
Mining NCBI Sequence Read Archive Database: An Untapped Source of Organelle Genomes for Taxonomic and Comparative Genomics Research

Abstract
:1. Introduction
2. Materials and Methods
2.1. Implementation
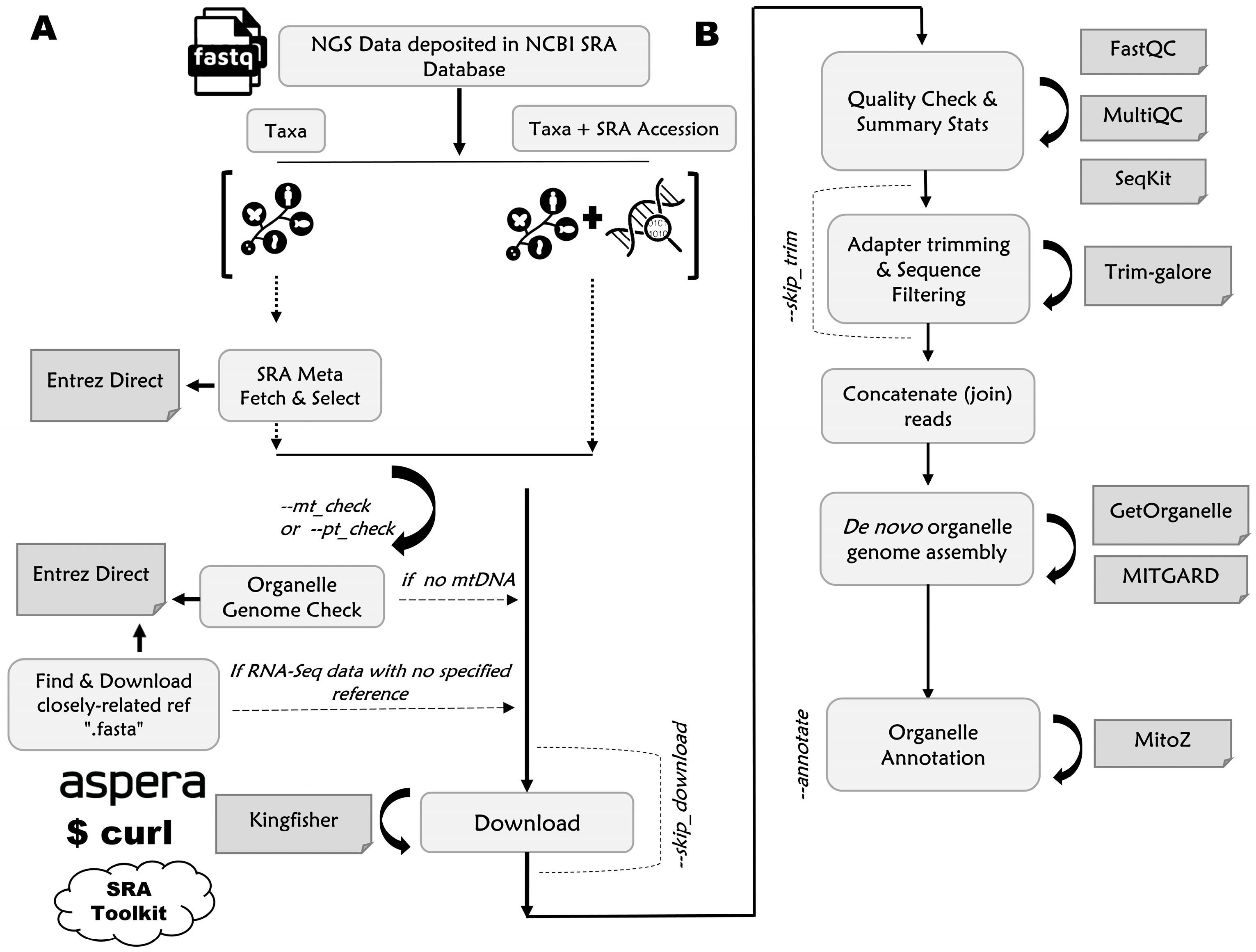
2.1.1. Input File Preparation
2.1.2. Downloading Unprocessed “.fastq” Files from SRA Database
2.1.3. Quality Assessment, De Novo Assembly, Annotation, and Outputting
2.2. Case Studies
2.3. Validation and Application of OrgaMiner Workflow across Diverse Clades
2.4. Code Availability
3. Results and Discussion
3.1. Sequence Characteristics of Datasets in Study
3.2. Molluscan mtDNA Annotation Findings and Implications
3.3. Applicability of OrgaMiner Workflow across Different Clades
3.4. Liminations of OrgaMiner Pipeline and Recommendations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lou, R.N.; Jacobs, A.; Wilder, A.P.; Therkildsen, N.O. A beginner’s guide to low-coverage whole genome sequencing for population genomics. Mol. Ecol. 2021, 30, 5966–5993. [Google Scholar] [CrossRef] [PubMed]
- Holmer, R.; van Velzen, R.; Geurts, R.; Bisseling, T.; de Ridder, D.; Smit, S. GeneNoteBook, a collaborative notebook for comparative genomics. Bioinformatics 2019, 35, 4779–4781. [Google Scholar] [CrossRef] [PubMed]
- Baxter, S.W.; Davey, J.W.; Johnston, J.S.; Shelton, A.M.; Heckel, D.G.; Jiggins, C.D.; Blaxter, M.L. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS ONE 2011, 6, e19315. [Google Scholar] [CrossRef] [PubMed]
- Berhe, M.; Dossa, K.; You, J.; Mboup, P.A.; Diallo, I.N.; Diouf, D.; Zhang, X.; Wang, L. Genome-wide association study and its applications in the non-model crop Sesamum indicum. BMC Plant Biol. 2021, 21, 283. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Liu, S.; Yao, J.; Zhang, Y.; Yuan, Z.; Jiang, C.; Chen, A.; Fu, Q.; Su, B.; Dunham, R.; et al. Transcriptome Display During Testicular Differentiation of Channel Catfish (Ictalurus punctatus) as Revealed by RNA-Seq Analysis. Biol. Reprod. 2016, 95, 19. [Google Scholar] [CrossRef] [PubMed]
- Eldem, V.; Zararsiz, G.; Erkan, M.; Bakir, Y. De novo assembly and comprehensive characterization of the skeletal muscle transcriptomes of the European anchovy (Engraulis encrasicolus). Mar. Genom. 2015, 20, 7–9. [Google Scholar] [CrossRef]
