

Comparative Analysis of Gut Microbial Community Structure of Three Tropical Sea Cucumber Species
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and PCR Amplification
2.3. High-Throughput Sequencing
2.4. Data Analysis
3. Results
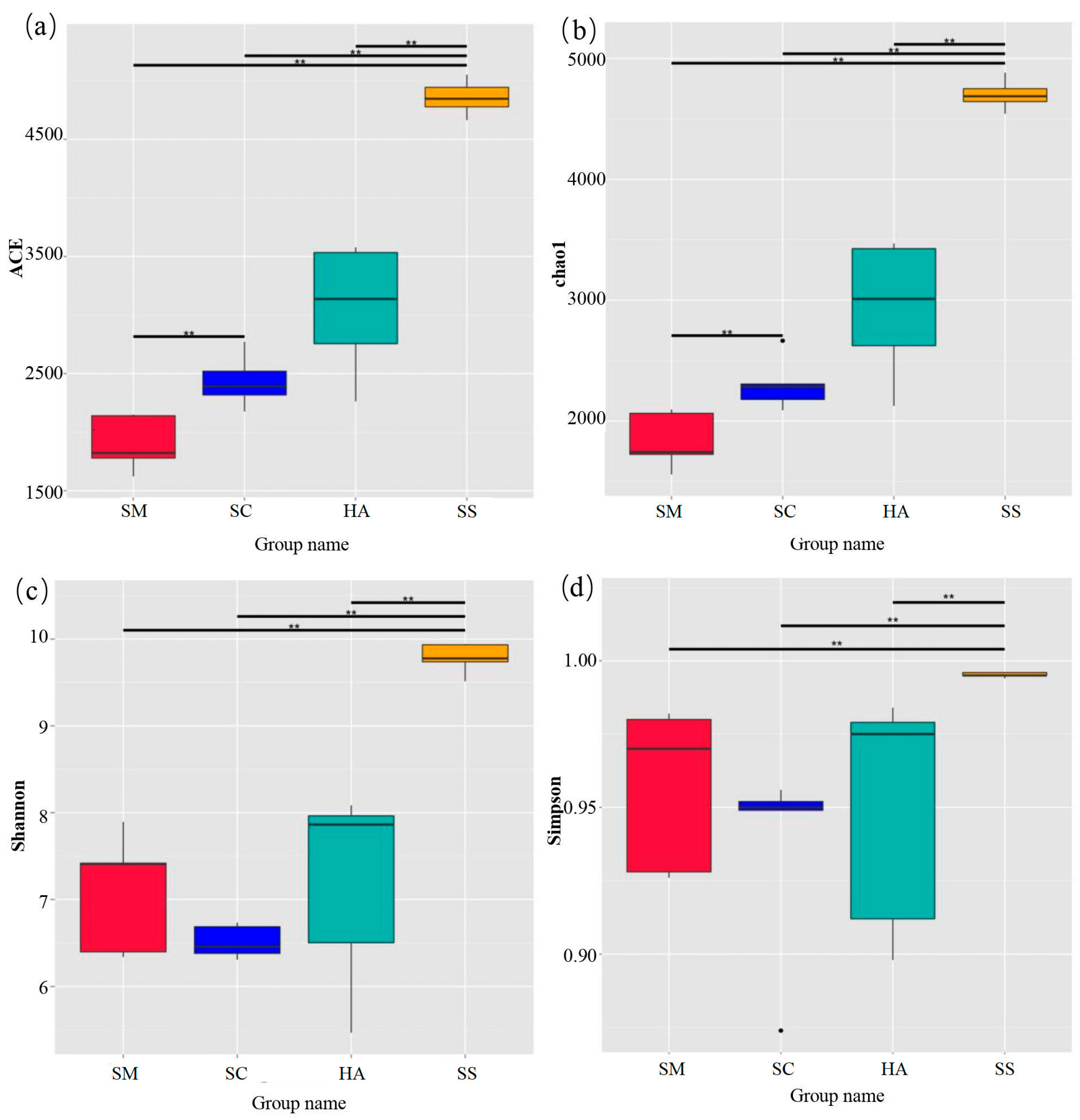
3.1. Sequencing Data Statistics and Analysis
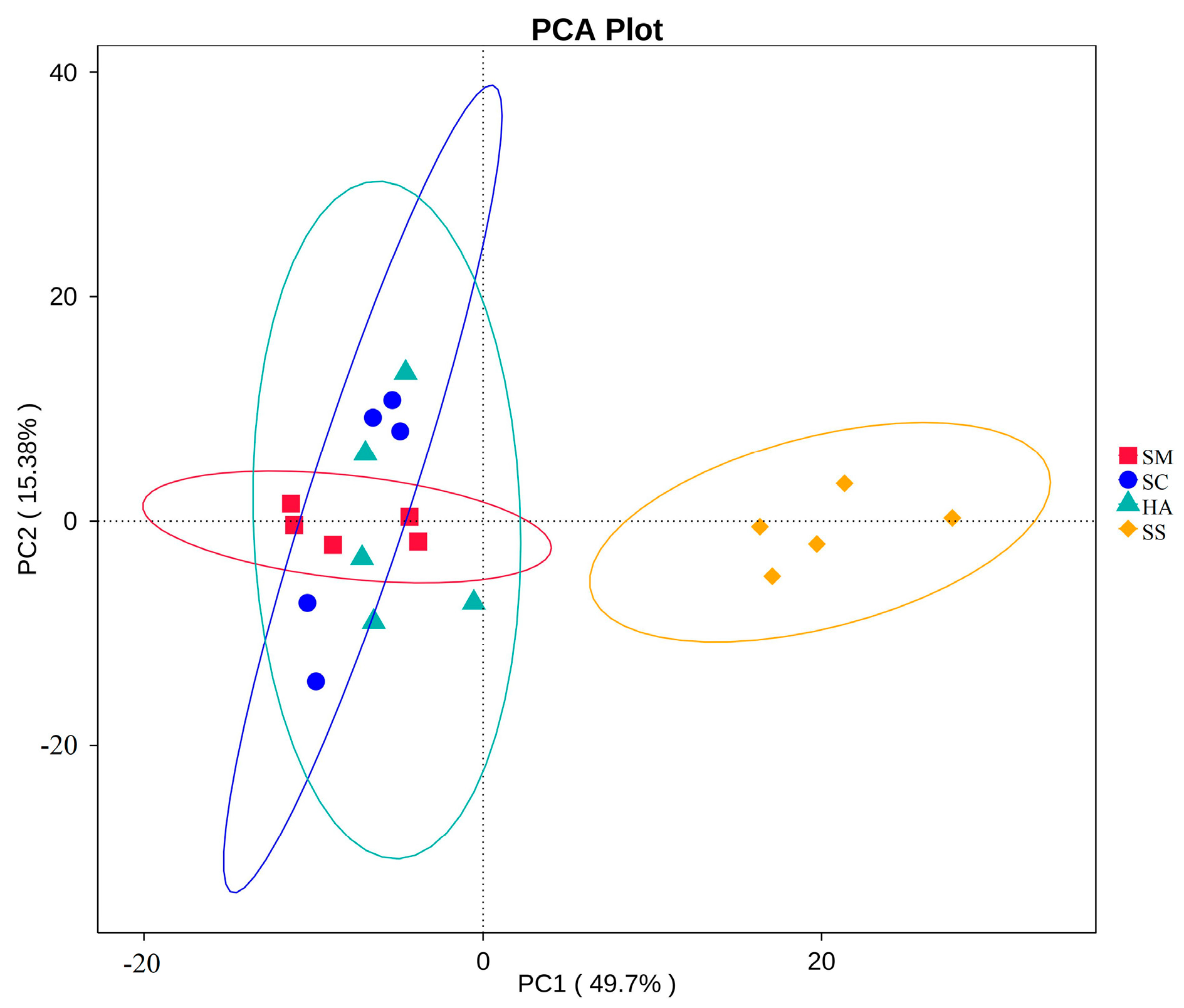
3.2. Relationships of Microbial Communities among the Gut and Sediment Samples
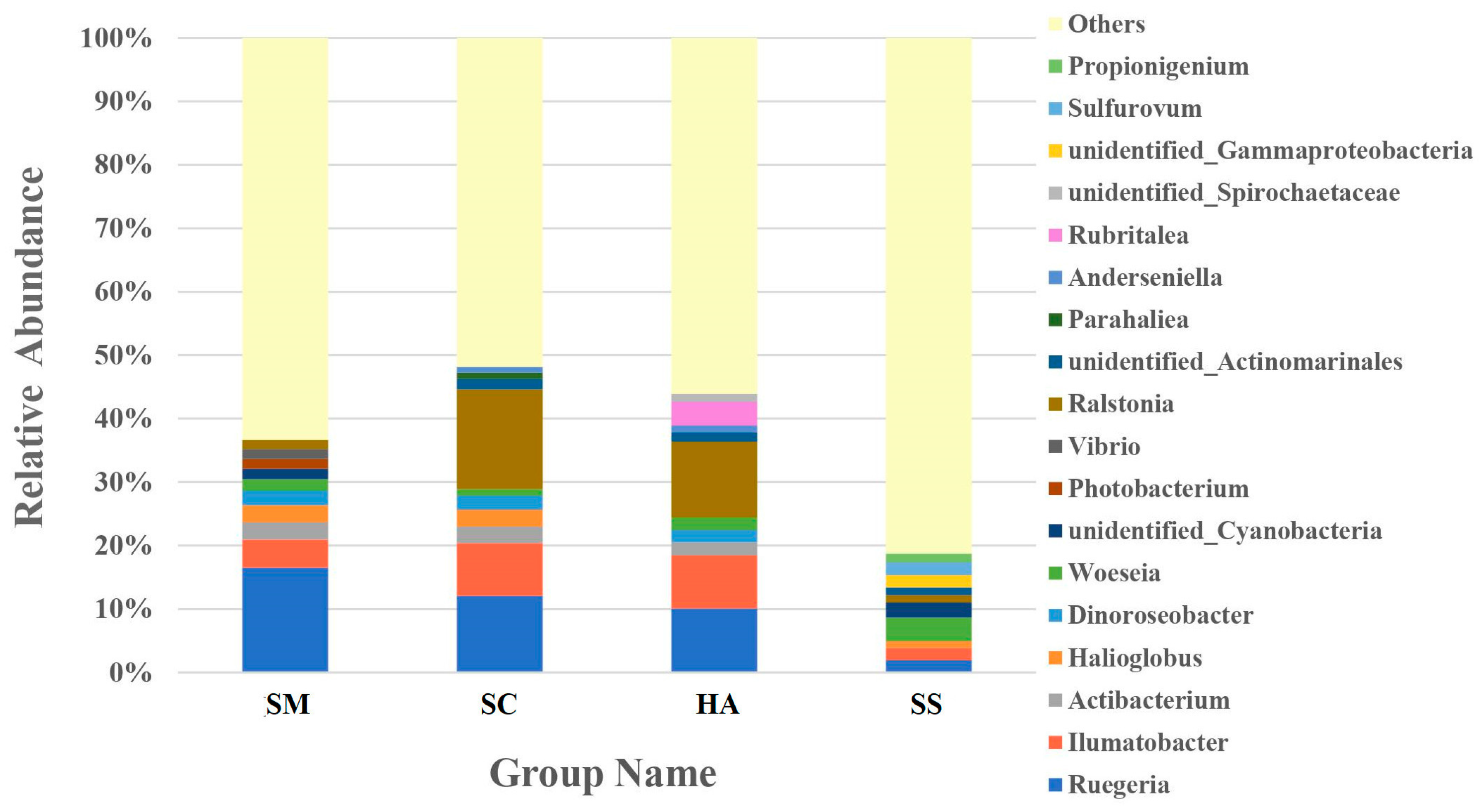
3.3. Relative Abundance of Microbial Communities
3.4. Annotated Analysis of Microbiota Function
4. Discussion
4.1. Dominant Bacteria in the Gut Microbiota
4.2. Potential Probiotics
4.3. Relationship between Microbial Communities in the Gut Contents and the Sediments
4.4. Relationships among Gut Microbial Communities of Three Sea Cucumber Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worms. Holothuroidea. Available online: https://www.marinespecies.org/aphia.php?p=browser&id=148744#f-ocus (accessed on 19 May 2022).
- Amon, R.; Herndl, G.J. Deposit feeding and sediment: II. Decomposition of fecal pellets of Holothuria tubulosa (Holothurioida, Echinodermata). Mar. Ecol. -Evol. Persp. 1991, 12, 175–184. [Google Scholar] [CrossRef]
- Liao, Y. Fauna Sinica: Echinoderma: Holothuroidea, 1st ed.; Science Press: Beijing, China, 1997; pp. 99–156. (In Chinese) [Google Scholar]
- Farjami, B.; Nematollahi, M.A.; Moradi, Y.; Nazemi, M. Derivation of extracts from Persian Gulf sea cucumber (Holothuria leucospilota) and assessment of its antifungal effect. Iran J. Fish. Sci. 2014, 13, 785–795. [Google Scholar]
- Saito, M.; Kunisaki, N.; Urano, N.; Kimura, S. Collagen as the major edible component of sea cucumber (Stichopus japonicus). J. Food Sci. 2002, 67, 1319–1322. [Google Scholar] [CrossRef]
- Sugawara, T.; Zaima, N.; Yamamoto, A.; Sakai, S.; Noguchi, R.; Hirata, T. Isolation of sphingoid bases of sea cucumber cerebrosides and their cytotoxicity against human colon cancer cells. Biosci. Biotech. Bioch. 2006, 70, 2906–2912. [Google Scholar] [CrossRef]
- Zhou, D.Y.; Chang, X.N.; Bao, S.S.; Song, L.; Zhu, B.W.; Dong, X.P.; Zong, Y.; Li, D.M.; Zhang, M.M.; Liu, Y.X.; et al. Purification and partial characterisation of a cathepsin L-like proteinase from sea cucumber (Stichopus japonicus) and its tissue distribution in body wall. Food Chem. 2014, 158, 192–199. [Google Scholar] [CrossRef]
- Nie, L.; Zhou, Q.; Qiao, Y.; Chen, J. Interplay between the gut microbiota and immune responses of ayu (Plecoglossus altivelis) during Vibrio anguillarum infection. Fish Shellfish. Immun. 2017, 68, 479–487. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Jian, X.; Leip, D.D.; Chen, C.H.; Westover, B.P.; Weatherford, J.; Buhler, J.D.; Gordon, J.I. Glycan foraging in vivo by an Intestine-Adapted bacterial symbiont. Science 2005, 307, 1955–1959. [Google Scholar] [CrossRef]
