Abstract
2,5-Disubstituted 1,3,4-oxadiazoles are privileged versatile scaffolds in medicinal chemistry that have exhibited diverse biological activities. Acetyl- (AChE) and butyrylcholinesterase (BChE) inhibitors are used, e.g., to treat dementias and myasthenia gravis. 5-Aryl-1,3,4-oxadiazoles decorated with dodecyl linked via nitrogen, sulfur or directly to this heterocycle have been designed as potential inhibitors of AChE and BChE. They were prepared from commercially available or in-house prepared hydrazides by reaction with dodecyl isocyanate to form hydrazine-1-carboxamides 2 (yields 67–98%) followed by cyclization using p-toluenesulfonyl chloride and triethylamine in 41–100% yields. Thiadiazole isostere was also synthesized. The derivatives were screened for inhibition of AChE and BChE using Ellman’s spectrophotometric method. The compounds showed a moderate dual inhibition with IC50 values of 12.8–99.2 for AChE and from 53.1 µM for BChE. All the heterocycles were more efficient inhibitors of AChE. The most potent inhibitor, N-dodecyl-5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine 3t, was subjected to advanced reversibility and type of inhibition evaluation. Structure–activity relationships were identified. Many oxadiazoles showed lower IC50 values against AChE than established drug rivastigmine. According to molecular docking, the compounds interact non-covalently with AChE and BChE and block entry into enzyme gorge and catalytic site, respectively.
1. Introduction
Acetylcholine (ACh) is a potent neurotransmitter in both the central nervous system and the peripheral nervous system, that plays an important role in performing the cognitive functions. Enzyme acetylcholinesterase (AChE), which cleaves choline esters, is a serine hydrolase found mainly at neuromuscular junctions and cholinergic brain synapses. The enzyme inactivation induced by various inhibitors leads to acetylcholine accumulation, hyperstimulation of nicotinic and muscarinic receptors, and disruption of neurotransmission [1]. Uncontrolled acetylcholine can lead to a massive disturbance in the cholinergic system, respiratory arrest, and death [2].
According to the mode of action, AChE inhibitors can be divided into two groups: irreversible and reversible. Reversible competitive or non-competitive inhibitors (donepezil, rivastigmine, galantamine) are the protagonists of pharmacotherapy of Alzheimer disease symptoms. Their therapeutic effect is based on maintaining the level of ACh by slowing down its hydrolysis rate. Reversible AChE inhibitors of various chemical structures have noteworthy pharmacological application in the treatment of neurological disorders such as myasthenia gravis, Lewy bodies and Parkinson’s disease dementia, and as well as prophylactics against nerve agent intoxication [3].
Butyrylcholinesterase (BChE) is a sister enzyme to AChE, which was discovered more than 80 years ago [4] and is found throughout the body [5]. BChE catalyzes the hydrolysis of numerous esters of choline such as succinylcholine, adipoyl choline, benzoyl choline, and neurotoxic peptides [6,7]. Although AChE is a primary enzyme for the hydrolysis of ACh, BChE is a putative backup enzyme for this reaction. Changes in the homeostasis of ACh and its destruction by AChE involve numerous impairments such as muscular dystrophy, motor neuron diseases such as amyotrophic lateral sclerosis [8], congenital myasthenias [9] and myasthenia gravis [10]. While the reason for the widespread presence of BChE in the body is not well understood [11], it can contribute to the inactivation of the neurotransmitter acetylcholine, degrading many neurotoxic organophosphate esters and natural poisons including solanidine, physostigmine, cocaine, and others [12]. Thus, it plays important pharmacological and toxicological roles and is thought to be involved in the pathological progression. Based on these facts, a dual inhibition strategy for both these enzymes has been proposed to increase the effectiveness of the treatment strategy and expand the indications [13].
The five-membered ring 1,3,4-oxadiazole, one of four isomers, is a thermally stable aromatic molecule that was first prepared by Ainsworth in 1965 by the thermolysis of ethyl (E)-N-formylformohydrazonate at atmospheric pressure [14]. Synthesis approaches are summarized in the review article of Patel and co-authors [15]. The replacement of two –CH= groups in furan by two nitrogens (–N=) reduces the aromaticity of the resulting oxadiazole ring to such an extent that the oxadiazole ring exhibits the character of conjugated diene [16]. They undergo several reactions such as photochemical, thermal, electrophilic, and nucleophilic substitutions [17]. Several molecules with this valued low-lipophilicity moiety plays significant role in medicinal chemistry due to their diverse biological activities. They are widely applied in the development of novel compounds with anticonvulsant, antidepressant, analgesic, anti-inflammatory, antiallergic, antipsychotic, antimicrobial, antimycobacterial, antitumor and antiviral activities [18,19,20,21]. 1,3,4-Oxadiazole heterocycles are bioisosteres of amides and esters, which can significantly contribute to increased pharmacological activity by participating in hydrogen bonding interactions with the receptors [22].
In this work, we were focused primarily on 5-(hetero)aryl-1,3,4-oxadiazol-2-amines. Based on the literature, the amino group can be secondary, monosubstituted by (hetero)aryl or (cyclo)alkyl, or it can be incorporated into a heterocycle as a tertiary amine. For example, 5-aryl-1,3,4-oxadiazoles were tethered with donepezil analogue N-benzylpiperidine via (methyl)amino linker (Figure 1, I). The presence of oxadiazole resulted in the enhanced binding affinity toward the peripheral anionic site and was responsible for the extension of the N-benzylpiperidine moiety deeper into the catalytic anionic site, which yielded stronger dual inhibitors. Moreover, they showed antioxidant and neuroprotective properties and also inhibition of β-secretase-1 (BACE-1) and aggregation of amyloid β-peptide (Aβ). Additionally, 4-(pyridin-2-yl)piperazin-1-yl oxadiazole analogues (Figure 1, II) exhibited similar promising behavior together with adequate physicochemical properties [23,24]. 5-Aryl-N-(4-pyridyl)-1,3,4-oxadiazol-2-amines (Figure 1, III) were found to be dual inhibitors that also interfered with Aβ aggregation, oxidative stress, were blood–brain barrier permeable and exhibited in vivo activity in murine model [25]. The type of enzyme inhibition is not consistent, in that some derivatives were mixed [24], while some were non-competitive [25] inhibitors. Additionally, other authors have reported 2,5-disubstituted 1,3,4-oxadiazoles as AChE and/or BChE inhibitors, e.g., [26,27,28]. Thus, this scaffold belongs to multitargeting compounds that represent leading and the most perspective strategy to combat two or more pathogenetic pathways of neurodegeneration. From another point of view, oxadiazoles can be considered as cyclic analogues of 1,2-diacylhydrazines (Figure 1, IV) with also known activity against AChE and BChE [28,29]. (Hetero)arenes decorated by a longer alkyl (Figure 1, V) have been described as cholinesterases inhibitors as well; the length of this alkyl was crucial for bioactivity [28,30]. Additionally, 1,3,4-oxadiazol-2-thiols [31] and 1,3,4-thiadiazoles [32] have been investigated as potential cholinesterases inhibitors.
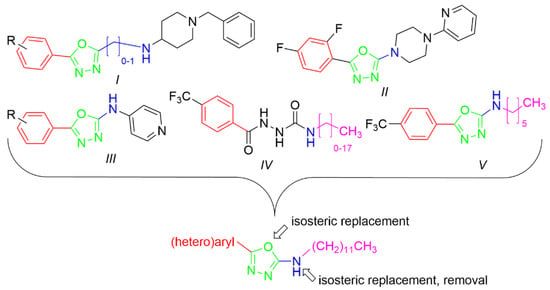
Figure 1.
Structural fragments involved in the structure of targeted derivatives and their design: 1,3,4-oxadiazoles, long alkyl, aryl, and amine/amide linker.
Considering the abovementioned facts, we designed, prepared, and evaluated N-dodecyl-5-substituted (hetero)aryl-1,3,4-oxadiazol-2-amines 3 and their isosteres 3s–3u as potential inhibitors AChE and BChE.
2. Results and Discussion
2.1. Synthesis
The synthetic scheme is illustrated in Figure 2. The synthesis of final oxadiazoles and their analogues 3a–3u started from aryl hydrazides that were either commercially available or prepared in-house from corresponding acid via Fisher esterification (with MeOH in the presence of a catalytic amount of sulfuric acid) and subsequent hydrazinolysis of the methyl ester. Then, hydrazide was reacted with a mild excess of dodecyl iso(thio)cyanate in acetonitrile. Crude 2-aryloylhydrazine-1-carboxamides 2 were purified by recrystallization with moderate to excellent yields of 67–98%. The lowest yields were for 2-bromopyridine derivative 2q, followed by 3,5-dinitrobenzohydrazide derivative 2k and thioamide 2t (for both 76%).
