
Resolving Rapid Radiation of Voles and Lemmings (Arvicolinae: Cricetinae, Rodentia) with QuaddRAD Sequencing and Transcriptome Analysis

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA and RNA Isolation, Library Preparation, and Sequencing
2.3. Phylogenetic-Tree Reconstruction Using the Transcriptomic Dataset
2.4. Phylogenetic-Tree Reconstruction Using the ddRADseq Dataset
2.5. Molecular Dating
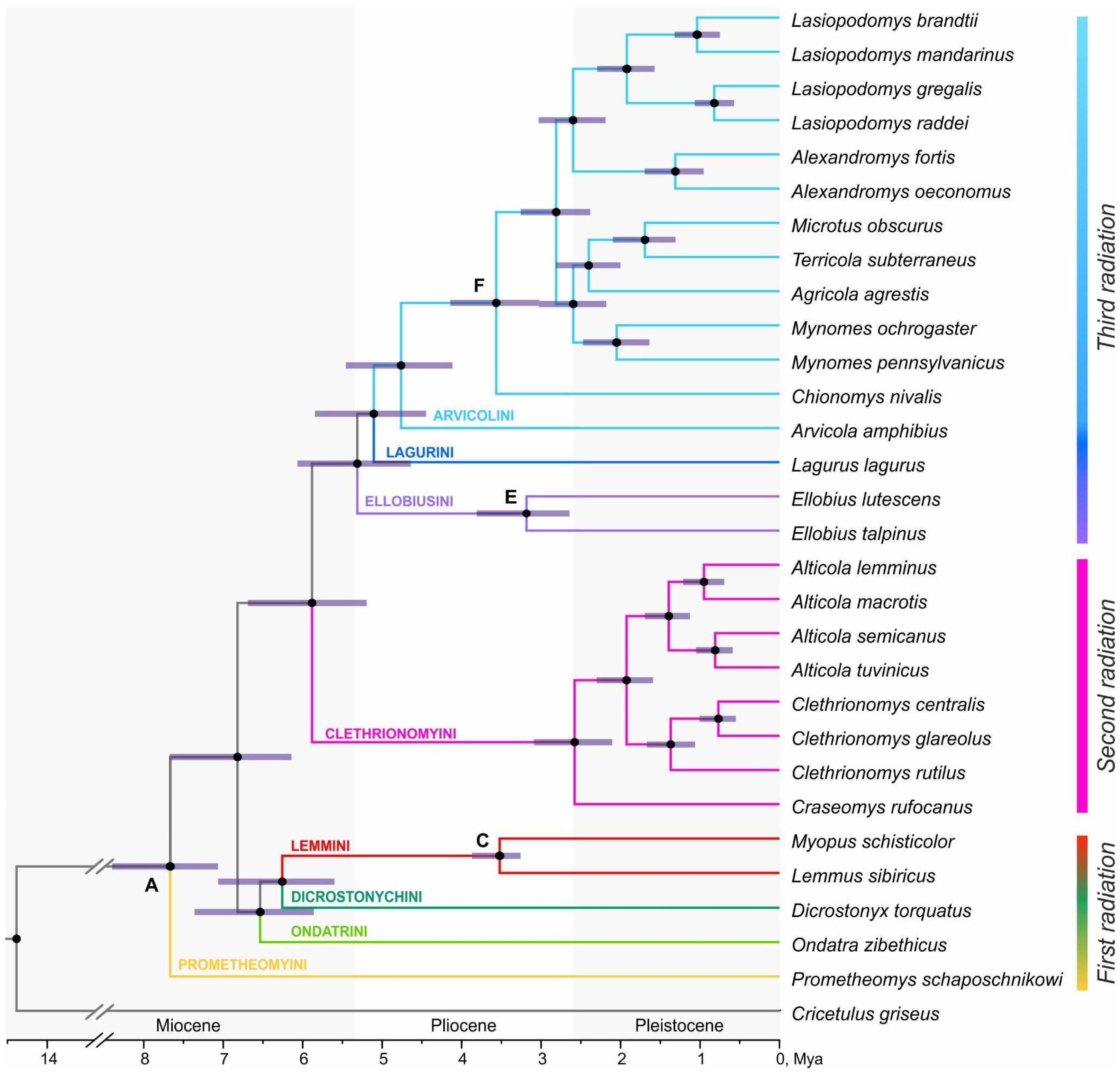
3. Results
3.1. Transcriptome Sequencing and Assembly
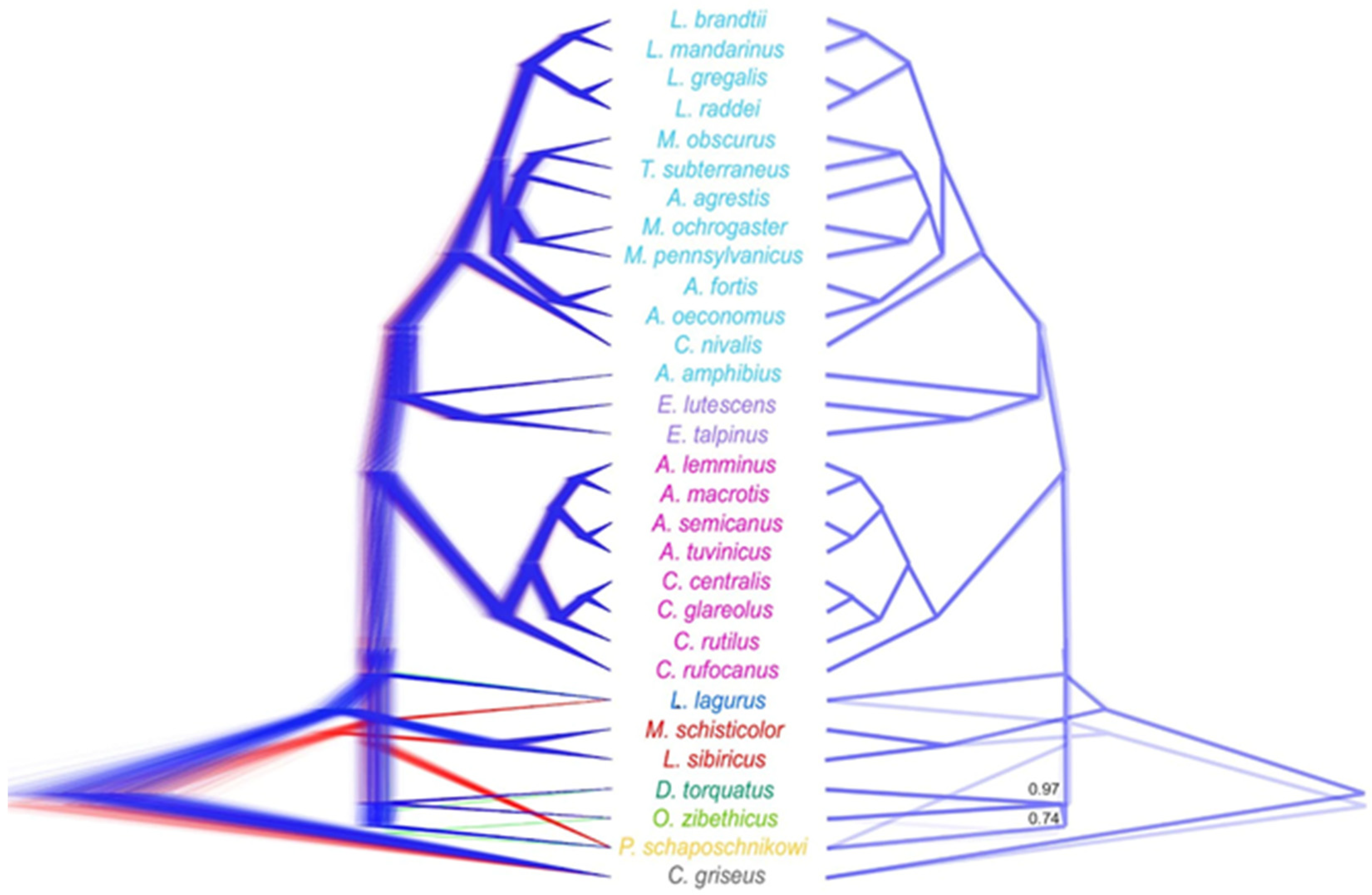
3.2. Phylogenetic Inference from the Transcriptomic Dataset
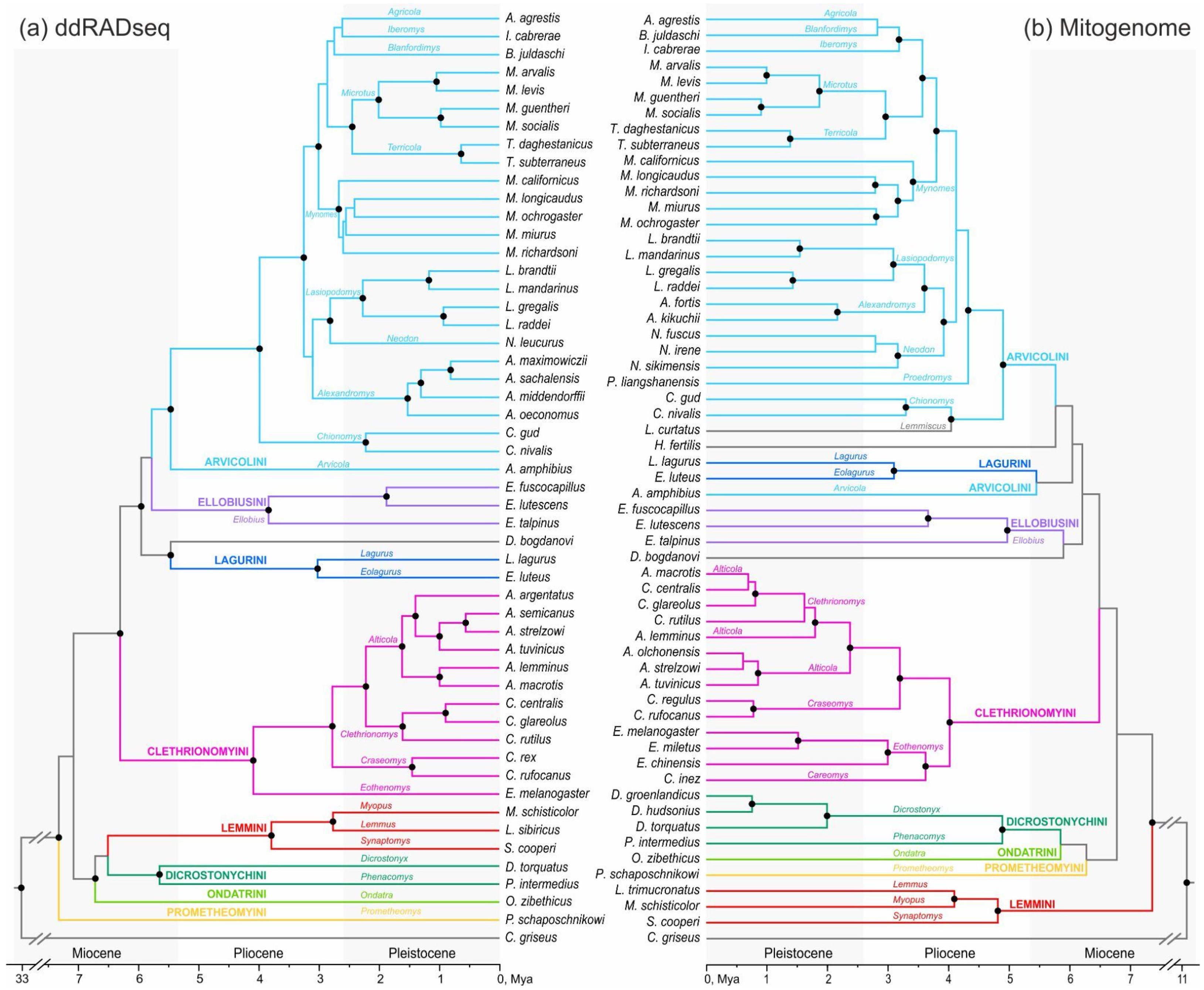
3.3. DdRADseq-Based Phylogenetic Reconstruction
3.4. Divergence Dating
4. Discussion
4.1. Features of the Different Radiation Waves Within Arvicolinae
4.2. Phylogenetic Relations of Supraspecies Taxa Within the First Radiation
4.3. Phylogenetic Relations Within the Third Radiation
4.4. Comparison of Divergence Dating Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fejfar, O.; Heinrich, W.-D.; Kordos, L.; Maul, L.C. Microtoid Cricetids and the Early History of Arvicolids (Mammalia, Rodentia). Palaeontol. Electron. 2011, 14, 1–38. [Google Scholar]
- Martin, R.A. Arvicolidae. In Evolution of Tertiary Mammals of North America; Janis, C.M., Gunnell, G.F., Uhen, M.D., Eds.; Cambridge University Press: Cambridge, UK, 2007; Volume 2, pp. 480–497. [Google Scholar]
- Conroy, C.J.; Cook, J.A. MtDNA Evidence for Repeated Pulses of Speciation Within Arvicoline and Murid Rodents. J. Mamm. Evol. 1999, 6, 221–245. [Google Scholar] [CrossRef]
- Abramson, N.I.; Lebedev, V.S.; Tesakov, A.S.; Bannikova, A.A. Supraspecies Relationships in the Subfamily Arvicolinae (Rodentia, Cricetidae): An Unexpected Result of Nuclear Gene Analysis. Mol. Biol. 2009, 43, 834–846. [Google Scholar] [CrossRef]
- Fabre, P.-H.; Hautier, L.; Dimitrov, D.; P Douzery, E.J. A Glimpse on the Pattern of Rodent Diversification: A Phylogenetic Approach. BMC Evol. Biol. 2012, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Steppan, S.J.; Schenk, J.J. Muroid Rodent Phylogenetics: 900-Species Tree Reveals Increasing Diversification Rates. PLoS ONE 2017, 12, e0183070. [Google Scholar] [CrossRef] [PubMed]
- Abramson, N.I.; Bodrov, S.Y.; Bondareva, O.V.; Genelt-Yanovskiy, E.A.; Petrova, T.V. A Mitochondrial Genome Phylogeny of Voles and Lemmings (Rodentia: Arvicolinae): Evolutionary and Taxonomic Implications. PLoS ONE 2021, 16, e0248198. [Google Scholar] [CrossRef] [PubMed]