- Du, X.; Wang, B.; Liu, X.; Liu, X.; He, Y.; Zhang, Q.; Wang, X. Comparative transcriptome analysis of ovary and testis reveals potential sex-related genes and pathways in spotted knifejaw Oplegnathus punctatus. Gene 2017, 637, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Ubrihien, R.P.; Ezaz, T.; Taylor, A.M.; Stevens, M.M.; Krikowa, F.; Foster, S.; Maher, W.A. The response of Isidorella newcombi to copper exposure: Using an integrated biological framework to interpret transcriptomic responses from RNA-seq analysis. Aquat. Toxicol. 2017, 185, 183–192. [Google Scholar] [CrossRef]
- Liang, Q.; Dong, B.; Li, A.; Wu, L.; Zhang, Y.; Han, T.; Liu, X. scRNA-seq analysis reveals toxicity mechanisms in shrimp hemocytes subjected to nitrite stress. Chemosphere 2023, 316, 137853. [Google Scholar] [CrossRef]
- Jiao, L.; Dai, T.; Jin, M.; Sun, P.; Zhou, Q. Transcriptome Analysis of the Hepatopancreas in the Litopenaeus vannamei Responding to the Lead Stress. Biol. Trace Elem. Res. 2021, 199, 1100–1109. [Google Scholar] [CrossRef]
- Eldem, V.; Zararsiz, G.; Taşçi, T.; Duru, I.P.; Bakir, Y.; Erkan, M. Transcriptome analysis for non-model organism: Current status and best-practices. Appl. RNA-Seq Omics Strateg.-Microorg. Hum. Health 2017, 1, 1–19. [Google Scholar]
- Ghazi, S.; Bourgeois, S.; Gomariz, A.; Bugarski, M.; Haenni, D.; Martins, J.R.; Nombela-Arrieta, C.; Unwin, R.J.; Wagner, C.A.; Hall, A.M.; et al. Multiparametric imaging reveals that mitochondria-rich intercalated cells in the kidney collecting duct have a very high glycolytic capacity. Faseb. J. 2020, 34, 8510–8525. [Google Scholar] [CrossRef]
- Jurss, K.; Bastrop, R. The Function of Mitochondria-Rich Cells (Chloride Cells) in Teleost Gills. Rev. Fish Biol. Fish. 1995, 5, 235–255. [Google Scholar] [CrossRef]
- Magoffin, D.A. Ovarian theca cell. Int. J. Biochem. Cell B 2005, 37, 1344–1349. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Bell, E. Mitochondrial genomes in invertebrate animals. In Molecular Life Sciences: An Encyclopedic Reference; Springer: New York, NY, USA, 2014; pp. 1–8. [Google Scholar]
- Gorska-Andrzejak, J.; Stowers, R.S.; Borycz, J.; Kostyleva, R.; Schwarz, T.L.; Meinertzhagen, I.A. Mitochondria are redistributed in Drosophila photoreceptors lacking milton, a kinesin-associated protein. J. Comp. Neurol. 2003, 463, 372–388. [Google Scholar] [CrossRef]
- Tunholi, V.M.; Tunholi-Alves, V.M.; Santos, A.T.; Garcia, J.D.; Maldonado, A.; da-Silva, W.S.; Rodrigues, M.D.D.; Pinheiro, J. Evaluation of the mitochondrial system in the gonad-digestive gland complex of Biomphalaria glabrata (Mollusca, Gastropoda) after infection by Echinostoma paraensei (Trematoda, Echinostomatidae). J. Invertebr. Pathol. 