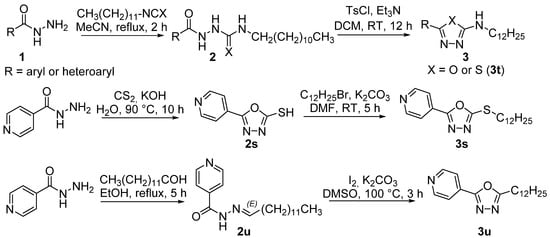
Figure 2.
Synthesis of the targeted derivatives 3 (DCM: dichloromethane, DMF: N,N-dimethylformamide, DMSO: dimethylsulfoxide, TsCl: p-toluenesulfonyl chloride, RT: room temperature.
2-Aryl-N-dodecylhydrazine-1-carboxamides 2 (or carbothioamide 2t) were treated with an excess of p-toluenesulfonyl chloride (3 eq.) and triethylamine (5 eq.) in dichloromethane (DCM). After purification, yields of 1,3,4-oxa(or thia)diazoles 3 were lower due to reduced conversion rate and/or less effective isolation (41–100%). In particular, oxadiazoles prepared from heterocyclic hydrazides were obtained in lower yields (derivatives of pyridazine 3r, picolinohydrazide 3n, 2-bromoisonicotinohydrazide 3q, and pyrazine 3p). The thiadiazole 3t was isolated with satisfactorily yield of 68%.
Two analogues were prepared in a different way. Dodecylsulfanyl derivative 3s was prepared from isonicotinoylhydrazide that was heated with an excess of carbon disulfide keeping a strong basic pH (73%). The 5-(pyridin-4-yl)-1,3,4-oxadiazol-2-thiol was treated with an over-stochiometric amount of 1-bromododecane as an alkylating agent and potassium carbonate in dimethylformamide. Thioether 3s was obtained quantitatively. 5-Dodecyl derivative 3u was synthesized from isoniazid by condensation reaction with tridecanal to form hydrazide-hydrazone 2u. This intermediate was cyclized to oxadiazole using a mild excess of iodine and potassium carbonate (76%).
The prepared compounds were characterized by 1H and 13C NMR, IR spectra and melting point. The purity was confirmed by thin-layer chromatography and elemental analysis. In the 1H NMR spectra, the signals of heteroaryls and substituted phenyls are clearly visible, while signals of aliphatic hydrogens from the dodecyl chain partially overlap. Here, we can distinguish signals of C1, C2 and C12, remaining hydrogen signals are merged. By analogy, the signals of most aliphatic carbons in the 13C NMR spectra are close together. As expected, both hydrazide hydrogen signals in the spectra of 2 disappeared in the spectra of 3 due to cyclization.
2.2. Inhibition of Acetyl- and Butyrylcholinesterase
The heterocycles 3 were screened for their ability to interfere with the function of AChE from electric eel (EeAChE) and BChE from equine serum (EqBChE) using Ellman’s method (Table 1). Their activities are expressed as the concentration producing 50% inhibition of enzymatic activity (IC50). In addition, we calculated selectivity indexes (SI) that quantify the selectivity for AChE as more inhibited cholinesterase. SI is the ratio of IC50 for BChE/IC50 for AChE. Clinically used AChE and BChE inhibitor rivastigmine was used as a comparator.

Table 1.
Inhibition of AChE and BChE and selectivity indexes of the heterocycles 3.
Most derivatives 3 caused dual inhibition of both cholinesterases (derivative 3s was an exception). AChE was inhibited more consistently with lower IC50 values of 12.8–99.2 µM with thiadiazole 3t being the most potent, followed by several 5-(4-substituted phenyl) derivatives: OMe (3c), Me (3b), F (3g), I (3j), Br (3i) and H (3a) with IC50 of 33.9–40.1 µM. More bulky substituents increased IC50 (tert-butyl, dimethylamino, 4-nitro). The conversion of thiadiazole to oxadiazole was detrimental (3t vs. 3l; 3.9× lower inhibition), as was the removal of secondary amine or its replacement with sulfide (3l vs. 3s and 3u). Focusing on 5-heteroaryls, 4-pyrimidinyl showed the best inhibition (3o, 45.5 µM) followed by various positional isomers of pyridine (4-pyridyl 3l, 2-pyridyl 3n and 3-pyridyl 3m; 50.2–64.2 µM). On the other hand, the pyrazine derivative 3p was the least active in this subgroup.
In contrast, BChE enzyme was inhibited at higher concentrations, and their range was wider. The IC50 of the dodecylsulfanyl compound 3s was higher than 500 µM. In fact, only the thiadiazole 3t and 5-(p-tolyl)-oxadiazole 3b were more potent inhibitors of BChE with IC50 of 53.05 and 89.97 µM, respectively. Remaining derivatives have IC50 ≥ 105 µM. Among the oxadiazoles derived from benzohydrazides 3a–3k, the optimal activity was associated with 4-methyl (3b), while its replacement by hydrogen (3a), methoxy (3c) or tert-butyl (3d) led to a decrease in activity. Halogens and dimethylamino group were also unfavorable. Interestingly, only the 4-NO2 group with completely different properties led to a comparably active derivative. Of the various 5-heteroaryls, the best inhibition is associated with 4-pyridyl (3l), but its bromination (3q) and isosteric replacement by pyridazine (3r) is tolerated. The switch of oxygen to sulfur (3l vs. 3t) boosted inhibitory potency 4.2×. On the other hand, replacement of exocyclic NH group with sulfur completely abolished the activity (3l vs. 3s), whereas its removal resulted in only a slight decrease in inhibition (3l vs. 3u).
We used the selectivity index values to describe the selectivity to particular ACh hydrolysing enzymes. All the derivatives were preferential inhibitors of AChE (SI within the range from 1.5 to 12.9). The most selective was iodinated derivative 3i; on the other hand, 4-nitro compound 3e was the most balanced inhibitor. Interestingly, an introduction of the second nitro group (3k) and increasing molecular weight of halogens (3g → 3j) resulted in a higher selectivity to AChE. Among the derivatives with a heterocycle on carbon 5, the least selective are brominated pyridine 3q and pyridazine 3r. Positional isomers (3m and 3n) of isoniazid-derived oxadiazole 3l are more selective to AChE. The removal of a heteroatom at the position 2 of oxadiazole led to more balanced inhibition (3l, 3s, and 3t vs. 3u).
Then, we tried to correlate activity with calculated lipophilicity (log Po/w; Table 1). However, there are no clear conclusive structure–activity relationships and differences in lipophilicity cannot fully explain differences in activity. For example, compounds with close log P values (3s–3u) showed different degrees of inhibition of both enzymes. The most potent AChE inhibitors were lowly lipophilic (3t, 3k), moderately (3c) and also highly lipophilic (3j). Analogously, compounds with the same log P values (3e, 3l–3n, and 3t) inhibited BChE over a wide concentration range of 53–369 µM.
When comparing the heterocycles 3 with the drug rivastigmine, none of the new compounds had lower IC50 values for BChE than this clinically used dual AChE/BChE inhibitor. By contrast, many of them (3a–3c, 3g–3k, 3l, 3o, and 3t) showed higher in vitro activities expressed as IC50 (up to 4.4× for 3t) against AChE.
2.2.1. Reversibility of the Inhibition
Based on the promising in vitro inhibition of AChE by thiadiazole 3t, we investigated the derivative to distinguish between reversible, pseudo-irreversible and irreversible inhibition on the basis of changes in enzyme activity over time in the presence of the inhibitor. In reversible inhibition, the activity of enzyme goes down immediately, but the inhibitory molecule is bound to the target for a short period of time, after which the inhibitor-enzyme complex dissociates, and enzyme activity is restored. In pseudo-irreversible inhibition, the inhibitor binds covalently to the enzyme, but the bond is broken down more slowly, delaying the return of enzyme activity to its initial state. In the case of irreversible inhibitors, the reaction between the enzyme and the inhibitor is not rapid because there is a time-dependent drop of activity. Irreversible inhibitors decrease enzyme activity gradually. The inhibitor permanently binds to the enzyme and prevents the enzyme from restoring its activity [33].
Three different concentrations were chosen for 3t according to its IC50 value for AChE. The first concentration was lower than IC50, the second was close to IC50 and the third was higher than IC50 (1, 10, and 100 µM, respectively). All obtained dependencies of A (expressed as residual activity on the Y-axis) versus time showed a very similar pattern (Figure 3). First, a rapid decrease in activity (for two lower concentrations, almost maximum inhibition is reached after 5 min), then a plateau phase followed by a recovery of enzyme activity over time.
