
Experimental Nuclear Medicine Meets Tumor Biology

 , ,
, ,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biomarkers
2.1. Biomarkers in the Context of Nuclear Medicine
2.1.1. Molecular Imaging of Biomarkers
2.1.2. Radiopharmaceuticals for Biomarker Imaging or Therapy
- endogenous ligands or derivatives, that map metabolic (dys)functions or
- drugs or (modified) model substances targeting specific disease-related proteins (e.g., enzymes, receptors, transporters) or
- antibodies or antibody constructs directed against specific disease-associated antigens [25].
2.2. Biomarkers in the Context of Pathology
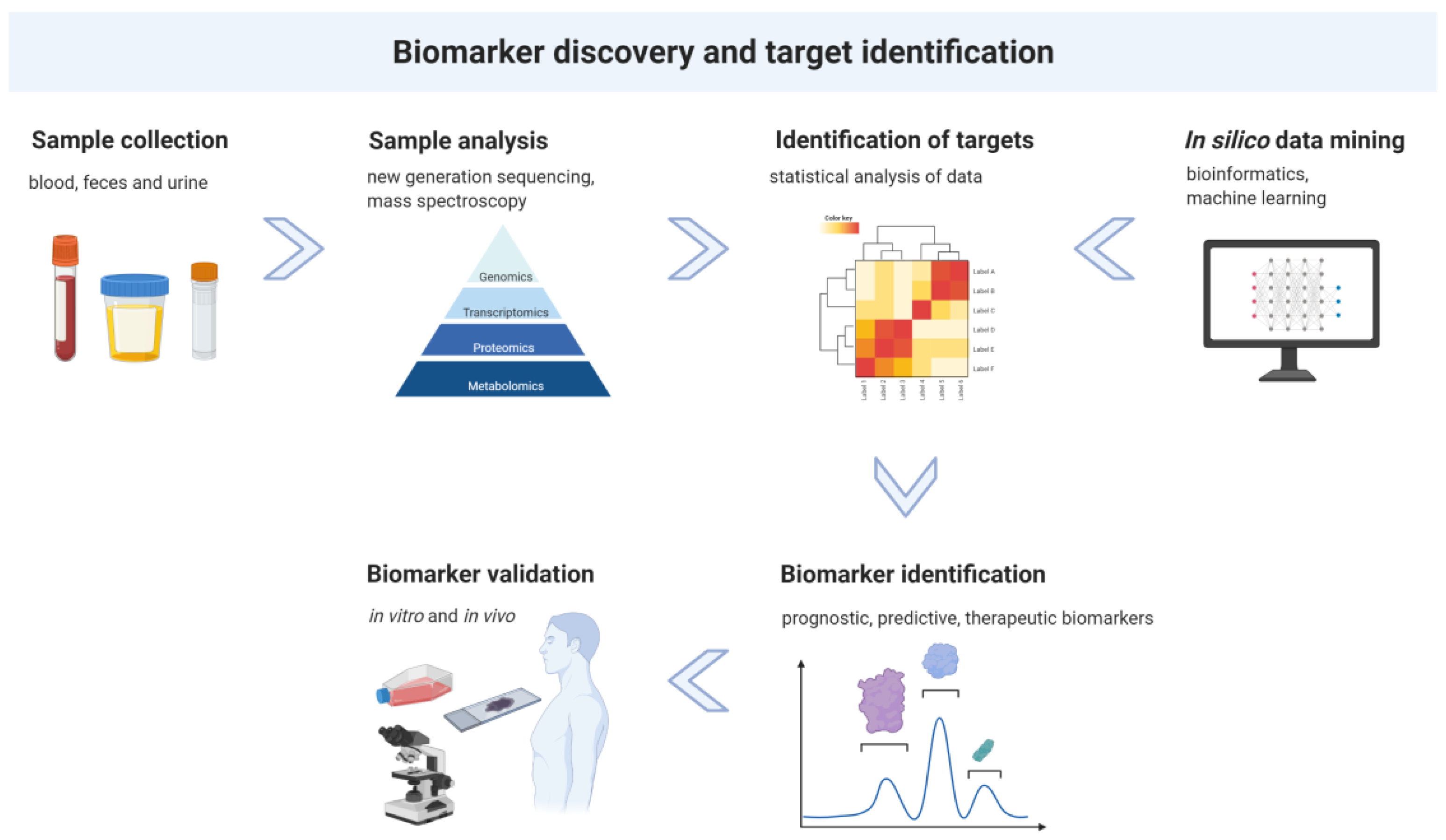
3. Biomarker Discovery and Target Identification
4. Experimental Nuclear Medicine
4.1. Binding Studies
4.2. In Vitro Models
4.3. In Vivo Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Zenner, H.P. Individual Biomarkers Using Molecular Personalized Medicine Approaches. ORL J. Otorhinolaryngol. Relat. Spec. 2017, 79, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.S.; Beyer, T.; Babayan, A.; Bergmann, M.; Brehme, M.; Buyx, A.; Czernin, J.; Egger, G.; Elenitoba-Johnson, K.S.J.; Gückel, B.; et al. Advancing Biomarker Development Through Convergent Engagement: Summary Report. In Proceedings of the 2nd International Danube Symposium on Biomarker Development, Molecular Imaging and Applied Diagnostics, Vienna, Austria, 14–16 March 2018; Molecular Imaging and Biology, MIB: The official publication of the Academy of Molecular Imaging; Springer: Cham, Switzerland, 2020; Volume 22, pp. 47–65. [Google Scholar]
- Sharp, P.; Hockfield, S. Convergence: The future of health. Science 2017, 355, 589. [Google Scholar] [CrossRef] [PubMed]
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and other Tools); Food and Drug Administration: Silver Spring, MD, USA, 2016. [Google Scholar]
- Nicolini, A. Biomarkers: A framework driving advances in oncology. Biomark. Med. 2015, 9, 303–306. [Google Scholar] [CrossRef]
- Carlomagno, N.; Incollingo, P.; Tammaro, V.; Peluso, G.; Rupealta, N.; Chiacchio, G.; Sandoval Sotelo, M.L.; Minieri, G.; Pisani, A.; Riccio, E.; et al. Diagnostic, Predictive, Prognostic, and Therapeutic Molecular Biomarkers in Third Millennium: A Breakthrough in Gastric Cancer. Biomed. Res. Int. 2017, 2017, 7869802. [Google Scholar] [CrossRef] [Green Version]
