q.; Lu, Y.; Wang, X.; Zhao, Y.; Chen, L. Residual levels of antimicrobial agents and heavy metals in 41 species of commonly consumed aquatic products in Shanghai, China, and cumulative exposure risk to children and teenagers. Food Control. 2021, 129, 108225. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, W.; Zhao, Z.; Ye, C.; Zhou, S.; Wu, S. Molecular characterization of carbapenem-resistant Klebsiella pneumoniae isolates with focus on antimicrobial resistance. BMC Genom. 2019, 20, 822. [Google Scholar] [CrossRef] [Green Version]
- Fortier, L.C.; Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, I.; Trastoy, R.; Blasco, L.; Fernandez-Cuenca, F.; Ambroa, A.; Fernandez-Garcia, L. Genomic analysis of 40 prophages located in the genomes of 16 carbapenemase-producing clinical strains of Klebsiella pneumoniae. Microb. Genom. 2020, 6, e000369. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.; Boucher, Y.; Labbate, M.; Holmes, A.; Krishnan, S.; Holley, M. The evolution of class 1 integrons and the rise of antibiotic resistance. J. Bacteriol. 2008, 190, 5095–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firoozeh, F.; Mahluji, Z.; Khorshidi, A.; Zibaei, M. Molecular characterization of class 1, 2 and 3 integrons in clinical multi-drug resistant Klebsiella pneumoniae isolates. Antimicrob. Resist. Infect. Control. 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Nzabarushimana, E.; Tang, H. Insertion sequence elements-mediated structural variations in bacterial genomes. Mob. DNA 2018, 9, 29. [Google Scholar] [CrossRef]
- Devi, V.; Harjai, K.; Chhibber, S. CRISPR-Cas systems: Role in cellular processes beyond adaptive immunity. Folia. Microbiol. 2022, 67, 837–850. [Google Scholar] [CrossRef]
- Wu, Y.; Battalapalli, D.; Hakeem, M.J.; Selamneni, V.; Zhang, P.; Draz, M.S. Engineered CRISPR-Cas systems for the detection and control of antibiotic-resistant infections. J. Nanobiotechnol. 2021, 19, 401. [Google Scholar] [CrossRef]
- Remya, P.A.; Shanthi, M.; Sekar, U. Characterisation of virulence genes associated with pathogenicity in Klebsiella pneumoniae. Indian J. Med. Microbiol. 2019, 37, 210–218. [Google Scholar] [CrossRef]
- Kuş, H.; Arslan, U.; Türk Dağı, H.; Fındık, D. Investigation of various virulence factors of Klebsiella pneumoniae strains isolated from nosocomial infections. Mikrobiyol. Bul. 2017, 51, 329–339. [Google Scholar] [CrossRef]
- van Duin, D.; Paterson, D.L. Multidrug-resistant bacteria in the community: An update. Infect. Dis. Clin. N. Am. 2020, 34, 709–722. [Google Scholar] [CrossRef]
- Tan, T.Y.; Ong, M.; Cheng, Y.; Ng, L.S.Y. Hypermucoviscosity, rmpA, and aerobactin are associated with community-acquired Klebsiella pneumoniae bacteremic isolates causing liver abscess in Singapore. J. Microbiol. Immunol. Infect. 2019, 52, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Candan, E.D.; Aksoz, N. Klebsiella pneumoniae: Characteristics of carbapenem resistance and virulence factors. Acta. Biochim. Pol. 2015, 62, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Ssekatawa, K.; Byarugaba, D.K.; Nakavuma, J.L.; Kato, C.D.; Ejobi, F.; Tweyongyere, R.; Eddie, W.M. Prevalence of pathogenic Klebsiella pneumoniae based on PCR capsular typing harbouring carbapenemases encoding genes in Uganda tertiary hospitals. Antimicrob. Resist. Infect. Control. 2021, 10, 57. [Google Scholar] [CrossRef] [PubMed]



| Genome Feature | K. pneumoniae Isolate | ||||||
|---|---|---|---|---|---|---|---|
| 7-5-4 | 7-10-14 | 7-13-2 | 7-17-8 | 8-1-12-1 | 8-2-5-4 | 8-2-10-5 | |
| Genome size (bp) | 5,857,823 | 5,376,532 | 5,412,275 | 5,438,640 | 5,593,530 | 5,432,731 | 5,256,522 |
| G + C (%) | 56.35 | 57.28 | 57.29 | 57.12 | 57.21 | 57.32 | 57.81 |
| DNA Scaffold | 81 | 113 | 79 | 50 | 74 | 64 | 45 |
| Total predicted gene | 5662 | 5142 | 5165 | 5189 | 5369 | 5143 | 4986 |
| Protein-coding gene | 5558 | 5044 | 5060 | 5089 | 5260 | 5042 | 4885 |
| RNA gene | 240 | 231 | 238 | 234 | 249 | 224 | 235 |
| Genes assigned to COG | 4986 | 4761 | 4757 | 4770 | 4916 | 4777 | 4639 |
| Genes with unknown function | 1363 | 1230 | 1288 | 1271 | 1357 | 1230 | 1188 |
| GI | 17 | 8 | 8 | 15 | 16 | 11 | 12 |
| Prophage | 2 | 2 | 2 | 2 | 3 | 1 | 2 |
| IN | 1 | 2 | 0 | 0 | 0 | 0 | 1 |
| IS | 2 | 2 | 6 | 2 | 4 | 4 | 2 |
| CRISPR-Cas repeat | 14 | 9 | 6 | 2 | 6 | 6 | 11 |
| Source | This study | This study | This study | This study | This study | [15] | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, H.; Xie, L.; Chen, L. Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals. Diversity 2023, 15, 527. https://doi.org/10.3390/d15040527
Guan H, Xie L, Chen L. Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals. Diversity. 2023; 15(4):527. https://doi.org/10.3390/d15040527
Chicago/Turabian StyleGuan, Huiqiong, Lu Xie, and Lanming Chen. 2023. "Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals" Diversity 15, no. 4: 527. https://doi.org/10.3390/d15040527
APA StyleGuan, H., Xie, L., & Chen, L. (2023). Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals. Diversity, 15(4), 527. https://doi.org/10.3390/d15040527