- Sugita, H.; Kawasaki, J.; Deguchi, Y. Production of amylase by the intestinal microflora in cultured freshwater fish. Lett. Appl. Microbiol. 1997, 24, 105–108. [Google Scholar] [CrossRef]
- Zhang, M.; Du, Z. Review and perspective: Function of intestinal microbiota in aquatic animals. J. East China Norm. Univ. (Nat. Sci.) 2016, 2016, 1–8. [Google Scholar]
- Roberts, D. Deposit-feeding mechanisms and resource partitioning in tropical holothurians. J. Exp. Mar. Biol. Ecol. 1979, 37, 43–56. [Google Scholar] [CrossRef]
- Moriarty, D. Feeding of Holothuria atra and Stichopus chloronotus on bacteria, organic carbon and organic nitrogen in sediments of the great barrier reef. Mar. Freshwater Res. 1982, 33, 255–263. [Google Scholar] [CrossRef]
- Amaro, T.; Witte, H.; Herndl, G.J.; Cunha, M.R.; Billett, D. Deep-sea bacterial communities in sediments and guts of deposit-feeding holothurians in Portuguese canyons (NE Atlantic). Deep. Sea Res. Part I Oceanogr. Res. Pap. 2009, 56, 1834–1843. [Google Scholar] [CrossRef]
- Deming, J.W.; Colwell, R.R. Barophilic bacteria associated with digestive tracts of abyssal holothurians. Appl. Environ. Microb. 1982, 44, 1222. [Google Scholar] [CrossRef]
- Fenchel, M.T. Detritus food chains of aquatic ecosystems: The role of bacteria. In Advances in Microbial Ecology, 1st ed.; Marshall, K.C., Ed.; Springer Science and Business Media: Boston, MA, USA, 1977; Volume 1, pp. 1–58. [Google Scholar] [CrossRef]
- Hatmanti, A.; Purwati, P. Bacteria associated holothurians: The key of habitat preference, diet, and function. J. Ilmu. Teknol. Kelaut. 2011, 3, 73–81. [Google Scholar] [CrossRef]
- Zhang, X.; Nakahara, T.; Miyazaki, M.; Nogi, Y.; Kudo, T. Diversity and function of aerobic culturable bacteria in the intestine of the sea cucumber Holothuria leucospilota. J. Gen. Appl. Microbiol. 2012, 58, 447–456. [Google Scholar] [CrossRef]
- Clements, K.D.; Angert, E.R.; Montgomery, W.L.; Choat, J.H. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ma, H.; Zhang, W.; Xu, W.; Liufu, Z.; Mai, K. Effects of potential probiotics on growth performance and immune response of the juvenile sea cucumber (Apostichopus japonicus). J. Fish. China 2010, 34, 775–783. [Google Scholar] [CrossRef]
- Ward-Rainey, N.; Rainey, F.A.; Stackebrandt, E. A study of the bacterial flora associated with Holothuria atra. J. Exp. Mar. Biol. Ecol. 1996, 203, 11–26. [Google Scholar] [CrossRef]
- Purcell, S.W.; Samyn, Y.; Conand, C. Commercially Important Sea Cucumbers of the World, 1st ed.; FAO: Rome, Italy, 2012; pp. 42–104. [Google Scholar]
- Liu, H.; Lei, W.; Mei, L.; Wang, B.; Jiang, K.; Ma, S.; Li, Q. The intestinal microbial diversity in Chinese shrimp (Fenneropenaeus chinensis) as determined by PCR–DGGE and clone library analyses. Aquaculture 2011, 317, 32–36. [Google Scholar] [CrossRef]
