
Phylogenetic Constraints and Ecological Implications of Gut Bacterial Communities in Necrophagous Flies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. DNA Extraction
2.3. PCR Amplification and Sequencing
2.4. Bioinformatics and Statistical Analysis
3. Results
3.1. Gut Bacterial Composition of Necrophagous Fly Species
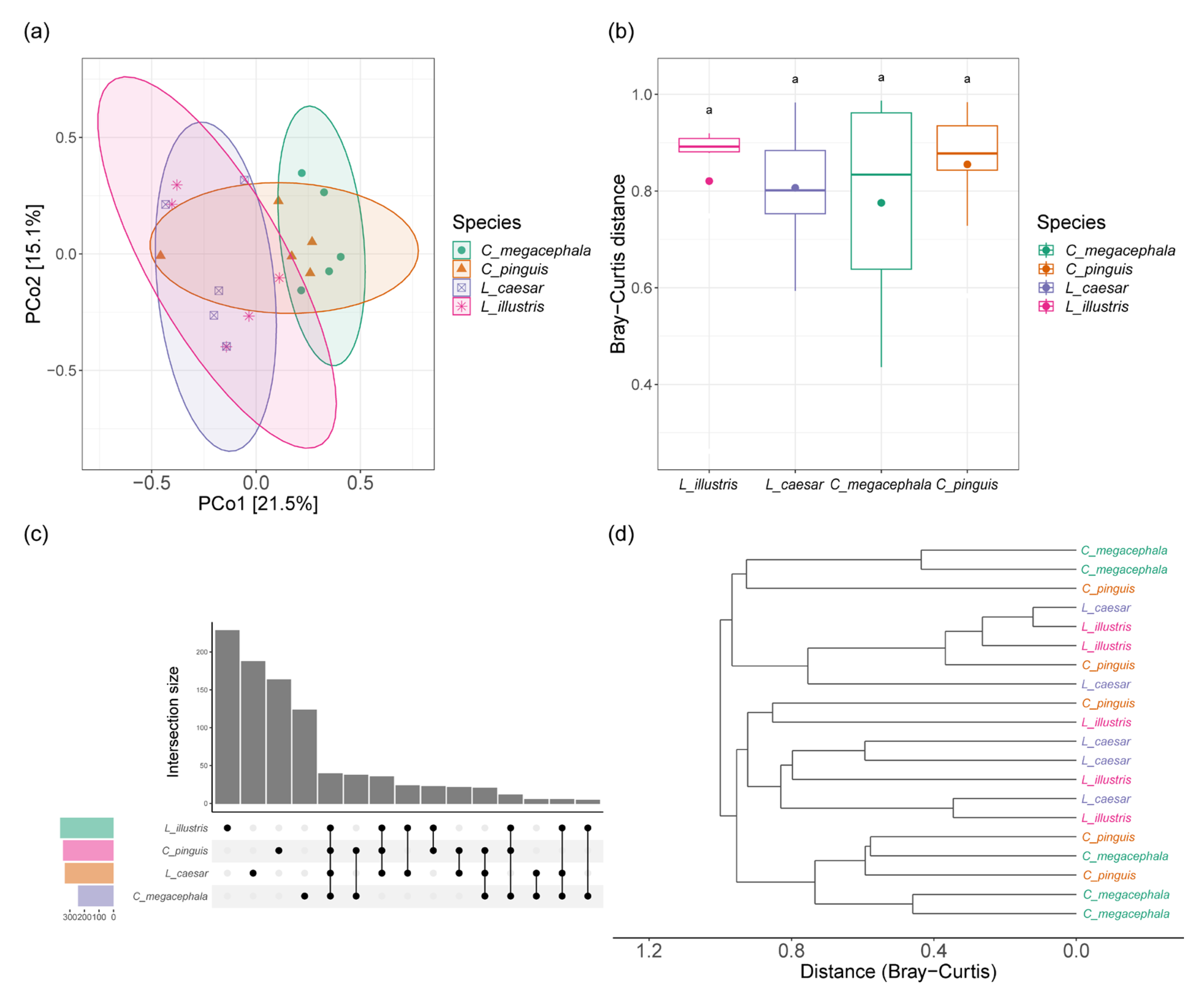
3.2. Alpha and Beta Diversity of Gut Bacterial Community of Necrophagous Fly Species
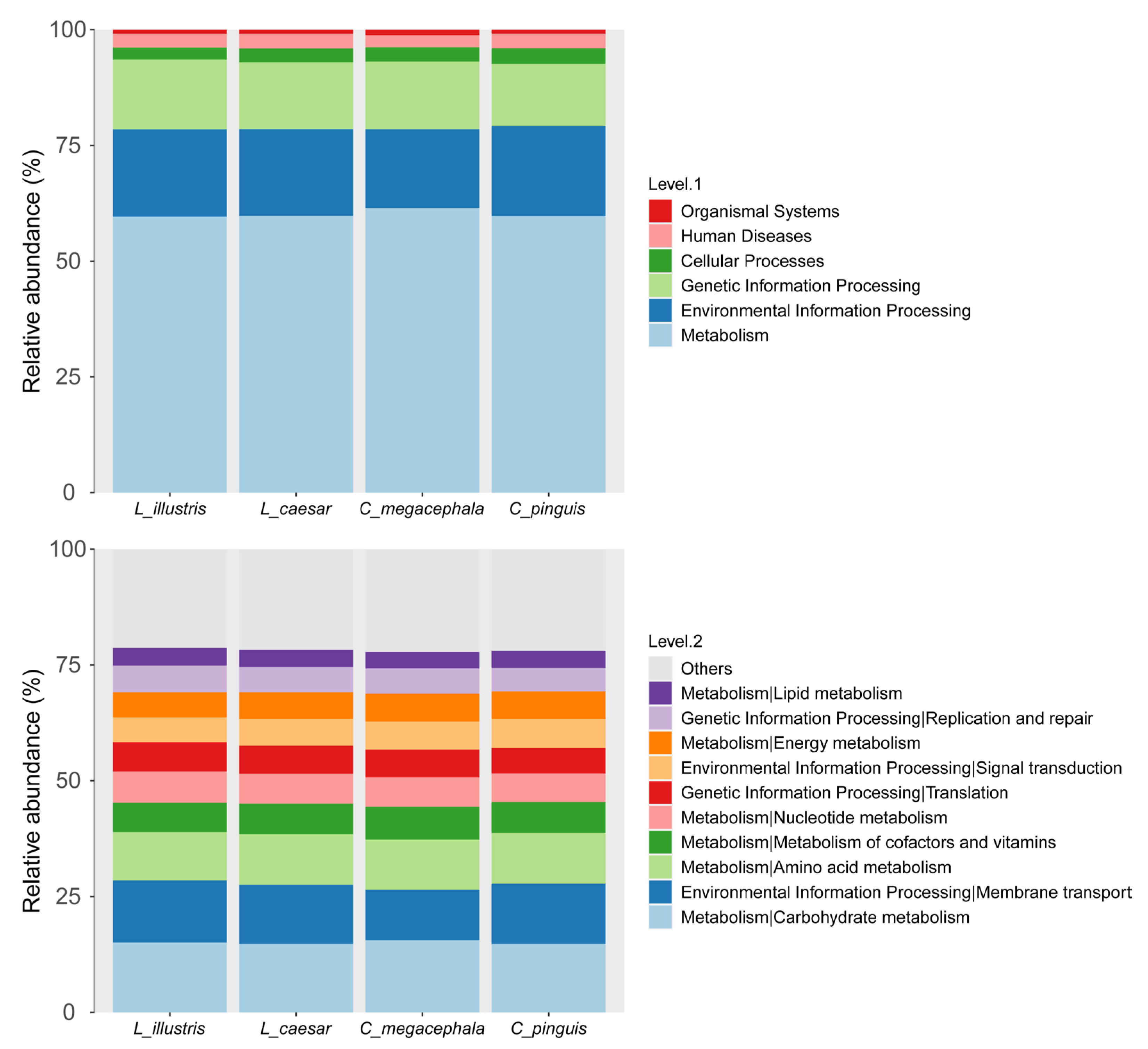
3.3. Gut Bacterial Community Functional Profile
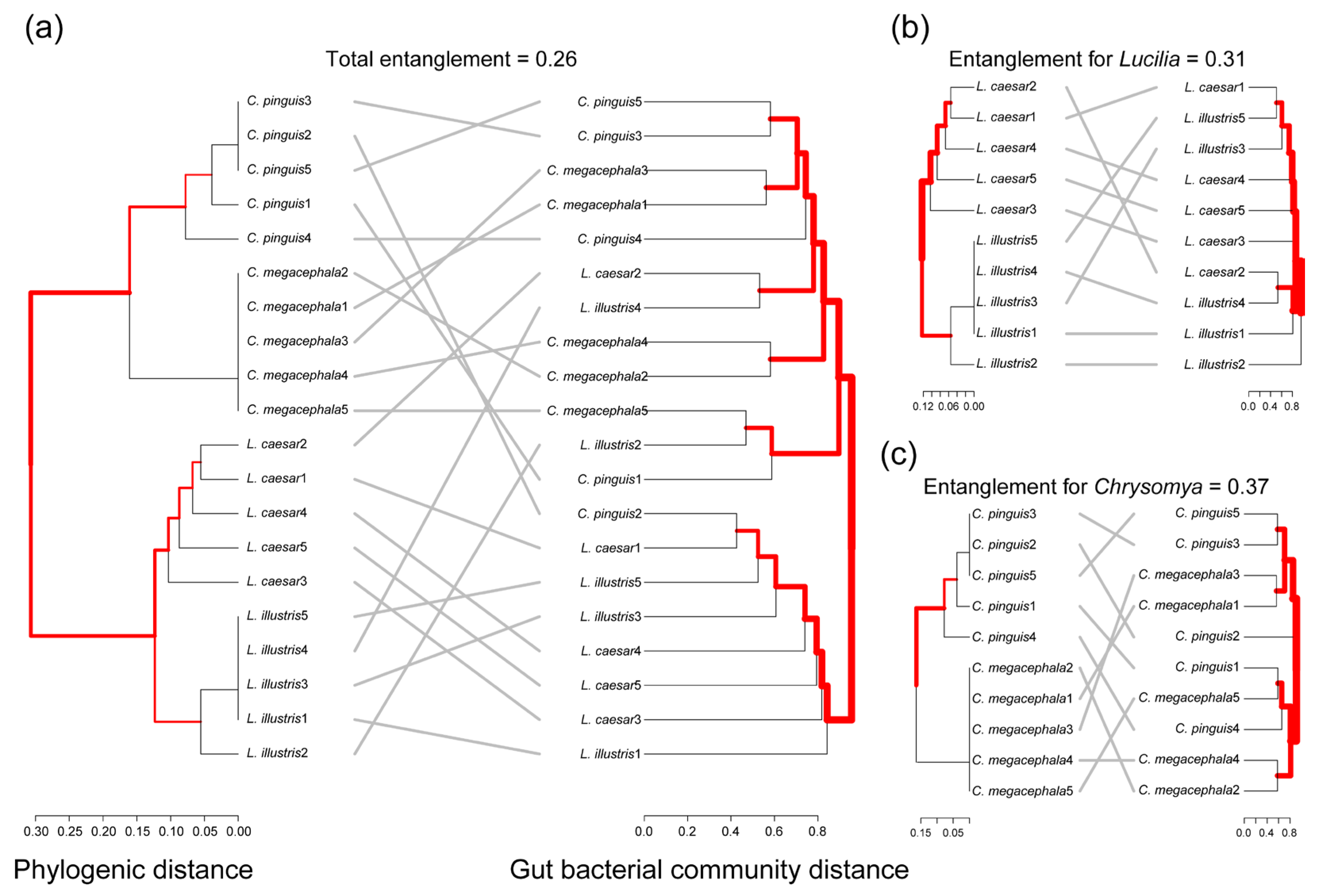
3.4. Correlation between Phylogenetic Distances and Gut Bacterial Community Distances
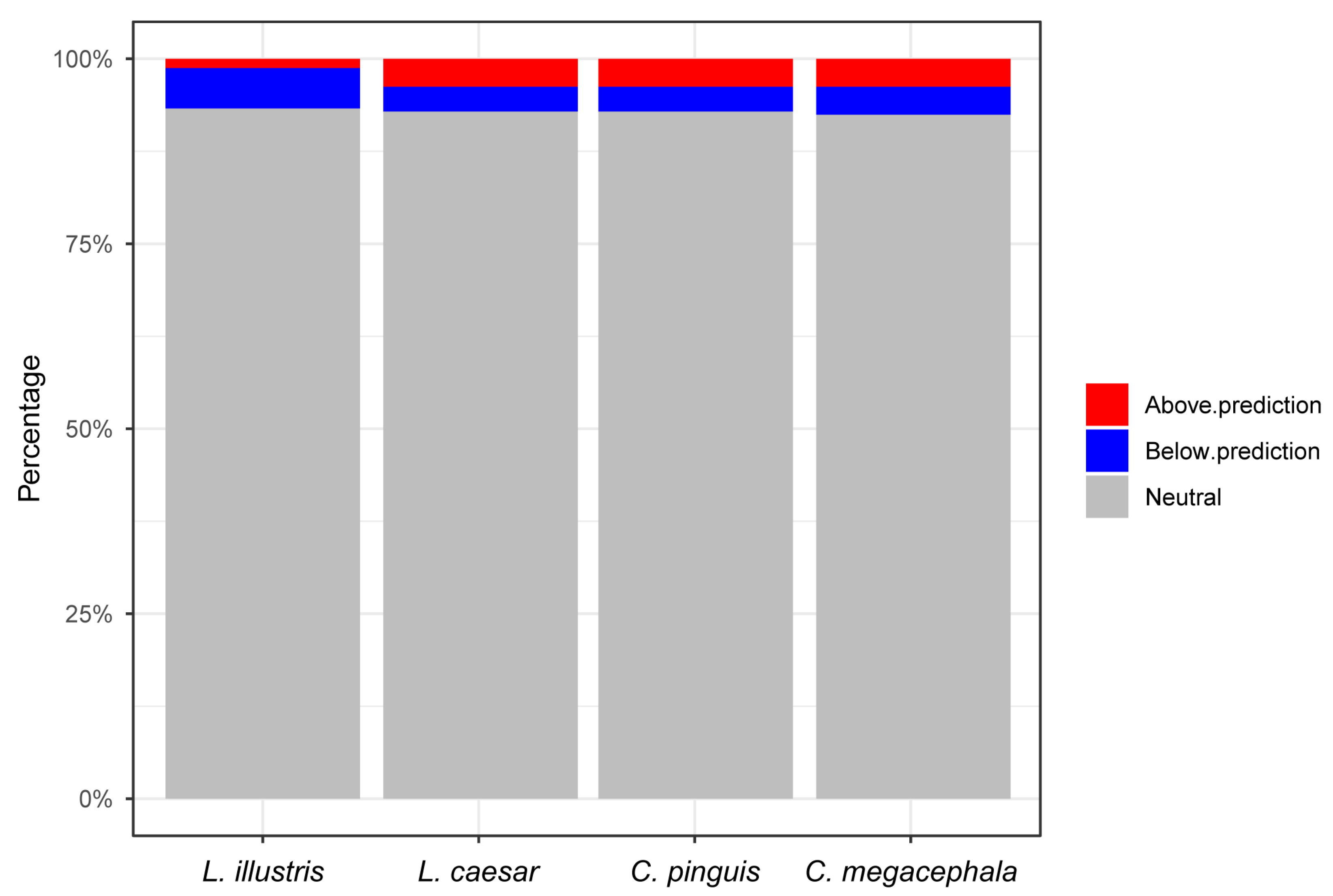
3.5. Sloan Neutral Model Analysis of Gut Bacterial Community
4. Discussion
4.1. Gut Bacterial Composition of Necrophagous Fly Species
4.2. Gut Bacterial Community Diversity and Functional Profile
4.3. Correlation between Phylogenetic Relationship and Gut Bacterial Community Distances
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Losey, J.E.; Vaughan, M.J.B. The economic value of ecological services provided by insects. Bioscience 2006, 56, 311–323. [Google Scholar] [CrossRef]
- Hung, K.-L.J.; Kingston, J.M.; Albrecht, M.; Holway, D.A.; Kohn, J.R. The worldwide importance of honey bees as pollinators in natural habitats. Proc. R. Soc. B 2018, 285, 20172140. [Google Scholar] [CrossRef] [PubMed]
- Brundage, A.; Bros, S.; Honda, J.Y. Seasonal and habitat abundance and distribution of some forensically important blow flies (Diptera: Calliphoridae) in Central California. Forensic Sci. Int. 2011, 212, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Turner, B. Spatial and temporal variability of necrophagous Diptera from urban to rural areas. Med. Vet. Entomol. 