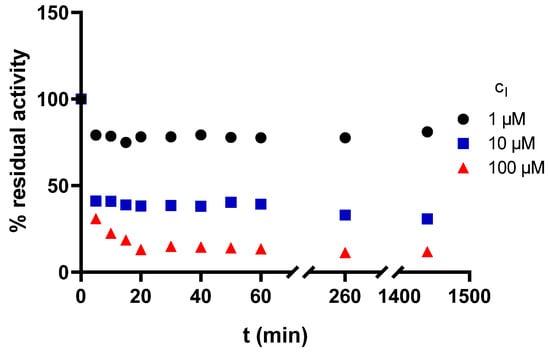
Figure 3.
The dependence of enzyme residual activity on time (enzyme: AChE, compound 3t).
In our opinion, we can conclude that the compound 3t acts as reversible AChE inhibitor.
2.2.2. Type of Inhibition
We also valuated the type of inhibition of 3t using AChE. In general, the reversible inhibitors can be classified as competitive, non-competitive, uncompetitive, or mixed type. The type of inhibition could be distinguished using the Lineweaver–Burk plot [34] and the corresponding comparison of two key kinetic parameters: maximum velocity (Vm) and Michaelis constant (KM) of the inhibited and uninhibited reactions. Based on the changes of these parameters and intercept of the lines in Lineweaver–Burk plot, the type of inhibition is uncovered.
The Lineweaver–Burk plot obtained for 3t and AChE is shown in Figure 4. Based on the plot, it can be concluded that this heterocycle causes a mixed type of inhibition. The inhibition is associated with a change in both KM and Vm and also their ratio is different compared to the uninhibited reaction. In the Lineweaver–Burk plot, the intersection of the lines is in quadrant III but not on axis.
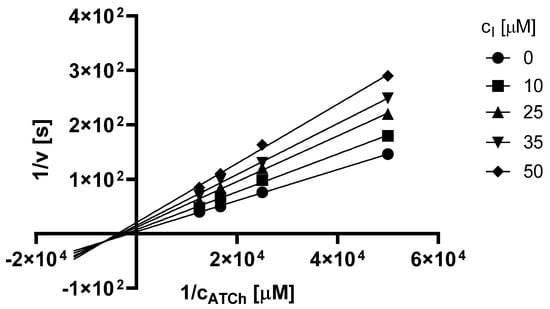
Figure 4.
Lineweaver–Burk plot for 3t and AChE inhibition (ATCh = acetylthiocholine).
2.3. Molecular Docking
For better visualization of the possible orientations of prepared compounds in enzymes, molecular modelling study was performed. Based on the obtained data, the structure–activity relationships were estimated.
The active site of AChE was studied extensively in the past and is now very well described. It is known to be at the bottom of a very narrow gorge penetrating deep under the surface of the enzyme. All the prepared most active ligands (3b, 3c, 3t) displayed similar orientation in the cavity of AChE (Figure 5) which corresponds with the results of in vitro testing, since the obtained activities were all in relatively small range. Variable 5-aryloxadiazole moiety was heading for the bottom of the gorge (though still quite far from the active site triad). Hydrophobic 2-dodecylamino moiety was convoluted, pointing out of the cavity, and blocking the entrance to the gorge. Several hydrophobic interactions with Trp286, Val294, Phe297, and Phe338 were observed. Interestingly, an important structural element seems to be the -NH- group at the dodecylamino moiety, which is stabilized at its position by hydrogen bonds (Asp74 or Tyr124). The replacement of the nitrogen with carbon or sulfur leads to decrease in the binding affinity (and also decrease in the in vitro inhibition data). The oxadiazole part of the molecules showed hydrogen bonding with Ser125. Aromatic/heteroaromatic moieties were located at the anionic site and displayed π-π stacking with Trp86 (Figure 6).
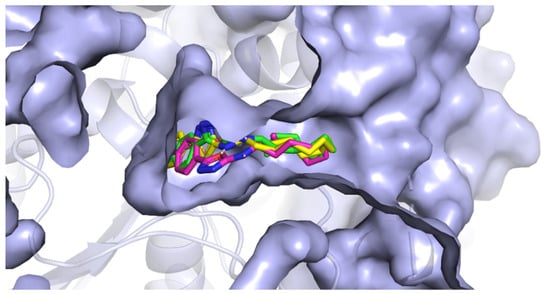
Figure 5.
The position of 3b, 3c, and 3t in the cavity of AChE.
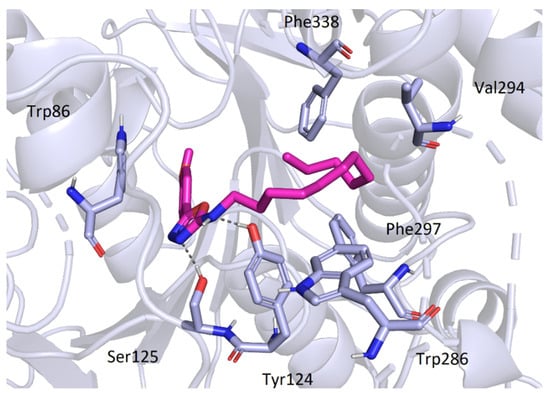
Figure 6.
Binding mode of the compound 3c in AChE.
Similarly to AChE, the active site of BChE is located at the bottom of the gorge. However, a considerable number of aromatic residues present in active center of AChE is replaced by smaller hydrophobic residues in BChE. This allows more spacious ligands to enter the cavity. The top score docking pose of 3t (being the most potent inhibitor in the series) showed the molecule deep within the cavity in considerable proximity to active site Ser198 and His438 (Figure 7). The ligand was stabilized in its conformation by several interactions (namely, hydrogen bonds of oxadiazole moiety with Gly116 and Gly117 and hydrophobic interactions of dodecylamino moiety with Trp82, Ala328, Phe329, and Trp430). Most of the investigated compounds exhibited the same or quite similar conformation, apart from 3s. Its dodecylamino moiety was placed in the same hydrophobic part of the cavity; however, the aryloxadiazole moiety was relatively far from Ser198 and His438 and showed only one interaction with Thr120 (Figure 8). This could stand behind its low ability to inhibit BChE.
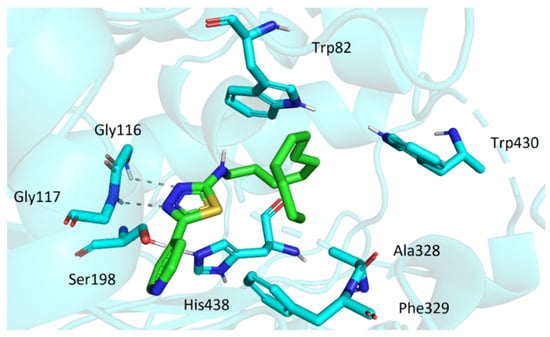
Figure 7.
The top score docking pose of 3t in active site of BChE.
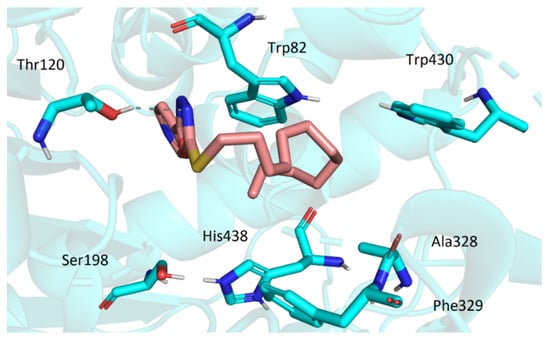
Figure 8.
The conformation of 3s in the active site of BChE.
3. Materials and Methods
3.1. Chemistry
3.1.1. General
All chemicals for synthesis and analysis were purchased from Merck KGaA (Darmstadt, Germany), Penta Chemicals Unlimited (Prague, Czech Republic), Avantor (Stříbrná Skalice, Czech Republic), and Lach-Ner (Neratovice, Czech Republic) and were used as received. The structures of the prepared substances were confirmed by 1H NMR and 13C NMR spectroscopy analysis. NMR spectra were measured in dimethylsulfoxide (DMSO-D6) as a solvent at ambient temperature or with tempering the sample (2b–2j, 2q, 3b–3k, 3m, 3o–3q, and 3u) at 60 °C by a JNM-ECZ 600R (600 MHz for 1H a 151 MHz for 13C; JEOL, Tokio, Japan) or a Varian VNMR S500 instrument (Varian Comp., Palo Alto, CA, USA). The chemical shifts δ are given in ppm and were referred indirectly to tetramethylsilane via residual signals of the solvent (2.50 for 1H and 39.51 for 13C spectra). The coupling constants (J) are reported in Hz. Infrared spectra were recorded by a Nicolet 6700 FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) in the range of 600–4000 cm−1, ATR (Ge) technique of measuring was used. Elemental analysis was performed on Vario MICRO Cube Element Analyzer (Elementar Analysensysteme, Hanau, Germany). Calculated as well as found values are given as percentages. Melting points were recorded using a Büchi B-545 apparatus (BÜCHI Labortechnik AG, Flawil, Switzerland) without corrections. Retention factors (Rf) of all prepared compounds, as well as reaction progresses, were analyzed by thin layer chromatography (TLC); the plates were coated with 0.2 mm Merck 60 F254 silica gel (Merck Millipore, Darmstadt, Germany) and were visualized by UV irradiation (254 nm). Dichloromethane (DCM)/methanol (93:7 v/v) mixture was used as the eluent.