- Susko, E.; Roger, A.J. Long Branch Attraction Biases in Phylogenetics. Syst. Biol. 2021, 70, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Koufos, G.D. The Villafranchian Mammalian Faunas and Biochronology of Greece. Boll. Della Soc. Paleontol. Ital. 2001, 40, 217–223. [Google Scholar]
- Jánossy, D. Pleistocene Vertebrate Faunas of Hungary; Developments in Palaeontology and Stratigraphy; Elsevier: Amsterdam, The Netherlands, 1986; ISBN 978-0-08-086849-3. [Google Scholar]
- Withnell, C.B.; Scarpetta, S.G. A New Perspective on the Taxonomy and Systematics of Arvicolinae (Gray, 1821) and a New Time-Calibrated Phylogeny for the Clade. PeerJ 2024, 12, e16693. [Google Scholar] [CrossRef]
- Thompson, R.S.; Fleming, R.F. Middle Pliocene Vegetation: Reconstructions, Paleoclimatic Inferences, and Boundary Conditions for Climate Modeling. Mar. Micropaleontol. 1996, 27, 27–49. [Google Scholar] [CrossRef]
- Kohli, B.A.; Speer, K.A.; Kilpatrick, C.W.; Batsaikhan, N.; Damdinbaza, D.; Cook, J.A. Multilocus Systematics and Non-Punctuated Evolution of Holarctic Myodini (Rodentia: Arvicolinae). Mol. Phylogenet. Evol. 2014, 76, 18–29. [Google Scholar] [CrossRef]
- Buzan, E.V.; Krystufek, B.; Hänfling, B.; Hutchinson, W.F. Mitochondrial Phylogeny of Arvicolinae Using Comprehensive Taxonomic Sampling Yields New Insights: Phylogeny of Arvicolinae. Biol. J. Linn. Soc. 2008, 94, 825–835. [Google Scholar] [CrossRef]
- Bodrov, S.Y.; Kostygov, A.Y.; Rudneva, L.V.; Abramson, N.I. Revision of the Taxonomic Position of the Olkhon Mountain Vole (Rodentia, Cricetidae). Biol. Bull. 2016, 43, 136–145. [Google Scholar] [CrossRef]
- Tryfonopoulos, G.; Thanou, E.; Chondropoulos, B.; Fraguedakis-Tsolis, S. mtDNA Analysis Reveals the Ongoing Speciation on Greek Populations of Microtus (Terricola) Thomasi (Arvicolidae, Rodentia): Ongoing Speciation in Greek Voles. Biol. J. Linn. Soc. 2008, 95, 117–130. [Google Scholar] [CrossRef]
- Paupério, J.; Herman, J.S.; Melo-Ferreira, J.; Jaarola, M.; Alves, P.C.; Searle, J.B. Cryptic Speciation in the Field Vole: A Multilocus Approach Confirms Three Highly Divergent Lineages in Eurasia. Mol. Ecol. 2012, 21, 6015–6032. [Google Scholar] [CrossRef] [PubMed]
- Musser, G.G.; Carleton, M.D. Superfamily Muroidea. In Mammal Species of the World: A Taxonomic and Geographic Reference; Wilson, D.E., Reeder, D.M., Eds.; The Johns Hopkins University Press: Baltimore, MD, USA, 2005; pp. 894–1531. [Google Scholar]