2005, 19, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Prado e Castro, C.; Serrano, A.; Martins da Silva, P.; García, M. Carrion flies of forensic interest: A study of seasonal community composition and succession in Lisbon, Portugal. Med. Vet. Entomol. 2012, 26, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Benbow, M.; Lewis, A.; Tomberlin, J.; Pechal, J. Seasonal necrophagous insect community assembly during vertebrate carrion decomposition. J. Med. Entomol. 2013, 50, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Crooks, E.R.; Bulling, M.T.; Barnes, K.M. Microbial effects on the development of forensically important blow fly species. Forensic Sci. Int. 2016, 266, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Durvasula, R.V.; Sundaram, R.K.; Kirsch, P.; Hurwitz, I.; Crawford, C.V.; Dotson, E.; Beard, C.B. Genetic transformation of a Corynebacterial symbiont from the Chagas disease vector Triatoma infestans. Exp. Parasitol. 2008, 119, 94–98. [Google Scholar] [CrossRef]
- Deel, H.L.; Montoya, S.; King, K.; Emmons, A.L.; Huhn, C.; Lynne, A.M.; Metcalf, J.L.; Bucheli, S.R. The microbiome of fly organs and fly-human microbial transfer during decomposition. Forensic Sci. Int. 2022, 340, 111425. [Google Scholar] [CrossRef]
- Wei, T.; Ishida, R.; Miyanaga, K.; Tanji, Y. Seasonal variations in bacterial communities and antibiotic-resistant strains associated with green bottle flies (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 2014, 98, 4197–4208. [Google Scholar] [CrossRef]