Lipophilicity was calculated in silico using a free web tool SwissADME (http://www.swissadme.ch/index.php, accessed on 16 March 2022). It is expressed as log Po/w (MLOGP).
3.1.2. Synthesis
Synthesis of 2-Aryloyl-N-Dodecylhydrazine-1-(thio)Carboxamides (2a–2r) and Other Precursors (2s–2u)
Appropriate aryl hydrazide (1.0 mmol) was suspended in 10 mL of acetonitrile and heated to initiate boiling of the mixture, then dodecyl isocyanate (or dodecyl isothiocyanate for precursor of 3t; 1.05 of equivalents, 1.05 mmol) was added quickly in one portion. The reaction mixture was refluxed continuously for 2 h, then left to cool down at RT and stored for 1 h at −20 °C. Solid material was filtered off and recrystallized from methanol to provide pure hydrazides 2. The progress of the reaction was monitored by TLC.
2-Benzoyl-N-dodecylhydrazine-1-carboxamide 2a. White solid; yield 98%; mp 137–137 °C. IR (ATR): 608, 661, 689, 749, 902, 1026, 1065, 1246, 1275, 1465, 1489, 1540, 1563, 1662, 1682, 2850, 2917, 2954, 3296 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.05 (1H, s, NH-CO-NH-CH2), 7.85 (2H, dd, J = 7.1, 1.7 Hz, H2, H6), 7.73 (1H, s, NH-CO-Ar), 7.53–7.49 (1H, m, H4), 7.43 (2H, t, J = 7.7 Hz, H3, H5), 6.40 (1H, t, J = 5.8 Hz, NH-CH2), 2.97 (2H, q, J = 6.6 Hz, NH-CH2), 1.34 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.82, 158.87, 133.32, 132.12, 128.79, 128.07, 31.84, 30.41, 29.61, 29.56, 29.38, 29.36, 29.34, 29.32, 29.26, 26.85, 22.63, 14.48. Elemental analysis for C20H33N3O2 (347.50); calculated: C, 69.13; H, 9.57; N, 12.09, found: C, 68.19; H, 9.44; N, 12.30. Rf: 0.66.
N-Dodecyl-2-(4-methylbenzoyl)hydrazine-1-carboxamide 2b. White solid; yield 95%; mp 121–122 °C. IR (ATR): 608, 661, 721, 746, 838, 911, 1191, 1252, 1265, 1338, 1469, 1495, 1542, 1583, 1613, 1642, 1665, 2849, 2917, 2954, 3294 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.96 (1H, s, NH-CO-NH-CH2), 7.75 (2H, d, J = 8.2 Hz, H2, H6), 7.69 (1H, s, NH-CO-Ar), 7.24 (2H, d, J = 8.1 Hz, H3, H5), 6.37 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.6 Hz, NH-CH2), 2.32 (3H, s, Ph-CH3), 1.38–1.29 (2H, m, NH-CH2-CH2), 1.21–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.73, 158.92, 142.05, 130.53, 129.31, 128.09, 31.84, 30.41, 29.61, 29.56, 29.42, 29.40, 29.38, 29.36, 29.26, 26.85, 22.63, 21.52, 14.48. Elemental analysis for C21H35N3O2 (361.53); calculated: C, 69.77; H, 9.76; N, 11.62, found: C, 69.89; H, 9.70; N, 11.73. Rf: 0.60.
N-Dodecyl-2-(4-methoxybenzoyl)hydrazine-1-carboxamide 2c. White solid; yield 95%; mp 158–159 °C. IR (ATR): 605, 726, 756, 841, 912, 1038, 1127, 1181, 1254, 1335, 1463, 1504, 1540, 1564, 1609, 1642, 2850, 2921, 2956, 3310 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.90 (1H, s, NH-CO-NH-CH2), 7.83 (2H, d, J = 9.1 Hz, H2, H6), 7.66 (1H, s, NH-CO-Ar), 6.96 (2H, d, J = 8.9 Hz, H3, H5), 6.37 (1H, t, J = 5.9 Hz, NH-CH2), 3.77 (3H, s, OCH3), 2.96 (2H, q, J = 6.7 Hz, NH-CH2), 1.37–1.30 (2H, m, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.36, 162.40, 159.00, 129.94, 125.49, 114.02, 55.90, 31.84, 30.41, 29.65, 29.63, 29.61, 29.59, 29.57, 29.38, 29.26, 26.85, 22.63, 14.48. Elemental analysis for C21H35N3O3 (377.53); calculated: C, 66.81; H, 9.34; N, 11.13, found: C, 66.85; H, 9.38; N, 11.14. Rf: 0.51.
2-[4-(tert-Butyl)benzoyl]-N-dodecylhydrazine-1-carboxamide 2d. White solid; yield 91%; mp 119–120 °C. IR (ATR): 621, 632, 718, 897, 1018, 1083, 1113, 1200, 1290, 1305, 1364, 1464, 1506, 1537, 1614, 1668, 2853, 2924, 2958, 3250 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.98 (1H, s, NH-CO-NH-CH2), 7.79 (2H, d, J = 8.7 Hz, H2, H6), 7.71 (1H, s, NH-CO-Ar), 7.44 (2H, d, J = 8.7 Hz, H3, H5), 6.37 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.7 Hz, NH-CH2), 1.34 (2H, q, J = 7.0 Hz, NH-CH2-CH2), 1.26 (9H, s, C(CH3)3), 1.23–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.68, 158.92, 154.96, 130.56, 127.94, 125.53, 35.17, 31.85, 31.45, 30.40, 29.68. 29.66, 29.64, 29.62, 29.57, 29.39, 29.27, 26.85, 22.64, 14.48. Elemental analysis for C24H41N3O2 (403.61); calculated: C, 71.42; H, 10.24; N, 10.41, found: C, 71.55; H, 10.29; N, 10.40. Rf: 0.62.
N-Dodecyl-2-(4-nitrobenzoyl)hydrazine-1-carboxamide 2e. White solid; yield 88%; mp 220–221 °C. IR (ATR): 625, 679, 713, 852, 868, 911, 1011, 1056, 1250, 1263, 1299, 1316, 1344, 1468, 1484, 1534, 1551, 1590, 1605, 1641, 1671, 2852, 2920, 3275 cm−1.1H NMR (600 MHz, DMSO-D6) δ 10.26 (1H, s, NH-CO-NH-CH2), 8.27 (2H, d, J = 8.8 Hz, H3, H5), 8.07 (2H, d, J = 8.8 Hz, H2, H6), 7.76 (1H, s, NH-CO-Ar), 6.35 (1H, t, J = 6.1 Hz, NH-CH2), 3.00 (2H, q, J = 6.6 Hz, NH-CH2), 1.37 (2H, q, J = 7.0 Hz, NH-CH2-CH2), 1.23–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.28, 158.52, 149.91, 139.22, 129.58, 123.91, 31.77, 30.33, 29.53, 29.50, 29.48, 29.46, 29.44, 29.30, 29.16, 26.83, 22.53, 14.33. Elemental analysis for C20H32N4O4 (392.50); calculated: C, 61.20; H, 8.22; N, 14.27, found: C, 61.29; H, 8.11; N, 14.36. Rf: 0.51.
2-[4-(Dimethylamino)benzoyl]-N-dodecylhydrazine-1-carboxamide 2f. White solid; yield 88%; mp 160–161 °C. IR (ATR): 609, 721, 763, 836, 950, 1068, 1173, 1211, 1265, 1288, 1340, 1375, 1468, 1515, 1540, 1556, 1607, 1637, 1685, 2850, 2918, 2952, 3292 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.72 (1H, s, NH-CO-NH-CH2), 7.75 (2H, d, J = 9.0 Hz, H2, H6), 7.59 (1H, s, NH-CO-Ar), 6.70 (2H, d, J = 8.9 Hz, H3, H5), 6.33 (1H, t, J = 5.8 Hz, NH-CH2), 3.02–2.95 (8H, m, N-CH3, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.42, 158.81, 152.51, 129.08, 119.38, 110.85, 39.85, 31.48, 30.06, 29.26, 29.25, 29.24, 29.21, 29.20, 29.02, 28.90, 26.49, 22.27, 14.12. Elemental analysis for C22H38N4O2 (390.57); calculated: C, 67.66; H, 9.81; N, 14.35, found: C, 67.75; H, 9.90; N, 14.34. Rf: 0.46.