- Abramson, N.I.; Lissovsky, A.A. Subfamily Arvicolinae. In The Mammals of Russia: A Taxonomic and Geographic Reference; Pavlinov, I.Y., Lissovsky, A.A., Eds.; KMK Scientific Press: Moscow, Russia, 2012; pp. 127–141. [Google Scholar]
- Romanenko, S.A.; Serdyukova, N.A.; Perelman, P.L.; Trifonov, V.A.; Golenishchev, F.N.; Bulatova, N.S.; Stanyon, R.; Graphodatsky, A.S. Multiple Intrasyntenic Rearrangements and Rapid Speciation in Voles. Sci. Rep. 2018, 8, 14980. [Google Scholar] [CrossRef] [PubMed]
- Kryštufek, B.; Shenbrot, G. Voles and Lemmings (Arvicolinae) of the Palaearctic Region; University of Maribor, University Press: Maribor, Slovenia, 2022; ISBN 978-961-286-611-2. [Google Scholar]
- De Queiroz, K.; Gauthier, J. Phylogenetic Taxonomy. Annu. Rev. Ecol. Syst. 1992, 23, 449–480. [Google Scholar] [CrossRef]
- De Queiroz, K.; Gauthier, J. Toward a Phylogenetic System of Biological Nomenclature. Trends Ecol. Evol. 1994, 9, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Galewski, T.; Tilak, M.; Sanchez, S.; Chevret, P.; Paradis, E.; Douzery, E.J. The Evolutionary Radiation of Arvicolinae Rodents (Voles and Lemmings): Relative Contribution of Nuclear and Mitochondrial DNA Phylogenies. BMC Evol. Biol. 2006, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A.; Bonillo, C.; Tshikung, D.; Pongombo Shongo, C.; Pourrut, X.; Kadjo, B.; Nakouné, E.; Tu, V.T.; Prié, V.; Goodman, S.M. Phylogeny of African Fruit Bats (Chiroptera, Pteropodidae) Based on Complete Mitochondrial Genomes. J. Zool. Syst. Evol. Res. 2020, 58, 1395–1410. [Google Scholar] [CrossRef]
- Hassanin, A.; Veron, G.; Ropiquet, A.; Jansen Van Vuuren, B.; Lécu, A.; Goodman, S.M.; Haider, J.; Nguyen, T.T. Evolutionary History of Carnivora (Mammalia, Laurasiatheria) Inferred from Mitochondrial Genomes. PLoS ONE 2021, 16, e0240770. [Google Scholar] [CrossRef]
- Kundu, S.; Kumar, V.; Tyagi, K.; Chandra, K. The Complete Mitochondrial Genome of the Endangered Assam Roofed Turtle, Pangshura Sylhetensis (Testudines: Geoemydidae): Genomic Features and Phylogeny. PLoS ONE 2020, 15, e0225233. [Google Scholar] [CrossRef]
- Barnett, R.; Larson, G. A Phenol–Chloroform Protocol for Extracting DNA from Ancient Samples. In Ancient DNA; Shapiro, B., Hofreiter, M., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 840, pp. 13–19. ISBN 978-1-61779-515-2. [Google Scholar]
- Wallis, G.P.; Cameron-Christie, S.R.; Kennedy, H.L.; Palmer, G.; Sanders, T.R.; Winter, D.J. Interspecific Hybridization Causes Long-term Phylogenetic Discordance between Nuclear and Mitochondrial Genomes in Freshwater Fishes. Mol. Ecol. 2017, 26, 3116–3127. [Google Scholar] [CrossRef] [PubMed]