2016, 136, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Lersten, N.R.; Curtis, J.D. Paraveinal Mesophyll in Calliandra Tweedii and C. emarginata (Leguminosae; Mimosoideae). Am. J. Bot. 1993, 80, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Procko, C.; Lee, T.; Borsuk, A.; Bargmann, B.O.R.; Dabi, T.; Nery, J.R.; Estelle, M.; Baird, L.; O’Connor, C.; Brodersen, C.; et al. Leaf cell-specific and single-cell transcriptional profiling reveals a role for the palisade layer in UV light protection. Plant Cell 2022, 34, 3261–3279. [Google Scholar] [CrossRef]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Duminil, J.; Besnard, G. Utility of the Mitochondrial Genome in Plant Taxonomic Studies. Methods Mol. Biol. 2021, 2222, 107–118. [Google Scholar]
- Shinzato, C.; Narisoko, H.; Nishitsuji, K.; Nagata, T.; Satoh, N.; Inoue, J. Novel Mitochondrial DNA Markers for Scleractinian Corals and Generic-Level Environmental DNA Metabarcoding. Front. Mar. Sci. 2021, 8, 758207. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, W.; Hua, W.; Liu, J. A large-scale population based organelle pan-genomes construction and phylogeny analysis reveal the genetic diversity and the evolutionary origins of chloroplast and mitochondrion in Brassica napus L. Bmc Genom. 2022, 23, 339. [Google Scholar] [CrossRef]
- Toparslan, E.; Karabag, K.; Bilge, U. A workflow with R: Phylogenetic analyses and visualizations using mitochondrial cytochrome b gene sequences. PLoS ONE 2020, 15, e0243927. [Google Scholar] [CrossRef]
- Cay, S.B.; Cinar, Y.U.; Kuralay, S.C.; Inal, B.; Zararsiz, G.; Ciftci, A.; Mollman, R.; Obut, O.; Eldem, V.; Bakir, Y.; et al. Genome skimming approach reveals the gene arrangements in the chloroplast genomes of the highly endangered Crocus L. species: Crocus istanbulensis (B.Mathew) Rukans. PLoS ONE 2022, 17, e0269747. [Google Scholar] [CrossRef]
- Nachtigall, P.G.; Grazziotin, F.G.; Junqueira-de-Azevedo, I.L.M. MITGARD: An automated pipeline for mitochondrial genome assembly in eukaryotic species using RNA-seq data. Brief. Bioinform. 2021, 22, bbaa429. [Google Scholar] [CrossRef]
- Smith, D.R. RNA-Seq data: A goldmine for organelle research. Brief. Funct. Genom. 2013, 12, 454–456. [Google Scholar] [CrossRef]
- Osuna-Mascaro, C.; de Casas, R.R.; Perfectti, F. Comparative assessment shows the reliability of chloroplast genome assembly using RNA-seq. Sci. Rep. 2018, 8, 17404. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic. Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Pouchon, C.; Boyer, F.; Roquet, C.; Denoeud, F.; Chave, J.; Coissac, E.; Alsos, I.G.; Lavergne, S.; Consortium, P.; Consortium, P. ORTHOSKIM: In silico sequence capture from genomic and transcriptomic libraries for phylogenomic and barcoding applications. Mol. Ecol. Resour. 2022, 22, 2018–2037. [Google Scholar] [CrossRef]