- Italiano, A. Prognostic or Predictive? It’s Time to Get Back to Definitions! J. Clin. Oncol. 2011, 29, 4718. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Liu, Y.; Zhou, S.; Jiang, H.; Zhu, K.; Wang, R. Predictive effect of PD-L1 expression for immune checkpoint inhibitor (PD-1/PD-L1 inhibitors) treatment for non-small cell lung cancer: A meta-analysis. Int. Immunopharmacol. 2020, 80, 106214. [Google Scholar] [CrossRef]
- Narayanan, S.; Kawaguchi, T.; Peng, X.; Qi, Q.; Liu, S.; Yan, L.; Takabe, K. Tumor Infiltrating Lymphocytes and Macrophages Improve Survival in Microsatellite Unstable Colorectal Cancer. Sci. Rep. 2019, 9, 13455. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef]
- Cimadamore, A.; Cheng, M.; Santoni, M.; López-Beltrán, A.; Battelli, N.; Massari, F.; Galosi, A.B.; Scarpelli, M.; Montironi, R. New Prostate Cancer Targets for Diagnosis, Imaging, and Therapy: Focus on Prostate-Specific Membrane Antigen. Front. Oncol. 2018, 8, 653. [Google Scholar] [CrossRef] [Green Version]
- Lucignani, G. Imaging biomarkers: From research to patient care—A shift in view. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1693–1697. [Google Scholar] [CrossRef]
- Townsend, D.W. Physical principles and technology of clinical PET imaging. Ann. Acad. Med. Singap. 2004, 33, 133–145. [Google Scholar]
- Cherry, S.R. Fundamentals of positron emission tomography and applications in preclinical drug development. J. Clin. Pharmacol. 2001, 41, 482–491. [Google Scholar] [CrossRef]
- Phelps, M.E. Positron emission tomography provides molecular imaging of biological processes. Proc. Natl. Acad. Sci. USA 2000, 97, 9226–9233. [Google Scholar] [CrossRef] [Green Version]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular Imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef]
- James, M.L.; Gambhir, S.S. A molecular imaging primer: Modalities, imaging agents, and applications. Physiol. Rev. 2012, 92, 897–965. [Google Scholar] [CrossRef] [Green Version]
- Rahmim, A.; Zaidi, H. PET versus SPECT: Strengths, limitations and challenges. Nucl. Med. Commun. 2008, 29, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Vaz, S.C.; Oliveira, F.; Herrmann, K.; Veit-Haibach, P. Nuclear medicine and molecular imaging advances in the 21st century. Br. J. Radiol. 2020, 93, 20200095. [Google Scholar] [CrossRef]