- Dvoyashov, I.A.; Bodrov, S.Y.; Mamaev, N.V.; Glagoleva, E.S.; Abramson, N.I. Inferring Phylogenetic Structure, Taxa Hybridization, and Divergence Times within Rock Voles of Subgenus Aschizomys (Cricetidae: Alticola) Using QuaddRAD Sequencing and a Cytb Dataset. Ecol. Evol. 2023, 13, e10742. [Google Scholar] [CrossRef]
- Da Fonseca, R.R.; Johnson, W.E.; O’Brien, S.J.; Ramos, M.J.; Antunes, A. The Adaptive Evolution of the Mammalian Mitochondrial Genome. BMC Genom. 2008, 9, 119. [Google Scholar] [CrossRef]
- Romero, P.E.; Weigand, A.M.; Pfenninger, M. Positive Selection on Panpulmonate Mitogenomes Provide New Clues on Adaptations to Terrestrial Life. BMC Evol. Biol. 2016, 16, 164. [Google Scholar] [CrossRef] [PubMed]
- Strohm, J.H.T.; Gwiazdowski, R.A.; Hanner, R. Fast Fish Face Fewer Mitochondrial Mutations: Patterns of dN/dS across Fish Mitogenomes. Gene 2015, 572, 27–34. [Google Scholar] [CrossRef]
- Lemmon, A.R.; Emme, S.A.; Lemmon, E.M. Anchored Hybrid Enrichment for Massively High-Throughput Phylogenomics. Syst. Biol. 2012, 61, 727–744. [Google Scholar] [CrossRef]
- Lemmon, E.M.; Lemmon, A.R. High-Throughput Genomic Data in Systematics and Phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 99–121. [Google Scholar] [CrossRef]
- Weitemier, K.; Straub, S.C.K.; Cronn, R.C.; Fishbein, M.; Schmickl, R.; McDonnell, A.; Liston, A. Hyb-Seq: Combining Target Enrichment and Genome Skimming for Plant Phylogenomics. Appl. Plant Sci. 2014, 2, 1400042. [Google Scholar] [CrossRef]
- Petrova, T.; Skazina, M.; Kuksin, A.; Bondareva, O.; Abramson, N. Narrow-Headed Voles Species Complex (Cricetidae, Rodentia): Evidence for Species Differentiation Inferred from Transcriptome Data. Diversity 2022, 14, 512. [Google Scholar] [CrossRef]
- Bondareva, O.; Petrova, T.; Bodrov, S.; Gavrilo, M.; Smorkatcheva, A.; Abramson, N. How Voles Adapt to Subterranean Lifestyle: Insights from RNA-Seq. Front. Ecol. Evol. 2023, 11, 1085993. [Google Scholar] [CrossRef]
- Franchini, P.; Monné Parera, D.; Kautt, A.F.; Meyer, A. quaddRAD: A New High-multiplexing and PCR Duplicate Removal ddRAD Protocol Produces Novel Evolutionary Insights in a Nonradiating Cichlid Lineage. Mol. Ecol. 2017, 26, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 September 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)Orthologs in Large-Scale Analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A. Phylogeny-Aware Alignment with PRANK. In Multiple Sequence Alignment Methods; Russell, D.J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1079, pp. 155–170. ISBN 978-1-62703-645-0. [Google Scholar]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE team. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Sipos, B.; Zhao, L. SeqKit2: A Swiss Army Knife for Sequence and Alignment Processing. iMeta 2024, 3, e191. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Morel, B.; Kozlov, A.M.; Stamatakis, A. ParGenes: A Tool for Massively Parallel Model Selection and Phylogenetic Tree Inference on Thousands of Genes. Bioinformatics 2019, 35, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Bouckaert, R.R. DensiTree: Making Sense of Sets of Phylogenetic Trees. Bioinformatics 2010, 26, 1372–1373. [Google Scholar] [CrossRef]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial Time Species Tree Reconstruction from Partially Resolved Gene Trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Mirarab, S. Weighting by Gene Tree Uncertainty Improves Accuracy of Quartet-Based Species Trees. Mol. Biol. Evol. 2022, 39, msac215. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Jiménez-Silva, C.L.; Bouckaert, R. StarBeast3: Adaptive Parallelized Bayesian Inference under the Multispecies Coalescent. Syst. Biol. 2022, 71, 901–916. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Ortiz, E.M. Vcf2phylip v2.0: Convert a VCF Matrix into Several Matrix Formats for Phylogenetic Analysis. 2019. Available online: https://github.com/edgardomortiz/vcf2phylip (accessed on 1 September 2019).
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLOS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.R.; Drummond, A.J. bModelTest: Bayesian Phylogenetic Site Model Averaging and Model Comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.B.; Lockhart, P.J. Deciphering Ancient Rapid Radiations. Trends Ecol. Evol. 2007, 22, 258–265. [Google Scholar] [CrossRef]
- Patel, S. Error in Phylogenetic Estimation for Bushes in the Tree of Life. J. Phylogenet. Evol. Biol. 2013, 01, 110. [Google Scholar] [CrossRef]