- White, O.; Hall, A.; Clark, M.; Williams, S. go_batch: A snakemake pipeline to assemble mitochondrial and ribosomal sequences from genome skims. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cai, L.M.; Zhang, H.R.; Davis, C.C. PhyloHerb: A high-throughput phylogenomic pipeline for processing genome skimming data. Appl. Plant Sci. 2022, 10, e11475. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Senthilkumar, S.; Ulaganathan, K.; Dasgupta, M.G. Reference-based assembly of chloroplast genome from leaf transcriptome data of Pterocarpus santalinus. 3 Biotech 2021, 11, 393. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Qu, J.Y.; Xu, Y.R.; Cui, Y.T.; Wu, S.; Wang, L.J.; Liu, X.M.; Xing, Z.K.; Guo, X.Y.; Wang, S.S.; Li, R.R.; et al. MODB: A comprehensive mitochondrial genome database for Mollusca. Database 2021, 2021, baab056. [Google Scholar] [CrossRef]
- Gomes-dos-Santos, A.; Lopes-Lima, M.; Castro, L.F.C.; Froufe, E. Molluscan genomics: The road so far and the way forward. Hydrobiologia 2020, 847, 1705–1726. [Google Scholar] [CrossRef]
- Uribe, J.E.; Sei, M.; Harasewych, M.G. The mitogenome of the sunken wood limpet: Insights into mitogenome evolution in Lepetellida (Gastropoda: Vetigastropoda). J. Mollus. Stud. 2022, 88, eyac009. [Google Scholar] [CrossRef]
- Rempel, E.M.; Marcus, J.M.; Detwiler, J.T. The complete mitochondrial genome of the file ramshorn snail (Mollusca: Gastropoda: Hygrophila: Planorbidae). Mitochondrial DNA B 2021, 6, 3181–3183. [Google Scholar] [CrossRef]
- Feng, J.; Guo, Y.; Yan, C.; Ye, Y.; Li, J.; Guo, B.; Lu, Z. Sequence comparison of the mitochondrial genomes in two species of the genus Nerita (Gastropoda: Neritimorpha: Neritidae): Phylogenetic implications and divergence time estimation for Neritimorpha. Mol. Biol. Rep. 2020, 47, 7903–7916. [Google Scholar] [CrossRef]
- Ghiselli, F.; Gomes-Dos-Santos, A.; Adema, C.M.; Lopes-Lima, M.; Sharbrough, J.; Boore, J.L. Molluscan mitochondrial genomes break the rules. Philos. Trans. R Soc. Lond B Biol. Sci. 2021, 376, 20200159. [Google Scholar] [CrossRef] [PubMed]
- Moreyra, N.N.; Mensch, J.; Hurtado, J.; Almeida, F.; Laprida, C.; Hasson, E. What does mitogenomics tell us about the evolutionary history of the cluster (group)? PLoS ONE 2019, 14, e0220676. [Google Scholar] [CrossRef]
- Grande, C.; Templado, J.; Zardoya, R. Evolution of gastropod mitochondrial genome arrangements. Bmc Evol. Biol. 2008, 8, 61. [Google Scholar] [CrossRef]