N-Dodecyl-2-(4-fluorobenzoyl)hydrazine-1-carboxamide 2g. White solid; yield 88%; mp 202–203 °C. IR (ATR): 610, 645, 726, 849, 914, 1011, 1164, 1232, 1262, 1339, 1466, 1502, 1544, 1569, 1605, 1638, 1661, 2851, 2921, 3301 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.08 (1H, s, NH-CO-NH-CH2), 7.95–7.89 (2H, m, H2, H6), 7.74 (1H, s, NH-CO-Ar), 7.31–7.24 (2H, m, H3, H5), 6.43 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.6 Hz, NH-CH2), 1.35–1.32 (2H, m, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.64 (d, J = 54.1 Hz), 163.81, 158.84, 130.77 (d, J = 8.9 Hz), 129.81 (d, J = 2.9 Hz), 115.76 (d, J = 21.8 Hz), 31.84, 30.40, 29.61, 29.58, 29.56, 29.38, 29.36, 29.34, 29.26, 26.85, 22.63, 14.47. Elemental analysis for C20H32FN3O2 (365.49); calculated: C, 65.72; H, 8.83; N, 11.50, found: C, 65.80; H, 8.92; N, 11.51. Rf: 0.48.
2-(4-Chlorobenzoyl)-N-dodecylhydrazine-1-carboxamide 2h. White solid; yield 91%; mp 215–216 °C. IR (ATR): 602, 680, 719, 754, 845, 911, 1011, 1092, 1252, 1266, 1339, 1470, 1485, 1587, 1599, 1639, 1664, 2850, 2918, 2954, 3299 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.13 (1H, s, NH-CO-NH-CH2), 7.86 (2H, d, J = 8.2 Hz, H2, H6), 7.76 (1H, s, NH-CO-Ar), 7.52 (2H, d, J = 8.3 Hz, H3, H5), 6.44 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.6 Hz, NH-CH2), 1.36–1.32 (2H, m, NH-CH2-CH2), 1.21–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.85, 158.77, 136.98, 132.11, 130.01, 128.92, 31.84, 30.39, 29.67, 29.65, 29.63, 29.61, 29.56, 29.37, 29.26, 26.84, 22.63, 14.48. Elemental analysis for C20H32ClN3O2 (381.95); calculated: C, 62.89; H, 8.45; N, 11.00, found: C, 62.81; H, 8.56; N, 11.02. Rf: 0.48.
2-(4-Bromobenzoyl)-N-dodecylhydrazine-1-carboxamide 2i. White solid; yield 90%; mp 214–215 °C. IR (ATR): 607, 681, 719, 752, 845, 855, 911, 1015, 1033, 1254, 1260, 1266, 1339, 1474, 1499, 1587, 1599, 1639, 1664, 2854, 2928, 2954, 3250 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.19 (1H, s, NH-CO-NH-CH2), 7.85 (2H, d, J = 8.3 Hz, H2, H6), 7.79 (1H, s, NH-CO-Ar), 7.70 (2H, d, J = 8.3 Hz, H3, H5), 6.46 (1H, t, J = 5.9 Hz, NH-CH2), 2.95 (2H, q, J = 6.6 Hz, NH-CH2), 1.37–1.33 (2H, m, NH-CH2-CH2), 1.22–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.62, 158.40, 132.10, 131.49, 129.83, 125.54, 31.48, 30.03, 29.27, 29.25, 29.23, 29.21, 29.17, 29.02, 28.90, 26.48, 22.28, 14.13. Elemental analysis for C20H32BrN3O2 (426.40); calculated: C, 56.34; H, 7.56; N, 9.85, found: C, 56.42; H, 7.66; N, 9.79. Rf: 0.51.
N-Dodecyl-2-(4-iodobenzoyl)hydrazine-1-carboxamide 2j. White solid; yield 94%; mp 216–217 °C. IR (ATR): 623, 635, 718, 750, 840, 908, 1005, 1066, 1252, 1268, 1339, 1470, 1479, 1507, 1542, 1590, 1636, 1663, 2850, 2917, 2951, 3303 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.11 (1H, s, NH-CO-NH-CH2), 7.84–7.82 (2H, m, H3, H5), 7.74 (1H, s, NH-CO-Ar), 7.63–7.61 (2H, m, H2, H6), 6.42 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (q, J = 6.8 Hz, NH-CH2), 1.36–1.31 (2H, m, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.25, 158.76, 137.71, 132.78, 130.01, 99.82, 31.84, 30.38, 29.65, 29.63, 29.61, 29.59, 29.56, 29.37, 29.26, 26.84, 22.64, 14.49. Elemental analysis for C20H32IN3O2 (473.40); calculated: C, 50.74; H, 6.81; N, 8.88, found: C, 50.86; H, 6.90; N, 8.95. Rf: 0.51.
2-(3,5-Dinitrobenzoyl)-N-dodecylhydrazine-1-carboxamide 2k. Greyish solid, yield 76%; mp 157–159 °C. IR (ATR): 717, 732, 920, 1084, 1241, 1350, 1466, 1476, 1538, 1569, 1615, 1653, 2848, 2924, 3099, 3216, 3357 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.81 (1H, s, NH-CO-NH-CH2), 9.06 (2H, d, J = 2.0 Hz, H2, H6), 8.99 (1H, t, J = 2.1 Hz, H4), 8.04 (1H, s, NH-CO-Ar), 6.67 (1H, s, NH-CH2), 3.02 (2H, q, J = 6.6 Hz, NH-CH2), 1.41–1.36 (2H, m, NH-CH2-CH2), 1.27–1.21 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 162.70, 158.10, 148.31, 135.70, 128.01, 121.41, 39.20, 31.49, 30.06, 29.27, 29.25, 29.22, 29.20, 29.03, 28.91, 26.49, 22.29, 14.14. Elemental analysis for C20H31N5O6 (437.50); calculated C, 54.91; H, 7.14; N, 16.01, found: C, 55.01; H, 7.21; N, 16.00. Rf: 0.50.
N-Dodecyl-2-isonicotinoylhydrazine-1-carboxamide 2l. The synthesis and characterization of the compound was published previously by our group [21].
N-Dodecyl-2-nicotinoylhydrazine-1-carboxamide 2m. White solid; yield 95%; mp 143–144 °C. IR (ATR): 632, 721, 826, 903, 1028, 1124, 1194, 1253, 1266, 1317, 1379, 1470, 1525, 1589, 1638, 1695, 2848, 2916, 2979, 3197, 3311 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.25 (1H, s, NH-CO-NH-CH2), 9.00 (1H, dd, J = 2.3, 0.9 Hz, H2), 8.69 (1H, dd, J = 4.8, 1.7 Hz, H4), 8.21–8.15 (1H, m, H6), 7.82 (1H, s, NH-CO-Ar), 7.48 (1H, ddd, J = 7.9, 4.8, 0.9 Hz, H5), 6.50 (t, J = 5.9 Hz, NH-CH2), 3.00–2.94 (2H, m, NH-CH2), 1.37–1.32 (2H, m, NH-CH2-CH2), 1.21–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.48, 158.70, 152.74, 149.17, 135.79, 128.98, 123.97, 31.84, 30.39, 29.61, 29.59, 29.57, 29.56, 29.55, 29.37, 29.25, 26.85, 22.63, 14.48. Elemental analysis for C19H32N4O2 (348.49); calculated: C, 65.48; H, 9.26; N, 16.08, found: C, 65.42; H, 9.15; N, 16.01. Rf: 0.24.
N-Dodecyl-2-picolinoylhydrazine-1-carboxamide 2n. White solid; yield 89%; mp 101–103 °C. IR (ATR): 613, 727, 747, 823, 1017, 1038, 1066, 1240, 1271, 1435, 1465, 1481, 1497, 1565, 1573, 1634, 1715, 2849, 2926, 2956, 3316, 3377 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 10.13 (1H, s, NH-CO-NH-CH2), 8.65 (1H, dd, J = 4.6, 1.4 Hz, H6), 8.03–7.97 (2H, m, H3, H4), 7.85 (1H, s, NH-CO-Ar), 7.62 (1H, ddd, J = 6.9, 4.7, 2.5 Hz, H5), 6.37 (1H, t, J = 5.7 Hz, NH-CH2), 2.99 (2H, q, J = 6.6 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 163.71, 158.08, 149.65, 148.67, 137.84, 126.95, 122.41, 39.40, 31.47, 29.99, 29.24, 29.22, 29.21, 29.19, 29.00, 28.89, 26.48, 22.26, 14.11. Elemental analysis for C19H32N4O2 (348.49); calculated: C, 65.48; H, 9.26; N, 16.08, found: C, 65.59; H, 9.19; N, 16.19. Rf: 0.49.