- Le, D.H.; Nguyen, N.T.; Dang, O.H.T.; Steinert, G.; Tran, T.T.; Vu, T.H.; Sipkema, D.; Chu, H.H. Characterization of bacterial community in the gut of Penaeus monodon and its culture water in shrimp ponds. Turk. J. Fish. Aquat. Sci. 2019, 19, 977–986. [Google Scholar] [CrossRef]
- Wei, S.; Li, L.; Huang, H.; Jiang, K.; Zhang, F.; Chen, X.; Zhao, M.; Ma, L. The gut microbial community of Antarctic fish detected by 16s rRNA gene sequence analysis. Biomed. Res. Int. 2016, 2016, 3241529. [Google Scholar] [CrossRef]
- Zhou, L.; Qu, Y.; Qin, J.G.; Chen, L.; Li, E. Deep insight into bacterial community characterization and relationship in the pond water, sediment and the gut of shrimp (Penaeus japonicus). Aquaculture 2021, 539, 736658. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, L.; Tang, X.; Xia, X.; Hu, W.; Zhou, P. Season and geography induced variation in sea cucumber (Stichopus japonicus) nutritional composition and gut microbiota. J. Food Compos. Anal. 2021, 101, 103838. [Google Scholar] [CrossRef]
- Wang, X.; Fan, Y.; Yu, X.; Wang, Y.; Ye, H.; Diao, J.; Wang, S.; Liu, H.; Xu, L.; Ma, D.; et al. Analysis of gut microbiota and immune-related genes during sea cucumber (Apostichopus japonicus) response to dietary supplementation with codonopsis pilosula. Isr. J. Aquacult. -Bamid. 2021, 73, 12p. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Q.; Liu, H.; Li, B.; Zhang, H. High-throughput sequencing of 16S rRNA amplicons characterizes gut microbiota shift of juvenile sea cucumber Apostichopus japonicus feeding with three antibiotics. J. Oceanol. Limnol. 2019, 37, 1714–1725. [Google Scholar] [CrossRef]
- Dissanayake, D.; Stefansson, G. Habitat preference of sea cucumbers: Holothuria atra and Holothuria edulis in the coastal waters of sri lanka. J. Mar. Biol. Assoc. UK 2012, 92, 581–590. [Google Scholar] [CrossRef]
- Xue, Y.; Gao, F.; Xu, Q.; Huang, D.; Wang, A.; Sun, T. Study on feeding selection of environmental sediment and digestive adaptability of Holothuria atra. Oceanol. Et Limnol. Sin. 2019, 50, 1070–1079. (In Chinese) [Google Scholar]
- Gao, F.; Li, F.; Tan, J.; Yan, J.; Sun, H. Bacterial community composition in the gut content and ambient sediment of sea cucumber Apostichopus japonicus revealed by 16s rRNA gene pyrosequencing. PLoS ONE 2014, 9, e100092. [Google Scholar] [CrossRef]
- Jia, C.; Zhang, Y.; Xu, Q.; Sun, C.; Wang, Y.; Gao, F. Comparative analysis of in situ eukaryotic food sources in three tropical sea cucumber species by metabarcoding. Animals 2022, 12, 2303. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. Available online: https://www.nature.com/articles/nmeth.f.303/ (accessed on 8 November 2022). [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- John, D. A direct approach to false discovery rates. J. R. Statist. Soc. B. 2002, 64, 479–498. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Forbes, J.; Van Domselaar, G.; Bernstein, C. Microbiome survey of the inflamed and noninflamed gut at different compartments within the gastrointestinal tract of inflammatory bowel disease patients. Inflamm. Bowel Dis. 2016, 22, 817–825. [Google Scholar] [CrossRef]