- Antoch, G.; Freudenberg, L.S.; Beyer, T.; Bockisch, A.; Debatin, J.F. To enhance or not to enhance? 18F-FDG and CT contrast agents in dual-modality 18F-FDG PET/CT. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2004, 45, 56s–65s. [Google Scholar]
- Rausch, I.; Quick, H.H.; Cal-Gonzalez, J.; Sattler, B.; Boellaard, R.; Beyer, T. Technical and instrumentational foundations of PET/MRI. Eur. J. Radiol. 2017, 94, A3–A13. [Google Scholar] [CrossRef]
- Wadsak, W.; Mitterhauser, M. Basics and principles of radiopharmaceuticals for PET/CT. Eur. J. Radiol. 2010, 73, 461–469. [Google Scholar] [CrossRef]
- Baillet, G.Y.; Mena, I.G.; Kuperus, J.H.; Robertson, J.M.; French, W.J. Simultaneous technetium-99m MIBI angiography and myocardial perfusion imaging. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1989, 30, 38–44. [Google Scholar]
- Citrin, D.L.; Bessent, R.G.; McGinley, E.; Gordon, D. Dynamic studies with 99mTc-HEDP in normal subjects and in patients with bone tumors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1975, 16, 886–890. [Google Scholar]
- Wiseman, G.A.; White, C.A.; Sparks, R.B.; Erwin, W.D.; Podoloff, D.A.; Lamonica, D.; Bartlett, N.L.; Anthony Parker, J.; Dunn, W.L.; Spies, S.M.; et al. Biodistribution and dosimetry results from a phase III prospectively randomized controlled trial of Zevalin radioimmunotherapy for low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. Crit. Rev. Oncol. Hematol. 2001, 39, 181–194. [Google Scholar] [CrossRef]
- Krenning, E.P.; Bakker, W.H.; Kooij, P.P.; Breeman, W.A.; Oei, H.Y.; de Jong, M.; Reubi, J.C.; Visser, T.J.; Bruns, C.; Kwekkeboom, D.J.; et al. Somatostatin receptor scintigraphy with indium-111-DTPA-D-Phe-1-octreotide in man: Metabolism, dosimetry and comparison with iodine-123-Tyr-3-octreotide. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1992, 33, 652–658. [Google Scholar]
- Yordanova, A.; Eppard, E.; Kürpig, S.; Bundschuh, R.A.; Schönberger, S.; Gonzalez-Carmona, M.; Feldmann, G.; Ahmadzadehfar, H.; Essler, M. Theranostics in nuclear medicine practice. OncoTargets Ther. 2017, 10, 4821–4828. [Google Scholar] [CrossRef] [Green Version]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Pichler, V.; Berroteran-Infante, N.; Philippe, C.; Vraka, C.; Klebermass, E.M.; Balber, T.; Pfaff, S.; Nics, L.; Mitterhauser, M.; Wadsak, W. An Overview of PET Radiochemistry, Part 1: The Covalent Labels (18)F, (11)C, and (13)N. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1350–1354. [Google Scholar]