- Nichols, R. Gene Trees and Species Trees Are Not the Same. Trends Ecol. Evol. 2001, 16, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Nesi, N.; Tsagkogeorga, G.; Tsang, S.M.; Nicolas, V.; Lalis, A.; Scanlon, A.T.; Riesle-Sbarbaro, S.A.; Wiantoro, S.; Hitch, A.T.; Juste, J.; et al. Interrogating Phylogenetic Discordance Resolves Deep Splits in the Rapid Radiation of Old World Fruit Bats (Chiroptera: Pteropodidae). Syst. Biol. 2021, 70, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Olave, M.; Meyer, A. Implementing Large Genomic Single Nucleotide Polymorphism Data Sets in Phylogenetic Network Reconstructions: A Case Study of Particularly Rapid Radiations of Cichlid Fish. Syst. Biol. 2020, 69, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P.; Knowles, L.L. Inferring Phylogeny Despite Incomplete Lineage Sorting. Syst. Biol. 2006, 55, 21–30. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.E.; Huang, H.; Knowles, L.L. Maximum Likelihood Estimates of Species Trees: How Accuracy of Phylogenetic Inference Depends upon the Divergence History and Sampling Design. Syst. Biol. 2009, 58, 501–508. [Google Scholar] [CrossRef]
- Heled, J.; Drummond, A.J. Bayesian Inference of Species Trees from Multilocus Data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef]
- Corl, A.; Ellegren, H. Sampling Strategies for Species Trees: The Effects on Phylogenetic Inference of the Number of Genes, Number of Individuals, and Whether Loci Are Mitochondrial, Sex-Linked, or Autosomal. Mol. Phylogenet. Evol. 2013, 67, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Alfaro-Ibáñez, M.; Lira-Garrido, J.; Cuenca-Bescós, G.; Pons, J.; Bover, P. Insights on the Evolution of the Tribe Pliomyini (Arvicolinae, Rodentia): Ancient DNA from the Extinct Pliomys Lenki. Palaeontol. Electron. 2024, 27, a47. [Google Scholar] [CrossRef]
- Wang, X.; Liang, D.; Wang, X.; Tang, M.; Liu, Y.; Liu, S.; Zhang, P. Phylogenomics Reveals the Evolution, Biogeography, and Diversification History of Voles in the Hengduan Mountains. Commun. Biol. 2022, 5, 1124. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, W.-D. Some Aspects of Evolution and Biostratigraphy of Arvicola (Mammalia, Rodentia) in the Central European Pleistocene. In Evolution, Phylogeny and Biostratigraphy of Arvicolids (Rodentia.Mammalia); Fejfar, O., Heinrich, W.-D., Eds.; Geological Survey: Prague, Czech Republic, 1990; pp. 165–182. [Google Scholar]
- Koenigswald, W.; van Kolfschoten, T. The Mimomys-Arvicola Boundary and the Enamel Thickness Quotient (SDQ) of Arvicola as Stratigraphic Markers in the Middle Pleistocene. In The Early Middle Pleistocene in Europe; Nurner, E., Ed.; Balkema: Rotterdam, The Netherlands, 1996; pp. 211–226. [Google Scholar]