- Fan, L.; Wang, Z.; Chen, M.; Qu, Y.; Li, J.; Zhou, A.; Xie, S.; Zeng, F.; Zou, J. Microbiota comparison of Pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 2019, 657, 1194–1204. [Google Scholar] [CrossRef]
- Whittaker, R.H. Vegetation of the Siskiyou mountains, Oregon and California. Ecol. Monogr. 1960, 30, 279–338. [Google Scholar] [CrossRef]
- Whittaker, R.H. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef]
- Chapman, M.G.; Underwood, A.J. Ecological patterns in multivariate assemblages: Information and interpretation of negative values in ANOSIM tests. Mar. Ecol. Prog. Ser. 1999, 180, 257–265. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Y.; Wu, P.; Chen, M.; He, L.; Xu, Q.; Wang, A. Bacterial community composition in gut content and ambient sediment of two tropical wild sea cucumbers (Holothuria atra and H. leucospilota). J. Oceonal. Limnol. 2022, 40, 360–372. [Google Scholar] [CrossRef]
- Pujalte, M.J.; Lucena, T.; Ruvira, M.; Arahal, D.R.; Macia, M.C. The family Rhodobacteraceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 439–512. [Google Scholar] [CrossRef]
- Sanudo-Wilhelmy, S.A.; Gomez-Consarnau, L.; Suffridge, C.; Webb, E.A. The role of b vitamins in marine biogeochemistry. Annu. Rev. Mar. Sci. 2014, 6, 339–367. [Google Scholar] [CrossRef]
- Madrid, V.M.; Aller, J.Y.; Aller, R.C.; Chistoserdov, A.Y. High prokaryote diversity and analysis of community structure in mobile mud deposits off french guiana: Identification of two new bacterial candidate divisions. FEMS Microbiol. Ecol. 2001, 37, 197–209. [Google Scholar] [CrossRef]
- Uchino, Y.; Yokota, A.; Sugiyama, J. Phylogenetic position of the marine subdivision of Agrobacterium species based on 16s rRNA sequence analysis. J. Gen. Appl. Microbiol. 1997, 43, 243–247. [Google Scholar] [CrossRef]
- Zhu, L.; Che, X.; Liu, X.; Liu, H.; Li, Y.; Wang, J.; Cheng, G.; Chen, J.; Tang, R.; Chen, X.; et al. Reducing carbon input improved the diversity of bacterial community in large-scale biofloc shrimp culture facilities. Diversity 2022, 14, 778. [Google Scholar] [CrossRef]
- Gao, F.; Gan, E.; Guo, H.; Wang, Y.; Wang, R.; Yan, M.; Dong, P.; Zhang, D. Screening of carbon sources for enrichment and directional isolation of Rhodobacteraceae from the gut of Litopenaeus vanname. Acta Microbiol. Sin. 2022, 62, 14. (In Chinese) [Google Scholar] [CrossRef]