- Mathis, C.A.; Wang, Y.; Holt, D.P.; Huang, G.-F.; Debnath, M.L.; Klunk, W. Synthesis and Evaluation of 11C-Labeled 6-Substituted 2-Arylbenzothiazoles as Amyloid Imaging Agents. J. Med. Chem. 2003, 46, 2740–2754. [Google Scholar] [CrossRef]
- Rösch, F. (68)Ge/ (68)Ga generators: Past, present, and future. Recent Results Cancer Res. Fortschr. Der Krebsforsch. Prog. Dans Les Rech. Sur Le Cancer 2013, 194, 3–16. [Google Scholar]
- Sanli, Y.; Garg, I.; Kandathil, A.; Kendi, T.; Zanetti, M.J.B.; Kuyumcu, S.; Subramaniam, R.M. Neuroendocrine Tumor Diagnosis and Management: (68)Ga-DOTATATE PET/CT. AJR Am. J. Roentgenol. 2018, 211, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, M.; Cardinale, J.; Aulsebrook, M.L.; Gasser, G.; Mindt, T.L. An Overview of PET Radiochemistry, Part 2: Radiometals. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1500–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasse, D.; Nonat, A. Radiometals: Towards a new success story in nuclear imaging? Dalton Trans. 2015, 44, 4845–4858. [Google Scholar] [CrossRef]
- Baum, R.P.; Kulkarni, H.R. THERANOSTICS: From Molecular Imaging Using Ga-68 Labeled Tracers and PET/CT to Personalized Radionuclide Therapy—The Bad Berka Experience. Theranostics 2012, 2, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-A.; Kim, J.Y. Recent advances in radiopharmaceutical application of matched-pair radiometals. Curr. Top. Med. Chem. 2013, 13, 458–469. [Google Scholar] [CrossRef]
- O’Hurley, G.; Sjöstedt, E.; Rahman, A.; Li, B.; Kampf, C.; Pontén, F.; Gallagher, W.M.; Lindskog, C. Garbage in, garbage out: A critical evaluation of strategies used for validation of immunohistochemical biomarkers. Mol. Oncol. 2014, 8, 783–798. [Google Scholar] [CrossRef]
- Tian, M.; He, X.; Jin, C.; He, X.; Wu, S.; Zhou, R.; Zhang, X.; Zhang, K.; Gu, W.; Wang, J.; et al. Transpathology: Molecular imaging-based pathology. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2338–2350. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Administration USFD. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools) 2021 [updated 26 July 2021]. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/nucleic-acid-based-tests (accessed on 27 September 2021).
- Administration USFD. Nucleic Acid Based Tests 2021 [updated 29 July 2021. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools (accessed on 31 July 2021).