N-Dodecyl-2-(pyrimidine-4-carbonyl)hydrazine-1-carboxamide 2o. White solid, yield 89%; mp 133–134 °C. IR (ATR): 605, 639, 664, 765, 870, 997, 1096, 1157, 1231, 1289, 1342, 1389, 1466, 1506, 1556, 1582, 1659, 1696, 2852, 2922, 3090, 2922, 3090, 3214, 3292 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.46 (1H, s, NH-CO-NH-CH2), 9.33 (1H, d, J = 1.5 Hz, H2), 9.07 (1H, d, J = 5.0 Hz, H5), 8.00 (1H, dd, J = 5.0, 1.4 Hz, H6), 7.95 (1H, s, NH-CO-Ar), 6.43 (1H, t, J = 5.7 Hz, NH-CH2), 2.99 (2H, q, J = 6.8 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 162.63, 159.69, 158.06, 157.82, 156.37, 119.01, 39.40, 31.46, 29.97, 29.23, 29.22, 29.19, 29.00, 28.88, 26.48, 22.26, 14.12. Elemental analysis for C18H31N5O2 (349.48); calculated C, 61.86; H, 8.94; N, 20.04, found: C, 61.95; H, 9.01; N, 20.15. Rf: 0.32.
N-Dodecyl-2-(pyrazine-2-carbonyl)hydrazine-1-carboxamide 2p. White solid, yield 79%; mp 128–132 °C. IR (ATR): 632, 725, 760, 915, 1021, 1072, 1117, 1170, 1224, 1263, 1268, 1408, 1466, 1490, 1567, 1644, 1698, 1717, 2849, 2923, 2955, 3310 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.35 (1H, s, NH-CO-NH-CH2), 9.16 (1H, d, J = 1.5 Hz, H3), 8.88 (1H, d, J = 2.5 Hz, H6), 8.74 (1H, dd, J = 2.4, 1.5 Hz, H5), 7.91 (1H, s, NH-CO-Ar), 6.42 (1H, t, J = 5.7 Hz, NH-CH2), 3.00 (2H, q, J = 6.8 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 162.98, 157.99, 147.84, 144.81, 143.86, 143.61, 39.41, 31.48, 30.00, 29.27, 29.25, 29.24, 29.20, 29.01, 28.90, 26.49, 22.27, 14.11. Elemental analysis for C18H31N5O2 (349.48); calculated C, 61.86; H, 8.94; N, 20.04, found: C, 62.00; H, 9.14; N, 20.07. Rf: 0.46.
2-(2-Bromoisonicotinoyl)-N-dodecylhydrazine-1-carboxamide 2q. White solid; yield 67%; mp 169–171 °C. IR (ATR): 634, 725, 739, 929, 1078, 1245, 1261, 1337, 1370, 1462, 1539, 1567, 1650, 2850, 2923, 2953, 3040, 3281 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.46 (1H, s, NH-CO-NH-CH2), 8.56 (1H, d, J = 5.1 Hz, H6), 8.03 (1H, d, J = 1.4 Hz, H3), 7.94 (1H, s, NH-CO-Ar), 7.81 (1H, dd, J = 5.1, 1.4 Hz, H5), 6.58 (1H, t, J = 5.9 Hz, NH-CH2), 3.00 (2H, q, J = 6.6 Hz, NH-CH2), 1.36 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 163.48, 158.06, 151.37, 143.17, 141.92, 126.05, 121.50, 39.36, 30.00, 29.31, 29.24, 29.20, 29.19, 19.11, 29.00, 28.89, 26.46, 22.27, 14.12. Elemental analysis for C19H31BrN4O2 (427.39); calculated: C, 53.40; H, 7.31; N, 13.11, found: C, 53.51; H, 7.22; N, 13.18. Rf: 0.33.
N-Dodecyl-2-(pyridazine-4-carbonyl)hydrazine-1-carboxamide 2r. White solid; yield 91%; mp 165–166 °C. IR (ATR): 644, 675, 716, 724, 753, 895, 979, 1052, 1247, 1265, 1288, 1351, 1464, 1481, 1545, 1569, 1620, 1662, 2849, 2922, 2954, 3080, 3177, 3349 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.60 (1H, s, NH-CO-NH-CH2), 9.56 (1H, dd, J = 2.3, 1.3 Hz, H3), 9.45 (1H, dd, J = 5.2, 1.3 Hz, H6), 8.02 (1H, dd, J = 5.3, 2.3 Hz, H5), 7.98 (1H, s, NH-CO-Ar), 6.60 (1H, t, J = 5.7 Hz, NH-CH2), 3.01 (2H, q, J = 6.6 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 163.44, 157.99, 152.29, 148.91, 130.31, 124.53, 39.87, 31.45, 29.98, 29.94, 29.21, 29.19, 29.17, 28.97, 28.86, 26.45, 22.24, 14.02. Elemental analysis for C18H31N5O2 (349.48); calculated: C, 61.86; H, 8.94; N, 20.04, found: C, 61.90; H, 9.02; N, 20.14. Rf: 0.13.
Isoniazid (10 mmol, 1.37 g) was dissolved in 50 mL of 5% aqueous solution of potassium hydroxide, then carbon disulfide (1.5 eq., 15 mmol, 1.14 g) was added in one portion. Reaction mixture was heated to 90 °C for 10 h. The mixture was cooled to room temperature and acidified to pH = 6 with 1M HCl. Residue precipitate was filtered off, washed with 10 mL of cold water and recrystallized from ethanol, gave pure 5-(pyridin-4-yl)-1,3,4-oxadiazol-2-thiol 2s [35].
5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-thiol 2s [35]. Yellow solid, yield 73%; mp 264–266 °C. IR (ATR): 693, 722, 734, 828, 887, 1010, 1051, 1076, 1147, 1218, 1236, 1311, 1331, 1368, 1421, 1496, 1553, 1597, 1846, 2385 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 8.82–8.79 (2H, m, H2, H6), 7.82–7.79 (2H, m, H3, H5). 13C NMR (151 MHz, DMSO-D6) δ 178.02, 158.94, 151.02, 129.95, 119.79. Elemental analysis for C7H5N3OS (179.20); calculated C, 46.92; H, 2.81; N, 23.45, found: C, 47.02; H, 2.95; N, 23.51. Rf: 0.46.
N-Dodecyl-2-isonicotinoylhydrazine-1-carbothioamide 2t. White solid, yield 76%; mp 199–201 °C. IR (ATR): 656, 692, 720, 752, 850, 908, 999, 1062, 1115, 1229, 1249, 1269, 1360, 1404, 1472, 1481, 1502, 1530, 1556, 1653, 2852, 2923, 3136, 3287 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.59 (1H, s, NH-CS-NH-CH2), 9.32 (1H, s, NH-CS-Ar), 8.77–8.74 (2H, m, H2, H6), 8.14 (1H, t, J = 5.7 Hz, NH-CH2), 7.82–7.79 (2H, m, H3, H5), 3.41 (2H, q, J = 6.8 Hz, NH-CH2), 1.47 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.29–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 201.01, 164.58, 150.32, 139.78, 121.82, 43.91, 31.47, 29.24, 29.19, 29.17, 28.99, 28.89, 28.88, 28.86, 26.42, 22.27, 14.12. Elemental analysis for C19H32N4OS (364.55); calculated C, 62.60; H, 8.85; N, 15.37, found: C, 62.69; H, 8.92; N, 15.48. Rf: 0.32.
Tridecanal (1 mmol, 198 mg) was dissolved in 20 mL of ethanol, 1 eq. of isoniazid (1 mmol, 137 mg) was added to the solution and reaction mixture was heated to reflux for 5 h. The mixture was reduced the volume in a rotary evaporator, and then stored in the freezer to crystalize (−20 °C). The solid product was filtered off and washed with methanol to provide pure hydrazone 2u [36].
(E)-N′-Tridecylideneisonicotinohydrazide 2u [37]. White solid, yield 90%; mp 90–92 °C. IR (ATR): 655, 670, 728, 754, 846, 877, 1043, 1295, 1367, 1412, 1460, 1548, 1598, 1624, 1655, 2848, 2923, 2955, 3070, 3257 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 11.62 (1H, s, NH), 8.75–8.73 (2H, m, H2, H6), 7.76–7.73 (3H, m, H3, H5, N=CH), 2.26 (2H, td, J = 7.4, 5.5 Hz, =CH-CH2), 1.47 (2H, p, J = 7.1 Hz, =CH-CH2-CH2), 1.32–1.19 (18H, m, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2, C12H2), 0.84 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 161.36, 153.98, 150.42, 140.82, 121.63, 32.18, 31.48, 29.24, 29.21, 29.14, 28.99, 28.90, 28.83, 26.12, 22.28, 14.12. Elemental analysis for C19H31N3O (317.48); calculated C, 71.88; H, 9.84; N, 13.24, found: C, 71.95; H, 9.99; N, 13.15. Rf: 0.31.