- Mitova, M.; Tommonaro, G.; Hentschel, U.; Müller, W.G.; Rosa, S.D. Exocellular cyclic dipeptides from a Ruegeria strain associated with cell cultures of Suberites domuncula. Mar. Biotechnol. 2004, 6, 95–103. [Google Scholar] [CrossRef]
- Gatesoupe, F.J. The use of probiotics in aquaculture. Aquaculture 1999, 180, 147–165. [Google Scholar] [CrossRef]
- Nayak, S.K. Multifaceted applications of probiotic Bacillus species in aquaculture with special reference to Bacillus subtilis. Rev. Aquacult. 2020, 13, 862–906. [Google Scholar] [CrossRef]
- Verschuere, L.; Rombaut, G.; Sorgeloos, P.; Verstraete, W. Probiotic bacteria as biological control agents in aquaculture. Microbiol. Mol. Biol. R. 2000, 64, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, T.; Hedayati, S.A.; Yavari, V.; Alizade, M.; Farzanfar, A. Growth, survival and gut microbial load of rainbow trout (Oncorhynchus mykiss) fry given diet supplemented with probiotic during the two months of first feeding. Turk. J. Fish. Aquat. Sci. 2008, 8, 43–48. Available online: https://dergipark.org.tr/en/pub/trjfas-ayrildi/issue/13282/160524 (accessed on 8 November 2022).
- Balcázar, J.L.; De-Blas, I.; Ruiz-Zarzuela, I.; Vendrell, D.; Gironés, O.; Muzquiz, J.L. Enhancement of the immune response and protection induced by probiotic lactic acid bacteria against furunculosis in rainbow trout (Oncorhynchus mykiss). FEMS Immunol. Med. Mic. 2007, 51, 185–193. [Google Scholar] [CrossRef]
- Banerjee, G.; Nandi, A.; Ray, A.K. Assessment of hemolytic activity, enzyme production and bacteriocin characterization of Bacillus subtilis LR1 isolated from the gastrointestinal tract of fish. Arch. Microbiol. 2017, 199, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ringø, E.; Li, X.; Doan, V.H.; Ghosh, K. Interesting probiotic bacteria other than the more widely used lactic acid bacteria and bacilli in finfish. Front. Mar. Sci. 2022, 9, 848037. [Google Scholar] [CrossRef]
- Hasan, K.N.; Banerjee, G. Recent studies on probiotics as beneficial mediator in aquaculture: A review. J. Basic Appl. Zool. 2020, 81, 53. Available online: https://basicandappliedzoology.springeropen.com/articles/10.1186/s41936-020-00190-y (accessed on 8 November 2022). [CrossRef]
- Wang, Y.M.; Wang, Y.G. Advance in the mechanisms and application of microecologics in aquaculture. Vet. Med. 2008, 29, 72–75. (In Chinese) [Google Scholar]
- Merrifield, D.L.; Carnevali, O. Probiotic modulation of the gut microbiota of fish. In Aquaculture Nutrition: Gut Health, Probiotics and Prebiotics, 1st ed.; Merrifield, D.L., Ringø, E., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2014; Volume 8, pp. 185–222. [Google Scholar] [CrossRef]
- Hou, C.; Zeng, X.; Yang, F.; Liu, H.; Qiao, S. Study and use of the probiotic Lactobacillus reuteri in pigs: A review. J. Anim. Sci. Biotechno. 2015, 6, 14. [Google Scholar] [CrossRef]