- Pardiñas, U.F.J.; Myers, P.; León-Paniagua, L.; Ordóñez Garza, N.; Cook, J.A.; Kryštufek, B. Family Cricetidae (True Hamsters, Voles, Lemmings and New World Rats and Mice). In Handbook of the Mammals of the World; Lynx Edicions: Barcelona, Spain, 2017; Volume 7, pp. 204–279. [Google Scholar]
- Villaverde, T.; Maguilla, E.; Luceño, M.; Hipp, A.L. Assessing the Sensitivity of Divergence Time Estimates to Locus Sampling, Calibration Points, and Model Priors in a RAD-seq Phylogeny of Carex Section Schoenoxiphium. J. Syst. Evol. 2021, 59, 687–697. [Google Scholar] [CrossRef]
- Near, T.J.; Sanderson, M.J. Assessing the Quality of Molecular Divergence Time Estimates by Fossil Calibrations and Fossil–Based Model Selection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 1477–1483. [Google Scholar] [CrossRef]
- Hug, L.A.; Roger, A.J. The Impact of Fossils and Taxon Sampling on Ancient Molecular Dating Analyses. Mol. Biol. Evol. 2007, 24, 1889–1897. [Google Scholar] [CrossRef]
- Sauquet, H.; Ho, S.Y.W.; Gandolfo, M.A.; Jordan, G.J.; Wilf, P.; Cantrill, D.J.; Bayly, M.J.; Bromham, L.; Brown, G.K.; Carpenter, R.J.; et al. Testing the Impact of Calibration on Molecular Divergence Times Using a Fossil-Rich Group: The Case of Nothofagus (Fagales). Syst. Biol. 2012, 61, 289–313. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, S.; Holmes, E.C.; Ho, S.Y.W. Analyses of Evolutionary Dynamics in Viruses Are Hindered by a Time-Dependent Bias in Rate Estimates. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140732. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MRCA | RNA-seq | ddRADseq | mt Complete [5] | mt RY-Masked [5] |
|---|---|---|---|---|
| Arvicolinae | 7.67 (7.07–8.53) | 7.33 (7.05–7.73) | 7.36 (7.04–7.78) | 7.33 (7.05–7.73) |
| Ondatrini/(Dicrostonychini + Lemmini) | 6.54 (5.86–7.36) | 6.73 (6.18–7.27) | n/a | n/a |
| Dicrostonychini/Lemmini | 6.26 (5.6–7.07) | 6.51 (5.94–7.03) | n/a | n/a |
| Dicrostonychini | n/a | 5.66 (4.97–6.32) | 4.89 (4.08–5.7) | 4.49 (3.23–5.86) |
| Lemmini | 3.52 (3.26–3.87) | 3.79 (3.32–4.32) | 4.81 (3.68–5.97) | 4.37 (3.31–5.71) |
| Clethrionomyini | 2.58 (2.11–3.09) | 4.09 (3.41–4.78) | 4.02 (3.33–4.72) | 4.46 (3.35–5.64) |
| Third radiation (Arvicolini, Ellobiusini, Lagurini, and Dinaromys *) | 5.31 (4.64–6.07) | 5.96 (5.37–6.54) | 6.2 (5.65–6.76) | 6.11 (5.17–6.92) |
| Lagurini and Dinaromys | n/a | 5.47 (4.89–6.06) | n/a | n/a |
| Arvicolini | 4.76 (4.11–5.46) | 5.47 (4.88–6.04) | n/a | n/a |
| Arvicolini s. str. * | 3.56 (3.03–4.14) | 3.99 (3.47–4.51) | 4.9 (4.33–5.47) | 5.02 (4.12–5.89) |