- Malkócs, T.; Viricel, A.; Becquet, V.; Evin, L.; Dubillot, E.; Pante, E. Complex mitogenomic rearrangements within the Pectinidae (Mollusca: Bivalvia). Bmc Ecol. Evol. 2022, 22, 29. [Google Scholar] [CrossRef]
- He, C.B.; Wang, J.; Gao, X.G.; Song, W.T.; Li, H.J.; Li, Y.F.; Liu, W.D.; Su, H. The complete mitochondrial genome of the hard clam Meretrix meretrix. Mol. Biol. Rep. 2011, 38, 3401–3409. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Kong, L.F.; Sasaki, T.; Li, Q. Phylogenomic resolution of Imparidentia (Mollusca: Bivalvia) diversification through mitochondrial genomes. Mar. Life Sci. Tech. 2023, 5, 326–336. [Google Scholar] [CrossRef]
- Li, F.P.; Liu, H.Y.; Heng, X.; Zhang, Y.; Fan, M.F.; Wang, S.S.; Liu, C.S.; Gu, Z.F.; Wang, A.M.; Yang, Y. The complete mitochondrial genome of (Bivalvia, Ostreoidea) indicates the genetic diversity within Gryphaeidae. Biodivers Data J. 2023, 11, e101333. [Google Scholar] [CrossRef]
- Gaitán-Espitia, J.D.; Quintero-Galvis, J.F.; Mesas, A.; D’Elía, G. Mitogenomics of southern hemisphere blue mussels (Bivalvia: Pteriomorphia): Insights into the evolutionary characteristics of the complex. Sci. Rep. 2016, 6, 26853. [Google Scholar] [CrossRef] [PubMed]
- Kinkar, L.; Gasser, R.B.; Webster, B.L.; Rollinson, D.; Littlewood, D.T.J.; Chang, B.C.H.; Stroehlein, A.J.; Korhonen, P.K.; Young, N.D. Nanopore Sequencing Resolves Elusive Long Tandem-Repeat Regions in Mitochondrial Genomes. Int. J. Mol. Sci. 2021, 22, 1811. [Google Scholar] [CrossRef]
- Uliano-Silva, M.; Ferreira, J.G.R.N.; Krasheninnikova, K.; Formenti, G.; Abueg, L.; Torrance, J.; Myers, E.W.; Durbin, R.; Blaxter, M.; McCarthy, S.A.; et al. MitoHiFi: A python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. Bmc Bioinform. 2023, 24, 288. [Google Scholar] [CrossRef]
- Cantatore, P.; Gadaleta, M.N.; Roberti, M.; Saccone, C.; Wilson, A.C. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 1987, 329, 853–855. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Tomecki, R.; Dmochowska, A.; Gewartowski, K.; Dziembowski, A.; Stepien, P.P. Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic. Acids Res. 2004, 32, 6001–6014. [Google Scholar] [CrossRef] [PubMed]
- Kühn, U.; Gündel, M.; Knoth, A.; Kerwitz, Y.; Rüdel, S.; Wahle, E. Poly(A) Tail Length Is Controlled by the Nuclear Poly(A)-binding Protein Regulating the Interaction between Poly(A) Polymerase and the Cleavage and Polyadenylation Specificity Factor. J. Biol. Chem. 2009, 284, 22803–22814. [Google Scholar] [CrossRef] [PubMed]