- Indrio, F.; Riezzo, G.; Raimondi, F.; Bisceglia, M.; Cavallo, L.; Francavilla, R. The effects of probiotics on feeding tolerance, bowel habits, and gastrointestinal motility in preterm newborns. J. Pediatr. 2008, 152, 801–806. [Google Scholar] [CrossRef]
- Mcfall-Ngai, M. Adaptive immunity: Care for the community. Nature 2007, 445, 153. [Google Scholar] [CrossRef]
- Spinler, J.K.; Taweechotipatr, M.; Rognerud, C.L.; Ou, C.N.; Tumwasorn, S.; Versalovic, J. Human-derived probiotic Lactobacillus reuteri demonstrate antimicrobial activities targeting diverse enteric bacterial pathogens. Anaerobe 2008, 14, 166–171. [Google Scholar] [CrossRef]
- Tubelius, P.; Stan, V.; Zachrisson, A. Increasing work-place healthiness with the probiotic Lactobacillus reuteri: A randomised, double-blind placebo-controlled study. Environ. Health 2005, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, B.; Viswanath, B.; Sai Gopal, D. Probiotics as antiviral agents in shrimp aquaculture. J. Pathog. 2013, 2013, 424123. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.K.; Swarnakumar, N.S.; Sivakumar, K.; Thangaradjou, T.; Kannan, L. Probiotics in aquaculture: Importance and future perspectives. Indian J. Microbiol. 2008, 48, 299–308. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Song, L.; Wang, J.; Hu, Y.; Yang, Q.; Cheng, P.; Li, J. Effects of dietary supplementation of Lactobacillus plantarum and Bacillus subtilis on growth performance, survival, immune response, antioxidant capacity and digestive enzyme activity in olive flounder (Paralichthys olivaceus). Aquac. Fish. 2021, 6, 283–288. [Google Scholar] [CrossRef]
- Luis-Balcázar, J.; Decamp, O.; Vendrell, D.; De-Blas, I.; Ruiz-Zarzuela, I. Health and nutritional properties of probiotics in fish and shellfish. Microb. Ecol. Health Dis. 2006, 18, 65–70. [Google Scholar] [CrossRef]
- Mamun, M.; Nasren, S.; Bari, S.M. Role of probiotics in aquaculture: Importance and future guidelines. J. Bangladesh Acad. Sci. 2018, 42, 105–109. [Google Scholar] [CrossRef]
- Roy, N.C.; Munni, M.J.; Chowdhury, M.; Akther, K.R. Probiotic supplements in aquaculture: Latest developments and future trends. In Biotechnological Advances in Aquaculture Health Management, 1st ed.; Gupta, S.K., Giri, S.S., Eds.; Springer: Singapore, 2021; pp. 345–367. [Google Scholar] [CrossRef]
- Zheng, C.N.; Wang, W. Effects of Lactobacillus pentosus on the growth performance, digestive enzyme and disease resistance of white shrimp, Litopenaeus vannamei (Boone, 1931). Aquac. Res. 2017, 48, 2767–2777. [Google Scholar] [CrossRef]
- Singhal, R.; Shah, Y.M. Oxygen battle in the gut: Hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J. Biol. Chem. 2020, 295, 10493–10505. [Google Scholar] [CrossRef]
- Cartwright, I.M.; Colgan, S.P. The hypoxic tissue microenvironment as a driver of mucosal inflammatory resolution. Front. Immunol. 2023, 14, 1124774. [Google Scholar] [CrossRef]