- Doll, S.; Gnad, F.; Mann, M. The Case for Proteomics and Phospho-Proteomics in Personalized Cancer Medicine. Proteom. Clin. Appl. 2019, 13, 1800113. [Google Scholar] [CrossRef]
- Shruthi, B.S.; Vinodhkumar, P. Selvamani Proteomics: A new perspective for cancer. Adv. Biomed. Res. 2016, 5, 67. [Google Scholar] [CrossRef]
- Kuhlmann, L.; Cummins, E.; Samudio, I.; Kislinger, T. Cell-surface proteomics for the identification of novel therapeutic targets in cancer. Expert Rev. Proteom. 2018, 15, 259–275. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Aretz, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef]
- Strausberg, R.L. The Cancer Genome Anatomy Project: New resources for reading the molecular signatures of cancer. J. Pathol. 2001, 195, 31–40. [Google Scholar] [CrossRef]
- Zhang, J.; Finney, R.P.; Rowe, W.; Edmonson, M.; Yang, S.H.; Dracheva, T.; Jen, J.; Struewing, J.P.; Buetow, K.H. Systematic analysis of genetic alterations in tumors using Cancer Genome WorkBench (CGWB). Genome Res. 2007, 17, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010, 39, D945–D950. [Google Scholar] [CrossRef] [Green Version]
- Asada, K.; Kaneko, S.; Takasawa, K.; Machino, H.; Takahashi, S.; Shinkai, N.; Shimoyama, R.; Komatsu, M.; Hamamoto, R. Integrated Analysis of Whole Genome and Epigenome Data Using Machine Learning Technology: Toward the Establishment of Precision Oncology. Front. Oncol. 2021, 11, 666937. [Google Scholar] [CrossRef]
- Echle, A.; Rindtorff, N.T.; Brinker, T.J.; Luedde, T.; Pearson, A.T.; Kather, J.N. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 2021, 124, 686–696. [Google Scholar] [CrossRef]
- Lambin, P.; Rios-Velazquez, E.; Leijenaar, R.; Carvalho, S.; van Stiphout, R.G.P.M.; Granton, P.; Zegers, C.M.L.; Gillies, R.; Boellard, R.; Dekker, A.; et al. Radiomics: Extracting more information from medical images using advanced feature analysis. Eur. J. Cancer 2012, 48, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balber, T. Predictive Value and Limitations of Preclinical Methods in PET-Tracer Development. Ph.D. Thesis, Universität Wien Fakultät für Lebenswissenschaften, Vienna, Austria, 2019. Available online: https://utheses.univie.ac.at/ (accessed on 12 September 2021).
- Hulme, E.C.; Trevethick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. J. Cereb. Blood Flow Metab. 2010, 161, 1219–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, P.; Michel, M.C.; Leineweber, K.; Wieland, T.; Wettschureck, N.; Offermanns, S. Receptor and Binding Studies. In Practical Methods in Cardiovascular Research; Springer: Berlin/Heidelberg, Germany, 2005; pp. 723–783. [Google Scholar]
- Zeilinger, M.; Pichler, F.; Nics, L.; Wadsak, W.; Spreitzer, H.; Hacker, M.; Mitterhauser, M. New approaches for the reliable in vitro assessment of binding affinity based on high-resolution real-time data acquisition of radioligand-receptor binding kinetics. EJNMMI Res. 2017, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björke, H.; Andersson, K. Measuring the affinity of a radioligand with its receptor using a rotating cell dish with in situ reference area. Appl. Radiat. Isot. Incl. Data Instrum. Methods Use Agric. Ind. Med. 2006, 64, 32–37. [Google Scholar] [CrossRef]
- Björke, H.; Andersson, K. Automated, high-resolution cellular retention and uptake studies in vitro. Appl. Radiat. Isot. Incl. Data Instrum. Methods Use Agric. Ind. Med. 2006, 64, 901–905. [Google Scholar] [CrossRef]
- Qume, M. Overview of ligand-receptor binding techniques. Methods Mol. Biol. 1999, 106, 3–23. [Google Scholar]
- Imaging Modalities for Biological and Preclinical Research: A Compendium; Walter, A.; Mannheim, J.G.; Caruana, C.J. (Eds.) IOP Publishing: Bristol, UK, 2021. [Google Scholar] [CrossRef]