- Wohlfahrt, D.; Woolf, M.S.; Singh, B. A survey of bacteria associated with various life stages of primary colonizers: Lucilia sericata and Phormia regina. Sci. Justice 2020, 60, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Fonseca, A.; Liu, W.; Fields, A.T.; Pimsler, M.L.; Spindola, A.F.; Tarone, A.M.; Crippen, T.L.; Tomberlin, J.K.; Wood, T.K. Proteus mirabilis interkingdom swarming signals attract blow flies. ISME J. 2012, 6, 1356–1366. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.G.; Bueno, E.; Blow, F.; Douglas, A.E. Genome-inferred correspondence between phylogeny and metabolic traits in the wild Drosophila gut microbiome. Genome Biol. Evol. 2021, 13, evab127. [Google Scholar] [CrossRef] [PubMed]
- Nayduch, D. Special collection: Filth fly–microbe interactions. Ann. Entomol. Soc. Am. 2017, 110, 2–5. [Google Scholar] [CrossRef]
- Kaltenpoth, M. Actinobacteria as mutualists: General healthcare for insects? Trends Microbiol. 2009, 17, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Longnecker, M.; Tarone, A.M.; Tomberlin, J.K. Responses of Lucilia sericata (Diptera: Calliphoridae) to compounds from microbial decomposition of larval resources. Anim. Behav. 2016, 115, 217–225. [Google Scholar] [CrossRef]
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. Microbiologyopen 2018, 7, e00611. [Google Scholar] [CrossRef]
- Allali, I.; Arnold, J.W.; Roach, J.; Cadenas, M.B.; Butz, N.; Hassan, H.M.; Koci, M.; Ballou, A.; Mendoza, M.; Ali, R. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Diniz-Filho, J.A.F.; Soares, T.N.; Lima, J.S.; Dobrovolski, R.; Landeiro, V.L.; Telles, M.P.d.C.; Rangel, T.F.; Bini, L.M. Mantel test in population genetics. Genet. Mol. Biol. 2013, 36, 475–485. [Google Scholar] [CrossRef]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Matos, R.C.; Leulier, F. Lactobacilli-Host mutualism: “learning on the fly”. Microb. Cell Factories 2014, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kleerebezem, M.; Boekhorst, J.; Van Kranenburg, R.; Molenaar, D.; Kuipers, O.P.; Leer, R.; Tarchini, R.; Peters, S.A.; Sandbrink, H.M.; Fiers, M.W. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Nati. Acad. Sci. USA 2003, 100, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Han, G.; Kim, J.W.; Jeon, C.O.; Hyun, S. Taxon-specific effects of Lactobacillus on Drosophila host development. Microb. Ecol. 2020, 79, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Iancu, L.; Angelescu, I.R.; Paun, V.I.; Henríquez-Castillo, C.; Lavin, P.; Purcarea, C. Microbiome pattern of Lucilia sericata (Meigen) (Diptera: Calliphoridae) and feeding substrate in the presence of the foodborne pathogen Salmonella enterica. Sci. Rep. 2021, 11, 15296. [Google Scholar] [CrossRef] [PubMed]
- Cirimotich, C.M.; Ramirez, J.L.; Dimopoulos, G. Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe 2011, 10, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.J.; Oh, D.-C.; Yuceer, M.C.; Klepzig, K.D.; Clardy, J.; Currie, C.R. Bacterial protection of beetle-fungus mutualism. Science 2008, 322, 63. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.; Ferreira, Á.; Ashburner, M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 2008, 6, e1000002. [Google Scholar] [CrossRef] [PubMed]
- Kaltenpoth, M.; Göttler, W.; Herzner, G.; Strohm, E. Symbiotic bacteria protect wasp larvae from fungal infestation. Curr. Biol. 2005, 15, 475–479. [Google Scholar] [CrossRef]