- Wright, K.E.; Williamson, C.; Grasby, S.E.; Spear, J.R.; Templeton, A.S. Metagenomic evidence for sulfur lithotrophy by Epsilonproteobacteria as the major energy source for primary productivity in a sub-aerial arctic glacial deposit, Borup Fiord Pass. Front. Microbiol. 2013, 4, 63. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.J.; Wang, Z.J.; Zhao, J.X.; Chen, G.J. Woeseia oceani gen. nov.; sp. nov.; a chemoheterotrophic member of the order Chromatiales, and proposal of Woeseiaceae fam. nov. Int. J. Syst. Evol. Micr. 2016, 66, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gao, F.; Tan, J.; Fan, C.; Sun, H.; Yan, J.; Chen, S.; Wang, X. Characterization and identification of enzyme-producing microflora isolated from the gut of sea cucumber Apostichopus japonicus. Chin. J. Oceanol. Limn. 2016, 34, 153–162. Available online: https://link.springer.com/article/10.1007/s00343-015-4149-z (accessed on 8 November 2022). [CrossRef]
- Porsby, C.H.; Nielsen, K.F.; Gram, L. Phaeobacter and Ruegeria species of the Roseobacter clade colonize separate niches in a Danish Turbot (Scophthalmus maximus)-rearing farm and antagonize Vibrio anguillarum under different growth conditions. Appl. Environ. Microb. 2008, 74, 7356–7364. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wu, Y.; Xu, X.; Huang, C.; Xu, Y.; Cheng, H.; Wang, C.; Wu, M.; Xu, X. Actibacterium pelagium sp. Nov.; A novel alphaproteobacterium, and emended description of the genus Actibacterium. Int. J. Syst. Evol. Micr. 2017, 67, 5080–5086. [Google Scholar] [CrossRef]
- Li, G.; Lai, Q.; Sun, F.; Du, Y.; Liu, X.; Li, G.; Xie, Y.; Shao, Z. Actibacterium atlanticum sp. Nov.; Isolated from surface seawater of the Atlantic Ocean. Anton. Leeuw. Int. J. G. 2014, 106, 325–330. [Google Scholar] [CrossRef]
- Mfilinge, P.L.; Tsuchiya, M. Changes in sediment fatty acid composition during passage through the gut of deposit feeding holothurians: Holothuria atra (Jaeger, 1883) and Holothuria leucospilota (Brandt, 1835). J. Lipids 2016, 2016, 4579794. [Google Scholar] [CrossRef]
- Gao, F.; Xu, Q.; Li, X.; He, L.; Wang, A. Habitat preference and ecological function of sea cucumber in the tropical coral reef ecosystem. Acta Ecol. Sin. 2022, 42, 4301–4312. (In Chinese) [Google Scholar] [CrossRef]
- Hauksson, E. Feeding biology of Stichopus tremulus, a deposit-feeding holothurian. Sarsia 1979, 64, 155–160. [Google Scholar] [CrossRef]
- Zhao, P. Basic Study on Feeding Selectivity of Sea Cucumber Apostichopus japonicus. Ph.D. Thesis, Graduate School of Chinese Academy of Sciences (Institute of Oceanology), Qingdao, China, 2010. (In Chinese). [Google Scholar]
- Zhang, Y.; Gao, F.; Xu, Q. Morphology and histology of feeding and digestive organs in juvenile Stichopus monotuberculatus. Mar. Sci. B. 2021, 40, 198–205. (In Chinese) [Google Scholar]