- Harrison, R.G.; Greenman, M.J.; Mall, F.P.; Jackson, C.M. Observations of the living developing nerve fiber. Anat. Rec. 1907, 1, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Costa, E.C.; Moreira, A.F.; Diogo, D.M.D.M.; Gaspar, V.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Holtfreter, J. A study of the mechanics of gastrulation. J. Exp. Zool. 1944, 95, 171–212. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- McMahon, K.M.; Volpato, M.; Chi, H.Y.; Musiwaro, P.; Poterlowicz, K.; Peng, Y.; Scally, A.J.; Patterson, L.H.; Phillips, R.M.; Sutton, C.W. Characterization of Changes in the Proteome in Different Regions of 3D Multicell Tumor Spheroids. J. Proteome Res. 2012, 11, 2863–2875. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Santini, M.T.; Rainaldi, G. Three-dimensional spheroid model in tumor biology. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 1999, 67, 148–157. [Google Scholar] [CrossRef]
- Booij, T.H.; Price, L.S.; Danen, E.H.J. 3D Cell-Based Assays for Drug Screens: Challenges in Imaging, Image Analysis, and High-Content Analysis. SLAS Discov. Adv. Sci. Drug Discov. 2019, 24, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Fan, N.; Yang, X.; Peng, B.; Jiang, H. New advances in microfluidic flow cytometry. Electrophoresis 2019, 40, 1212–1229. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.E16. [Google Scholar] [CrossRef]
- Schachtschneider, K.; Schwind, R.; Newson, J.; Kinachtchouk, N.; Rizko, M.; Mendoza-Elias, N.; Grippo, P.; Principe, D.R.; Park, A.; Overgaard, N.H.; et al. The Oncopig Cancer Model: An Innovative Large Animal Translational Oncology Platform. Front. Oncol. 2017, 7, 190. [Google Scholar] [CrossRef]
- Watson, A.L.; Carlson, D.; Largaespada, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Engineered Swine Models of Cancer. Front. Genet. 2016, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Karkampouna, S.; La Manna, F.; Benjak, A.; Kiener, M.; De Menna, M.; Zoni, E.; Grosjean, J.; Klima, I.; Garofoli, A.; Bolis, M.; et al. Patient-derived xenografts and organoids model therapy response in prostate cancer. Nat. Commun. 2021, 12, 1117. [Google Scholar] [CrossRef]
- Hason, M.; Bartůněk, P. Zebrafish Models of Cancer-New Insights on Modeling Human Cancer in a Non-Mammalian Vertebrate. Genes 2019, 10, 935. [Google Scholar] [CrossRef] [Green Version]
- Brown, H.K.; Schiavone, K.; Tazzyman, S.; Heymann, D.; Chico, T.J. Zebrafish xenograft models of cancer and metastasis for drug discovery. Expert Opin. Drug Discov. 2017, 12, 379–389. [Google Scholar] [CrossRef]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM). A multifaceted experimental model. Mech. Dev. 2016, 141, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Rosenbruch, M. The sensitivity of chicken embryos in incubated eggs. ALTEX 1997, 14, 111–113. [Google Scholar]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]
- DeBord, L.C.; Pathak, R.R.; Villaneuva, M.; Liu, H.-C.; A Harrington, D.; Yu, W.; Lewis, M.T.; Sikora, A.G. The chick chorioallantoic membrane (CAM) as a versatile patient-derived xenograft (PDX) platform for precision medicine and preclinical research. Am. J. Cancer Res. 2018, 8, 1642–1660. [Google Scholar]
- Wörsdörfer, P.; Dalda, N.; Kern, A.; Krüger, S.; Wagner, N.; Kwok, C.K.; Henke, E.; Ergün, S. Generation of complex human organoid models including vascular networks by incorporation of mesodermal progenitor cells. Sci. Rep. 2019, 9, 15663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garreta, E.; Prado, P.; Tarantino, C.; Oria, R.; Fanlo, L.; Martí, E.; Zalvidea, D.; Trepat, X.; Roca-Cusachs, P.; Gavaldà-Navarro, A.; et al. Fine tuning the extracellular environment accelerates the derivation of kidney organoids from human pluripotent stem cells. Nat. Mater. 2019, 18, 397–405. [Google Scholar] [CrossRef]