Procedure for Synthesis of 2-Aryloyl-N-Dodecyl-1,3,4-Oxadiazol-2-Amines (3a–3r) and Their Analogues (3s–3u)
An amount of 0.5 mmol of the appropriate 2-aryl-N-dodecylhydrazine-1-carboxamide (or N-dodecyl-2-isonicotinoylhydrazine-1-carbothioamide) 2 was suspended in 30 mL of DCM. Thereafter, 1.5 mmol (3 eq., 286 mg) of p-toluenesulfonyl chloride was added to the solution. After a complete dissolution of p-toluenesulfonyl chloride, 2.5 mmol of triethylamine (5 eq., 348 µL) was added dropwise. The mixture was allowed to react at room temperature for 12 h. The reaction mixture was evaporated to dryness on a rotary evaporator and transferred to a separatory funnel with 30 mL of ethyl acetate (EtOAc) and 30 mL of demineralized water. The organic phase was washed with 30 mL of a saturated NaHCO3 solution followed by 30 mL of a saturated brine. The organic phase was dried over anhydrous sodium sulfate for 45 min, then the desiccant was separated, and the mixture was evaporated to dryness. The crude product was overlaid with dry DCM and stored at −20 °C overnight. The solid product was filtered off and washed twice with 5 mL of ice-cold DCM. The progress of the reaction was monitored by TLC using a mixture of DCM with MeOH (93:7 v/v) as a mobile phase.
N-Dodecyl-5-phenyl-1,3,4-oxadiazol-2-amine 3a. White solid, yield 100%; mp 114–115 °C. IR (ATR): 631, 635, 722, 750, 829, 833, 838, 1036, 1050, 1111, 1184, 1270, 1301, 1400, 1484, 1505, 1588, 1674, 2851, 2924, 3240 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.87–7.67 (2H, m, H2, H6), 7.55–7.38 (3H, m, H3, H5, NH), 3.22 (2H, q, J = 6.6 Hz, NH-CH2), 1.56 (2H, p, J = 7.2 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.35, 158.05, 130.77, 129.62, 125.65, 125.13, 43.30, 31.74, 29.48, 29.46, 29.44, 29.41, 29.39, 29.14, 29.11, 26.71, 22.48, 14.26. Elemental analysis for C20H31N3O (329.49); calculated C, 72.91; H, 9.48; N, 12.75, found: C, 73.00; H, 9.59; N, 12.64. Rf: 0.77.
N-Dodecyl-5-(p-tolyl)-1,3,4-oxadiazol-2-amine 3b. White solid, yield 91%; mp 121–122 °C. IR (ATR): 630, 724, 741, 829, 838, 1030, 1052, 1113, 1173, 1266, 1300, 1441, 1469, 1506, 1587, 1614, 2850, 2920, 3268 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.66 (2H, d, J = 7.8 Hz, H2, H6), 7.41–7.39 (1H, m, NH), 7.29 (2H, d, J = 7.9 Hz, H3, H5), 3.20 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 2.33 (3H, s, Ph-CH3), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.24–1.17 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.17, 158.16, 140.72, 130.19, 125.65, 122.41, 43.28, 31.74, 29.47, 29.44, 29.42, 29.40, 29.38, 29.13, 29.11, 26.70, 22.48, 21.45, 14.27. Elemental analysis for C21H33N3O (343.52); calculated C, 73.43; H, 9.68; N, 12.23, found: C, 73.52; H, 9.60; N, 12.33. Rf: 0.75.
N-Dodecyl-5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-amine 3c. White solid, yield 90%; mp 116–117 °C. IR (ATR): 606, 634, 724, 739, 903, 828, 838, 956, 1029, 1049, 1173, 1266, 1299, 1469, 1506, 1587, 1614, 2850, 2921, 3266 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.70 (2H, d, J = 8.4 Hz, H2, H6), 7.35–7.32 (1H, m, NH), 7.03 (2H, d, J = 8.4 Hz, H3, H5), 3.80 (3H, s, OCH3), 3.19 (2H, q, J = 6.6 Hz, NH-CH2), 1.55 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.7 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.03, 161.56, 158.01, 127.41, 117.72, 115.28, 55.99, 43.30, 31.74, 29.48, 29.46, 29.44, 29.43, 29.41, 29.14, 29.11, 26.71, 22.48, 14.27. Elemental analysis for C21H33N3O2 (359.51); calculated C, 70.16; H, 9.25; N, 11.69, found: C, 70.11; H, 9.15; N, 11.81. Rf: 0.69.
5-[4-(tert-Butyl)phenyl]-N-dodecyl-1,3,4-oxadiazol-2-amine 3d. White solid, yield 87%; mp 97–98 °C. IR (ATR): 614, 636, 725, 836, 852, 902, 1016, 1049, 1114, 1269, 1363, 1468, 1500, 1544, 1613, 2851, 2921, 3261 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.70 (2H, d, J = 8.5 Hz, H3, H5), 7.50 (2H, d, J = 8.5 Hz, H2, H6), 7.41 (1H, s, NH), 3.20 (2H, td, J = 7.1, 5.7 Hz, NH-CH2), 1.56 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.28 (9H, s, C(CH3)3), 1.25–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.46, 157.36, 132.75, 127.53, 124.27, 124.12, 43.28, 31.74, 29.48, 29.44, 29.43, 29.41, 29.39, 29.37, 29.36, 29.34, 29.12, 26.68, 22.49, 14.27. Rf: 0.77.
N-Dodecyl-5-(4-nitrophenyl)-1,3,4-oxadiazol-2-amine 3e. White solid, yield 90%; mp 127–128 °C. IR (ATR): 680, 719, 733, 857, 953, 1013, 1047, 1072, 1296, 1350, 1479, 1535, 1507, 1562, 1605, 2849, 2916, 3250 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 8.32–8.29 (2H, m, H2, H6), 8.01–7.99 (2H, m, H3, H5), 7.76 (1H, t, J = 5.8 Hz, NH), 3.25 (2H, q, J = 6.1 Hz, NH-CH2), 1.57 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.26–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.98, 156.77, 148.72, 130.52, 126.61, 124.97, 43.30, 31.74, 29.48, 29.46, 29.44, 29.43, 29.41, 29.33, 29.12, 26.68, 22.48, 14.25. Elemental analysis for C20H30N4O3 (374.49); calculated C, 64.15; H, 8.08; N, 14.96, found: C, 64.26; H, 8.00; N, 15.08. Rf: 0.85.
5-[4-(Dimethylamino)phenyl]-N-dodecyl-1,3,4-oxadiazol-2-amine 3f. White solid, yield 60%; mp 118–121 °C. IR (ATR): 612, 664, 721, 739, 815, 952, 1047, 1064, 1164, 1196, 1227, 1270, 1369, 1468, 1515, 1578, 1614, 1687, 2848, 2916, 3237 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 7.77 (2H, d, J = 8.8 Hz, H3, H5), 7.59 (2H, d, J = 8.9 Hz, H2, H6), 6.80 (1H, t, J = 5.7 Hz, NH), 3.19 (2H, q, J = 6.6 Hz, NH-CH2), 2.97 (6H, s, N-CH3), 1.54 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.33–1.16 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 161.84, 160.94, 152.33, 127.63, 112.08, 111.90, 42.28, 31.49, 29.23, 29.19, 29.13, 29.05, 28.91, 28.70, 27.07, 26.12, 22.29, 14.15. Elemental analysis for C22H36N4O (372.56); calculated C, 70.93; H, 9.74; N, 15.04, found: C, 80.02; H, 9.81; N, 14.95. Rf: 0.74.
N-Dodecyl-5-(4-fluorophenyl)-1,3,4-oxadiazol-2-amine 3g. White solid, yield 81%; mp 104–105 °C. IR (ATR): 609, 660, 737, 820, 841, 1029, 1050, 1158, 1249, 1268, 1286, 1412, 1471, 1505, 1609, 1623, 2849, 2917, 2954, 3271 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.83–7.79 (2H, m, H2, H6), 7.45 (1H, s, NH), 7.31–7.27 (2H, m, H3, H5), 3.21 (2H, td, J = 7.1, 5.7 Hz, NH-CH2), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.27–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.47 (d, J = 32.7 Hz), 162.93, 157.32, 128.10 (d, J = 8.8 Hz), 121.78 (d, J = 3.2 Hz), 116.77 (d, J = 22.4 Hz), 43.29, 31.74, 29.48, 29.44, 29.43, 29.41, 29.37, 29.14, 29.11, 26.70, 22.48, 14.24. Elemental analysis for C20H30FN3O (347.48); calculated C, 69.13; H, 8.70; N, 12.09, found: C, 69.24; H, 8.69; N, 11.55. Rf: 0.71.