- Brownlie, J.C.; Johnson, K.N. Symbiont-mediated protection in insect hosts. Trends Microbiol. 2009, 17, 348–354. [Google Scholar] [CrossRef]
- Su, W.; Liu, J.; Bai, P.; Ma, B.; Liu, W. Pathogenic fungi-induced susceptibility is mitigated by mutual Lactobacillus plantarum in the Drosophila melanogaster model. BMC Microbiol. 2019, 19, 302. [Google Scholar] [CrossRef]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Tomberlin, J.K.; Crippen, T.L.; Tarone, A.M.; Chaudhury, M.F.; Singh, B.; Cammack, J.A.; Meisel, R.P. A review of bacterial interactions with blow flies (Diptera: Calliphoridae) of medical, veterinary, and forensic importance. Ann. Entomol. Soc. Am. 2017, 110, 19–36. [Google Scholar] [CrossRef]
- De Smet, J.; Wynants, E.; Cos, P.; Van Campenhout, L. Microbial community dynamics during rearing of black soldier fly larvae (Hermetia illucens) and impact on exploitation potential. Appl. Environ. Microbiol. 2018, 84, e02717–e02722. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.R.; Brogan, R.S.; Scheifele, L.Z.; Rivers, D.B. Bacterial interactions with necrophagous flies. Ann. Entomol. Soc. Am. 2013, 106, 799–809. [Google Scholar] [CrossRef]
- Ridley, E.V.; Wong, A.C.; Westmiller, S.; Douglas, A.E. Impact of the resident microbiota on the nutritional phenotype of Drosophila melanogaster. PLoS ONE 2012, 7, e36765. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009, 23, 38–47. [Google Scholar] [CrossRef]
- Deguenon, J.M.; Travanty, N.; Zhu, J.; Carr, A.; Denning, S.; Reiskind, M.H.; Watson, D.W.; Michael Roe, R.; Ponnusamy, L. Exogenous and endogenous microbiomes of wild-caught Phormia regina (Diptera: Calliphoridae) flies from a suburban farm by 16S rRNA gene sequencing. Sci. Rep. 2019, 9, 20365. [Google Scholar] [CrossRef]