- Uthicke, S. Distribution patterns and growth of two reef flat holothurians, Holothuria atra and Stichopus chloronotus. In The Echinoderms through Time, 1st ed.; Uthicke, S., Ed.; CRC Press: London, UK, 1994; pp. 569–576. [Google Scholar]
- Uthicke, V. Sediment bioturbation and impact on benthic microalgae by Holothuria atra and Stichopus chloronotus, two sediment feeding holothurians. Bull. Mar. Sci. 1999, 64, 129–141. [Google Scholar]
- Sun, Y.; Chen, D. The microbial composition of Stichopus japonicus and its physiological property. Oceanol. Et Limnol. Sin. 1989, 20, 300–307. (In Chinese) [Google Scholar]
- Paltzat, D.L.; Pearce, C.M.; Barnes, P.A.; Mckinley, R.S. Growth and production of California sea cucumbers (Parastichopus californicus Stimpson) co-cultured with suspended Pacific oysters (Crassostrea gigas Thunberg). Aquaculture 2008, 275, 124–137. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group * | Sample ID | OTU | Good’s Coverage | Group * | Sample ID | OTU | Good’s Coverage |
|---|---|---|---|---|---|---|---|
| SM | SM1 | 1551 | 99.30% | SC | SC1 | 1659 | 98.70% |
| SM2 | 1502 | 99.60% | SC2 | 1909 | 98.80% | ||
| SM3 | 1481 | 99.30% | SC3 | 2351 | 98.80% | ||
| SM4 | 1869 | 99.20% | SC4 | 1782 | 99.00% | ||
| SM5 | 1739 | 99.00% | SC5 | 1858 | 98.90% | ||
| HA | HA1 | 1792 | 98.90% | SS | SS1 | 4224 | 98.00% |
| HA2 | 2923 | 98.30% | SS2 | 4272 | 98.30% | ||
| HA3 | 2942 | 98.40% | SS3 | 4086 | 98.20% | ||
| HA4 | 2621 | 98.70% | SS4 | 4210 | 97.80% | ||
| HA5 | 2169 | 98.70% | SS5 | 4017 | 97.90% |
| Group | R-Value | p-Value | Group | R-Value | p-Value |
|---|---|---|---|---|---|
| SM-SS | 1.000 | 0.008 | SC-SM | 0.500 | 0.009 |
| SC-SS | 1.000 | 0.010 | HA-SM | 0.628 | 0.009 |
| HA-SS | 0.948 | 0.015 | HA-SC | 0.020 | 0.310 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, Y.; Jia, C.; Xu, Q.; Rong, Y.; Xu, Z.; Wang, Y.; Gao, F. Comparative Analysis of Gut Microbial Community Structure of Three Tropical Sea Cucumber Species. Diversity 2023, 15, 855. https://doi.org/10.3390/d15070855
Wang Y, Zhang Y, Jia C, Xu Q, Rong Y, Xu Z, Wang Y, Gao F. Comparative Analysis of Gut Microbial Community Structure of Three Tropical Sea Cucumber Species. Diversity. 2023; 15(7):855. https://doi.org/10.3390/d15070855
Chicago/Turabian StyleWang, Yanan, Yue Zhang, Chenghao Jia, Qiang Xu, Yun Rong, Zening Xu, Yuanhang Wang, and Fei Gao. 2023. "Comparative Analysis of Gut Microbial Community Structure of Three Tropical Sea Cucumber Species" Diversity 15, no. 7: 855. https://doi.org/10.3390/d15070855
APA StyleWang, Y., Zhang, Y., Jia, C., Xu, Q., Rong, Y., Xu, Z., Wang, Y., & Gao, F. (2023). Comparative Analysis of Gut Microbial Community Structure of Three Tropical Sea Cucumber Species. Diversity, 15(7), 855. https://doi.org/10.3390/d15070855