- Honarmand, S.; Shoubridge, E.A. Poly (A) tail length of human mitochondrial mRNAs is tissue-specific and a mutation in LRPPRC results in transcript-specific patterns of deadenylation. Mol. Genet. Metab. Rep. 2020, 25, 100687. [Google Scholar] [CrossRef]
- Qu, X.J.; Zou, D.; Zhang, R.Y.; Stull, G.W.; Yi, T.S. Progress, challenge and prospect of plant plastome annotation. Front. Plant Sci. 2023, 14, 1166140. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Class | Family | Species | Coding Genes | tRNA Genes | rRNA Genes | Missing Genes | Total Genes | mtDNA Length |
|---|---|---|---|---|---|---|---|---|
| Bivalvia | Mytilidae | Botula fusca | 12 | 21 | 2 | 2 | 35 | 19,595 |
| Bivalvia | Unionidae | Elliptio hopetonensis | 13 | 22 | 2 | 0 | 37 | 15,775 |
| Bivalvia | Tellinidae | Macoma nasuta | 12 | 22 | 2 | 1 | 36 | 17,348 |
| Bivalvia | Unionidae | Megalonaias nervosa | 13 | 22 | 2 | 0 | 37 | 16,026 |
| Bivalvia | Anomiidae | Pododesmus macrochisma | 13 | 22 | 2 | 0 | 37 | 15,080 |
| Bivalvia | Veneridae | Saxidomus gigantea | 13 | 22 | 2 | 0 | 37 | 19,754 |
| Cephalopoda | Octopodidae | Muusoctopus eicomar | 13 | 22 | 2 | 0 | 37 | 16,168 |
| Cephalopoda | Octopodidae | Muusoctopus leioderma * | 13 | 22 | 2 | 0 | 37 | 17,006 |
| Cephalopoda | Octopodidae | Muusoctopus longibrachus | 13 | 22 | 2 | 0 | 37 | 16,192 |
| Cephalopoda | Octopodidae | Octopus americanus | 13 | 22 | 2 | 0 | 37 | 15,655 |
| Cephalopoda | Octopodidae | Amphioctopus burryi | 13 | 22 | 2 | 0 | 37 | 15,883 |
| Cephalopoda | Sepiolidae | Rondeletiola minor | 13 | 22 | 2 | 0 | 37 | 15,800 |
| Cephalopoda | Loliginidae | Doryteuthis pealeii | 13 | 21 | 2 | 1 | 36 | 16,674 |
| Gastropoda | Onchidorididae | Corambe burchi | 13 | 22 | 2 | 0 | 37 | 14,308 |
| Gastropoda | Neomphalidae | Cyathermia naticoides * | 13 | 22 | 2 | 0 | 37 | 16,156 |
| Gastropoda | Ovulidae | Cyphoma gibbosum | 13 | 20 | 2 | 2 | 35 | 16,638 |
| Gastropoda | Dironidae | Dirona albolineata | 13 | 23 | 2 | 0 | 38 | 14,651 |
| Gastropoda | Dorididae | Doris verrucosa * | 13 | 22 | 2 | 0 | 37 | 14,518 |
| Gastropoda | Plakobranchidae | Elysia diomedea | 13 | 21 | 2 | 1 | 36 | 14,158 |
| Gastropoda | Chromodorididae | Goniobranchus kuniei | 13 | 23 | 2 | 0 | 38 | 14,738 |
| Gastropoda | Haliotidae | Haliotis corrugata | 13 | 22 | 2 | 0 | 37 | 16,951 |
| Gastropoda | Haliotidae | Haliotis discus discus | 13 | 22 | 2 | 0 | 37 | 16,805 |
| Gastropoda | Haliotidae | Haliotis fulgens | 13 | 22 | 2 | 0 | 37 | 16,376 |
| Gastropoda | Haliotidae | Haliotis gigantea | 13 | 22 | 2 | 0 | 37 | 16,539 |