- Rous, P.; Murphy, J.B. TUMOR IMPLANTATIONS IN THE DEVELOPING EMBRYO. J. Am. Med. Assoc. 1911, LVI, 741–742. [Google Scholar]
- Würbach, L.; Heidrich, A.; Opfermann, T.; Gebhardt, P.; Saluz, H.P. Insights into Bone Metabolism of Avian Embryos In Ovo Via 3D and 4D 18F-fluoride Positron Emission Tomography. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging. 2012, 14, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, P.; Würbach, L.; Heidrich, A.; Heinrich, L.; Walther, M.; Opfermann, T.; Sørensen, B.; Saluz, H.P. Dynamic behaviour of selected PET tracers in embryonated chicken eggs. Rev. Esp. De Med. Nucl. E Imagen Mol. 2013, 32, 371–377. [Google Scholar]
- Haller, S.; Ametamey, S.M.; Schibli, R.; Müller, C. Investigation of the chick embryo as a potential alternative to the mouse for evaluation of radiopharmaceuticals. Nucl. Med. Biol. 2015, 42, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Löffler, J.; Hamp, C.; Scheidhauer, E.; Di Carlo, D.; Solbach, C.; Abaei, A.; Hao, L.; Glatting, G.; Beer, A.J.; Rasche, V.; et al. Comparison of Quantification of Target-Specific Accumulation of [(18)F]F-siPSMA-14 in the HET-CAM Model and in Mice Using PET/MRI. Cancers 2021, 13, 4007. [Google Scholar] [CrossRef]
- Warnock, G.; Turtoi, A.; Blomme, A.; Bretin, F.; Bahri, M.A.; Lemaire, C.; Libert, L.C.; Seret, A.E.; Luxen, A.; Castronovo, V.; et al. In Vivo PET/CT in a Human Glioblastoma Chicken Chorioallantoic Membrane Model: A New Tool for Oncology and Radiotracer Development. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2013, 54, 1782–1788. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Koch, A.B.F.; Löffler, J.; Lindén, M.; Solbach, C.; Abaei, A.; Li, H.; Glatting, G.; Beer, A.J.; Rasche, V. Multi-Modal PET and MR Imaging in the Hen’s Egg Test-Chorioallantoic Membrane (HET-CAM) Model for Initial in Vivo Testing of Target-Specific Radioligands. Cancers 2020, 12, 1248. [Google Scholar] [CrossRef]
- Swindle, M.M.; Makin, A.; Herron, A.J.; Clubb, F.J., Jr.; Frazier, K.S. Swine as Models in Biomedical Research and Toxicology Testing. Vet. Pathol. 2011, 49, 344–356. [Google Scholar] [CrossRef]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef]
- Sieren, J.; Meyerholz, D.; Wang, X.-J.; Davis, B.T.; Newell, J.D., Jr.; Hammond, E.; Rohret, J.A.; Rohret, F.A.; Struzynski, J.T.; Goeken, J.A.; et al. Development and translational imaging of a TP53 porcine tumorigenesis model. J. Clin. Investig. 2014, 124, 4052–4066. [Google Scholar] [CrossRef] [Green Version]
- Sorace, J.; Aberle, D.R.; Elimam, D.; Lawvere, S.; Tawfik, O.; Wallace, W.D. Integrating pathology and radiology disciplines: An emerging opportunity? BMC Med. 2012, 10, 100. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balber, T.; Tran, L.; Benčurová, K.; Raitanen, J.; Egger, G.; Mitterhauser, M. Experimental Nuclear Medicine Meets Tumor Biology. Pharmaceuticals 2022, 15, 227. https://doi.org/10.3390/ph15020227
Balber T, Tran L, Benčurová K, Raitanen J, Egger G, Mitterhauser M. Experimental Nuclear Medicine Meets Tumor Biology. Pharmaceuticals. 2022; 15(2):227. https://doi.org/10.3390/ph15020227
Chicago/Turabian StyleBalber, Theresa, Loan Tran, Katarína Benčurová, Julia Raitanen, Gerda Egger, and Markus Mitterhauser. 2022. "Experimental Nuclear Medicine Meets Tumor Biology" Pharmaceuticals 15, no. 2: 227. https://doi.org/10.3390/ph15020227
APA StyleBalber, T., Tran, L., Benčurová, K., Raitanen, J., Egger, G., & Mitterhauser, M. (2022). Experimental Nuclear Medicine Meets Tumor Biology. Pharmaceuticals, 15(2), 227. https://doi.org/10.3390/ph15020227