- Gibbons, S.M. Keystone taxa indispensable for microbiome recovery. Nat. Microbiol. 2020, 5, 1067–1068. [Google Scholar] [CrossRef]
- Singh, B.; Crippen, T.L.; Zheng, L.; Fields, A.T.; Yu, Z.; Ma, Q.; Wood, T.K.; Dowd, S.E.; Flores, M.; Tomberlin, J.K. A metagenomic assessment of the bacteria associated with Lucilia sericata and Lucilia cuprina (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 2015, 99, 869–883. [Google Scholar] [CrossRef]
- Chandler, J.A.; Morgan Lang, J.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial communities of diverse Drosophila species: Ecological context of a host–microbe model system. PLoS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Cram, J.A.; Needham, D.M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol. 2015, 13, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D.; Martiny, J.B. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef] [PubMed]
- Preston-Mafham, J.; Boddy, L.; Randerson, P.F. Analysis of microbial community functional diversity using sole-carbon-source utilisation profiles—A critique. FEMS Microbiol. Ecol. 2002, 42, 1–14. [Google Scholar] [PubMed]
- Pechal, J.L.; Crippen, T.L.; Tarone, A.M.; Lewis, A.J.; Tomberlin, J.K.; Benbow, M.E. Microbial community functional change during vertebrate carrion decomposition. PLoS ONE 2013, 8, e79035. [Google Scholar] [CrossRef] [PubMed]