| Gastropoda | Haliotidae | Haliotis kamtschatkana | 13 | 22 | 2 | 0 | 37 | 16,892 |
| Gastropoda | Haliotidae | Haliotis madaka | 13 | 22 | 2 | 0 | 37 | 16,745 |
| Gastropoda | Haliotidae | Haliotis midae | 13 | 22 | 2 | 0 | 37 | 16,530 |
| Gastropoda | Haliotidae | Haliotis sorenseni | 13 | 22 | 2 | 0 | 37 | 16,711 |
| Gastropoda | Glaucidae | Hermissenda crassicornis | 13 | 21 | 2 | 1 | 36 | 14,750 |
| Gastropoda | Lepetodrilidae | Lepetodrilus galriftensis * | 13 | 22 | 2 | 0 | 37 | 19,339 |
| Gastropoda | Lepetodrilidae | Lepetodrilus gordensis | 13 | 22 | 2 | 0 | 37 | 16,455 |
| Gastropoda | Littorinidae | Littorina arcana | 13 | 22 | 2 | 0 | 37 | 16,301 |
| Gastropoda | Littorinidae | Littorina compressa | 13 | 22 | 2 | 0 | 37 | 16,349 |
| Gastropoda | Lottiidae | Lottia persona | 12 | 22 | 2 | 1 | 36 | 17,106 |
| Gastropoda | Peltospiridae | Peltospira delicata | 13 | 23 | 2 | 0 | 38 | 15,523 |
| Gastropoda | Tateidae | Potamopyrgus kaitunuparaoa | 13 | 23 | 2 | 0 | 38 | 15,332 |
| Gastropoda | Lepetodrilidae | Pseudorimula midatlantica * | 13 | 22 | 2 | 0 | 37 | 16,411 |
| Gastropoda | Lymnaeidae | Radix swinhoei | 13 | 19 | 2 | 3 | 34 | 14,998 |
| Gastropoda | Scyllaeidae | Scyllaea pelagica | 13 | 22 | 2 | 0 | 37 | 14,759 |
| Gastropoda | Lymnaeidae | Stagnicola palustris | 12 | 21 | 2 | 2 | 35 | 14,261 |
| Gastropoda | Tritoniidae | Tritonia tetraquetra | 13 | 22 | 2 | 0 | 37 | 15,087 |
| Gastropoda | Tylodinidae | Tylodina fungina | 13 | 21 | 2 | 2 | 36 | 14,649 |
| Gastropoda | Aegiretidae | Aegires albopunctatus | 13 | 21 | 3 | 1 | 37 | 13,947 |
| Gastropoda | Aeolidiidae | Aeolidia papillosa | 12 | 22 | 2 | 2 | 36 | 16,696 |
| Gastropoda | Goniodorididae | Ancula gibbosa | 13 | 22 | 2 | 0 | 37 | 14,532 |
| Polyplacophora | Mopaliidae | Mopalia ciliata * | 13 | 21 | 2 | 1 | 36 | 13,987 |
| Polyplacophora | Mopaliidae | Mopalia kennerleyi * | 13 | 22 | 2 | 0 | 37 | 14,290 |
| Polyplacophora | Mopaliidae | Mopalia muscosa * | 13 | 22 | 2 | 0 | 37 | 14,976 |
| Polyplacophora | Mopaliidae | Mopalia swanii * | 13 | 22 | 2 | 0 | 37 | 14,969 |
| Polyplacophora | Mopaliidae | Mopalia vespertina * | 13 | 22 | 2 | 0 | 37 | 14,987 |
| Polyplacophora | Chitonidae | Acanthopleura granulata | 13 | 22 | 2 | 0 | 37 | 15,618 |
| Solenogastres | Gymnomeniidae | Wirenia argentea | 13 | 20 | 2 | 2 | 35 | 16,443 |
| Class | Family | Species | Coding Genes | tRNA Genes | rRNA Genes | Missing Genes | Total Genes | mtDNA Length |
|---|---|---|---|---|---|---|---|---|
| Bivalvia | Mactridae | Mactra antiquata | 13 | 18 | 2 | 5 | 33 | 16,429 |