| Lagurini and Arvicolini | 5.11 (4.45–5.85) | n/a | 6.04 (5.5–6.61) | 6.03 (5.12–6.86) |
| Ellobiusini and Arvicolini | n/a | 5.78 (5.22–6.38) | n/a | n/a |
| Ellobiusini | 3.18 (2.54–3.81) | 3.84 (3.14–4.56) | 4.97 (4.21–5.69) | 4.58 (3.42–5.68) |
| Lagurini | n/a | 3.02 (2.58–3.52) | 3.1 (2.59–3.75) | 3.05 (2.56–3.75) |
| Chionomys | n/a | 2.22 (1.66–2.8) | 3.29 (2.5–4.04) | 3.45 (2.13–4.67) |
| Microtus ** (Microtus, Terricola, Agricola, Blanfordimys *, and Iberomys *) | 2.4 (2–2.81) | 2.86 (2.49–3.25) | 3.8 (3.31–4.3) | 3.87 (3.07–4.63) |
| Mynomes | 2.05 (1.64–2.48) | 2.67 (2.29–3.08) | 3.41 (2.89–3.91) | 3.32 (2.48–4.11) |
| Microtus + Terricola | 1.69 (1.31–2.1) | 2.45 (2.08–2.81) | 2.96 (2.46–3.5) | 3.18 (2.35–3.95) |
| Microtus | n/a | 2.01 (1.67–2.36) | 1.87 (1.43–2.33) | 2.06 (1.31–2.8) |
| Terricola | n/a | 0.64 (0.42–0.87) | 1.38 (0.89–1.93) | 1.46 (0.7–2.32) |
| Iberomys + Agricola + Blanfordimys | n/a | 2.75 (2.39–3.15) | 3.18 (2.66–3.71) | 3.11 (2.23–3.99) |
| Alexandromys, Lasiopodomys, and Neodon * | 2.6 (2.19–3.03) | 3.11 (2.71–3.51) | 3.92 (3.4–4.4) | 4.12 (3.31–4.9) |
| Lasiopodomys | 1.92 (1.57–2.3) | 2.27 (1.91–2.65) | 3.09 (2.59–3.56) | 3.07 (2.27–3.85) |
| Alexandromys | 1.31 (0.95–1.7) | 1.53 (1.23–1.84) | 2.16 (1.55–2.81) | 2.2 (1.27–3.22) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abramson, N.; Skalon, E.; Bondareva, O.; Bodrov, S.; Petrova, T.; Dvoyashov, I. Resolving Rapid Radiation of Voles and Lemmings (Arvicolinae: Cricetinae, Rodentia) with QuaddRAD Sequencing and Transcriptome Analysis. Diversity 2025, 17, 61. https://doi.org/10.3390/d17010061
Abramson N, Skalon E, Bondareva O, Bodrov S, Petrova T, Dvoyashov I. Resolving Rapid Radiation of Voles and Lemmings (Arvicolinae: Cricetinae, Rodentia) with QuaddRAD Sequencing and Transcriptome Analysis. Diversity. 2025; 17(1):61. https://doi.org/10.3390/d17010061
Chicago/Turabian StyleAbramson, Natalia, Elizaveta Skalon, Olga Bondareva, Semen Bodrov, Tatyana Petrova, and Ivan Dvoyashov. 2025. "Resolving Rapid Radiation of Voles and Lemmings (Arvicolinae: Cricetinae, Rodentia) with QuaddRAD Sequencing and Transcriptome Analysis" Diversity 17, no. 1: 61. https://doi.org/10.3390/d17010061
APA StyleAbramson, N., Skalon, E., Bondareva, O., Bodrov, S., Petrova, T., & Dvoyashov, I. (2025). Resolving Rapid Radiation of Voles and Lemmings (Arvicolinae: Cricetinae, Rodentia) with QuaddRAD Sequencing and Transcriptome Analysis. Diversity, 17(1), 61. https://doi.org/10.3390/d17010061