- Malacrinò, A. Host species identity shapes the diversity and structure of insect microbiota. Mol. Ecol. 2022, 31, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Brucker, R.M.; Bordenstein, S.R. The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution 2012, 66, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.-N.; Dobson, A.J.; Douglas, A.E. Gut microbiota dictates the metabolic response of Drosophila to diet. J. Exp. Biol. 2014, 217, 1894–1901. [Google Scholar]
- Lim, S.h.; Park, J.K.; Park, W.B.; Won, D.H.; Kim, M.S.; Hong, S.; Do, Y. Gut microbiome of three species of Odonata. Entomol. Res. 2023, 53, 167–172. [Google Scholar] [CrossRef]
- Lee, Y.H. Overview of Mendelian randomization analysis. J. Rheum. Dis. 2020, 27, 241–246. [Google Scholar] [CrossRef]
- Liu, X.; Tong, X.; Zou, Y.; Lin, X.; Zhao, H.; Tian, L.; Jie, Z.; Wang, Q.; Zhang, Z.; Lu, H.; et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat. Genet. 2022, 54, 52–61. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, W.-B.; Park, J.-K.; Do, Y. Phylogenetic Constraints and Ecological Implications of Gut Bacterial Communities in Necrophagous Flies. Diversity 2023, 15, 970. https://doi.org/10.3390/d15090970
Park W-B, Park J-K, Do Y. Phylogenetic Constraints and Ecological Implications of Gut Bacterial Communities in Necrophagous Flies. Diversity. 2023; 15(9):970. https://doi.org/10.3390/d15090970
Chicago/Turabian StylePark, Woong-Bae, Jun-Kyu Park, and Yuno Do. 2023. "Phylogenetic Constraints and Ecological Implications of Gut Bacterial Communities in Necrophagous Flies" Diversity 15, no. 9: 970. https://doi.org/10.3390/d15090970
APA StylePark, W.-B., Park, J.-K., & Do, Y. (2023). Phylogenetic Constraints and Ecological Implications of Gut Bacterial Communities in Necrophagous Flies. Diversity, 15(9), 970. https://doi.org/10.3390/d15090970