| Bivalvia | Unionidae | Uniomerus tetralasmus | 13 | 18 | 2 | 4 | 33 | 15,247 |
| Bivalvia | Mytilidae | Mytilus planulatus | 13 | 22 | 2 | 1 | 37 | 16,727 |
| Bivalvia | Vesicomyidae | Archivesica packardana † | 14 | 22 | 2 | 0 | 38 | 16,467 |
| Bivalvia | Pharidae | Ensis directus | 13 | 23 | 2 | 0 | 38 | 16,925 |
| Bivalvia | Ostreidae | Saccostrea palmula | 13 | 19 | 2 | 4 | 34 | 16,130 |
| Bivalvia | Mytilidae | Gigantidas horikoshii | 12 | 20 | 2 | 3 | 34 | 17,504 |
| Bivalvia | Thyasiridae | Conchocele bisecta | 12 | 22 | 2 | 2 | 36 | 17,181 |
| Cephalopoda | Sepiolidae | Rossia pacifica † | 14 | 18 | 2 | 4 | 34 | 14,897 |
| Cephalopoda | Octopodidae | Enteroctopus megalocyathus † | 14 | 20 | 2 | 2 | 36 | 16,027 |
| Gastropoda | Ranellidae | Monoplex corrugatus | 13 | 18 | 2 | 4 | 33 | 16,178 |
| Gastropoda | Planorbidae | Biomphalaria alexandrina | 13 | 19 | 2 | 3 | 34 | 13,570 |
| Gastropoda | Conidae | Conus ammiralis | 13 | 19 | 2 | 3 | 34 | 15,459 |
| Gastropoda | Conidae | Conus purpurascens | 13 | 19 | 2 | 3 | 34 | 15,509 |
| Gastropoda | Facelinidae | Facelina rubrovittata | 13 | 19 | 2 | 3 | 34 | 14,481 |
| Gastropoda | Chromodorididae | Verconia verconis | 13 | 19 | 2 | 3 | 34 | 14,560 |
| Gastropoda | Nacellidae | Cellana rota † | 14 | 19 | 2 | 3 | 35 | 16,042 |
| Gastropoda | Semisulcospiridae | Semisulcospira reiniana | 13 | 20 | 2 | 4 | 35 | 15,291 |
| Gastropoda | Tritoniidae | Tritoniopsis frydis † | 13 | 20 | 2 | 5 | 35 | 14,481 |
| Gastropoda | Turbinidae | Angaria nodosa | 14 | 20 | 2 | 3 | 36 | 19,389 |
| Gastropoda | Conidae | Conus bayani † | 14 | 20 | 2 | 2 | 36 | 15,525 |
| Gastropoda | Nacellidae | Nacella polaris | 13 | 21 | 2 | 1 | 36 | 16,752 |
| Gastropoda | Conidae | Conus chaldaeus | 13 | 22 | 2 | 0 | 37 | 15,442 |
| Polyplacophora | Chitonidae | Tonicia schrammi | 14 | 18 | 2 | 4 | 34 | 14,909 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eldem, V.; Balcı, M.A. Mining NCBI Sequence Read Archive Database: An Untapped Source of Organelle Genomes for Taxonomic and Comparative Genomics Research. Diversity 2024, 16, 104. https://doi.org/10.3390/d16020104
Eldem V, Balcı MA. Mining NCBI Sequence Read Archive Database: An Untapped Source of Organelle Genomes for Taxonomic and Comparative Genomics Research. Diversity. 2024; 16(2):104. https://doi.org/10.3390/d16020104
Chicago/Turabian StyleEldem, Vahap, and Mehmet Ali Balcı. 2024. "Mining NCBI Sequence Read Archive Database: An Untapped Source of Organelle Genomes for Taxonomic and Comparative Genomics Research" Diversity 16, no. 2: 104. https://doi.org/10.3390/d16020104