5-(4-Chlorophenyl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3h. White solid, yield 92%; mp 120–121 °C. IR (ATR): 621, 719, 736, 827, 836, 955, 1014, 1051, 1074, 1099, 1287, 1406, 1487, 1521, 1607, 1621, 2849, 2918, 2953, 3246 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.78–7.75 (2H, m, H2, H6), 7.55–7.51 (3H, m, H3, H5, NH), 3.21 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 1.59–1.52 (2H, m, NH-CH2-CH2), 1.25–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.45, 157.27, 135.53, 129.81, 127.35, 123.93, 43.28, 31.74, 29.48, 29.44, 29.43, 29.41, 29.36, 29.13, 29.11, 26.69, 22.48, 14.26. Elemental analysis for C20H30ClN3O (363.93); calculated C, 66.01; H, 8.31; N, 11.55, found: C, 66.12; H, 8.30; N, 11.67. Rf: 0.75.
5-(4-Bromophenyl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3i. White solid, yield 96%; mp 129–130 °C. IR (ATR): 620, 720, 832, 1011, 1030, 1050, 1070, 1156, 1269, 1287, 1402, 1471, 1483, 1600, 1618, 2848, 2917, 2951, 3253 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.71–7.66 (4H, m, H2, H3, H5, H6), 7.54–7.51 (1H, m, NH), 3.21 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.29–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.46, 157.36, 132.75, 127.53, 124.27, 124.12, 43.28, 31.74, 29.48, 29.46, 29.44, 29.43, 29.41, 29.36, 29.12, 26.68, 22.49, 14.27. Elemental analysis for C20H30BrN3O (408.38); calculated C, 58.82; H, 7.40; N, 10.29, found: C, 58.97; H, 7.49; N, 10.37. Rf: 0.75.
N-Dodecyl-5-(4-iodophenyl)-1,3,4-oxadiazol-2-amine 3j. White solid, yield 74%; mp 150–152 °C. IR (ATR): 625, 711, 720, 736, 829, 1007, 1049, 1062, 1156, 1269, 1287, 1339, 1472, 1480, 1524, 1598, 1621, 2849, 2920, 2955, 3240 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.87–7.84 (2H, m, H3, H5), 7.55–7.51 (3H, m, H2, H6, NH), 3.21 (2H, q, J = 6.6 Hz, NH-CH2), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.24–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.44, 157.53, 138.60, 127.39, 124.56, 97.15, 43.28, 31.74, 29.47, 29.46, 29.44, 29.42, 29.40, 29.35, 29.11, 26.68, 22.49, 14.28. Elemental analysis for C20H30IN3O (455.38); calculated C, 52.75; H, 6.64; N, 9.23, found: C, 52.89; H, 6.71; N, 9.17. Rf: 0.77.
5-(3,5-Dinitrophenyl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3k. Yellow solid, yield 92%; mp 131–133 °C. IR (ATR): 685, 728, 911, 1027, 1072, 1135, 1343, 1352, 1469, 1545, 1558, 1568, 1671, 2850, 2921, 3100 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.86 (2H, d, J = 2.1 Hz, H2, H6), 8.76 (1H, t, J = 2.1 Hz, H4), 8.13 (1H, t, J = 5.7 Hz, NH-CH2), 3.27 (2H, q, J = 6.7 Hz, NH-CH2), 1.58 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.37–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.47, 154.91, 148.89, 127.01, 124.66, 119.33, 42.75, 31.48, 29.23, 29.20, 29.17, 29.16, 28.90, 28.87, 28.85, 26.33, 22.28, 14.12. Elemental analysis for C20H29N5O5 (419.48); calculated C, 57.27; H, 6.97; N, 16.70, found: C, 57.34; H, 7.00; N, 16.78. Rf: 0.62.
N-Dodecyl-5-(pyridin-4-yl)-1,3,4-oxadiazol-2-amine 3l. The synthesis and characterization of the compound was published previously by our group [21].
N-Dodecyl-5-(pyridin-3-yl)-1,3,4-oxadiazol-2-amine 3m. White solid, yield 87%; mp 121–122 °C. IR (ATR): 625, 706, 718, 733, 820, 961, 1016, 1056, 1079, 1390, 1430, 1471, 1479, 1551, 1619, 2851, 2917, 2953, 3269 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 8.95 (1H, d, J = 2.2 Hz, H2), 8.64 (1H, dd, J = 4.8, 1.6 Hz, H6), 8.11 (1H, dt, J = 8.0, 2.0 Hz, H4), 7.62–7.58 (1H, m, NH), 7.50 (1H, ddd, J = 8.0, 4.8, 0.9 Hz, H5), 3.23 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 1.56 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.27–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.64, 156.07, 151.45, 146.49, 132.99, 124.58, 121.46, 43.31, 31.74, 29.47, 29.45, 29.43, 29.40, 29.35, 29.13, 29.10, 26.69, 22.48, 14.26. Elemental analysis for C19H30N4O (330.48); calculated C, 69.05; H, 9.15; N, 16.95, found: C, 69.14; H, 9.14; N, 17.08. Rf: 0.47.
N-Dodecyl-5-(pyridin-2-yl)-1,3,4-oxadiazol-2-amine 3n. White solid, yield 47%; mp 114–116 °C. IR (ATR): 622, 685, 717, 741, 786, 961,1096, 1149, 1288, 1392, 1439, 1460, 1479, 1559, 1591, 1632, 2852, 2918, 3266 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 10.37 (1H, s, NH-CH2), 8.65 (1H, dd, J = 4.4, 1.3 Hz, H6), 7.98–7.91 (2H, m, H3, H4), 7.49 (1H, ddd, J = 6.8, 4.6, 1.8 Hz, H5), 3.23 (2H, q, J = 6.7 Hz, NH-CH2), 1.56 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.33–1.17 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.36, 157.51, 150.01, 143.71, 137.64, 125.08, 121.35, 45.50, 42.70, 31.46, 29.21, 29.17, 29.13, 28.87, 28.82, 26.32, 22.25, 14.11, 8.59. Elemental analysis for C19H30N4O (330.48); calculated C, 69.05; H, 9.15; N, 16.95, found: C, 69.15; H, 9.26; N, 16.81. Rf: 0.75.
N-Dodecyl-5-(pyrimidin-4-yl)-1,3,4-oxadiazol-2-amine 3o. Pinkish solid, yield 76%; mp 154–155 °C. IR (ATR): 665, 683, 723, 845, 971, 989, 1060, 1074, 1156, 1292, 1397, 1470, 1481, 1582, 1625, 2850, 2916, 2954, 3032, 3269 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 9.26 (1H, s, H2), 8.93 (1H, d, J = 5.2 Hz, H5), 8.03 (1H, s, NH-CH2), 7.96 (1H, d, J = 5.2 Hz, H6), 3.28 (2H, t, J = 7.0 Hz, NH-CH2), 1.60 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.38–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.71, 158.76, 158.19, 155.89, 149.85, 117.16, 42.54, 31.05, 28.77, 28.74, 28.73, 28.71, 28.70, 28.57, 28.41, 25.95, 21.80, 13.60. Elemental analysis for C18H29N5O (331.46); calculated C, 65.23; H, 8.82; N, 21.13, found: C, 65.31; H, 8.90; N, 21.11. Rf: 0.58.
N-Dodecyl-5-(pyrazin-2-yl)-1,3,4-oxadiazol-2-amine 3p. White solid, yield 61%; mp 130–131 °C. IR (ATR): 716, 737, 756, 850, 967, 1015, 1041, 1058, 1098, 1175, 1288, 1389, 1423, 1472, 1523, 1627, 2852, 2918, 3294 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 9.16 (1H, d, J = 1.5 Hz, H3), 8.73–8.70 (2H, m, H5, H6), 7.87 (1H, s, NH-CH2), 3.28 (2H, q, J = 6.6 Hz, NH-CH2), 1.60 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.38–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.45, 155.48, 145.02, 144.33, 142.09, 139.64, 42.58, 31.04, 28.77, 28.74, 28.72, 28.70, 28.68, 28.59, 28.41, 25.96, 21.79, 13.58. Elemental analysis for C18H29N5O (331.46); calculated C, 65.23; H, 8.82; N, 21.13, found: C, 65.20; H, 8.74; N, 21.00. Rf: 0.60.
5-(2-Bromopyridin-4-yl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3q. White solid, yield 50%; mp 92–93 °C. IR (ATR): 672, 689, 724, 736, 747, 850, 872, 1005, 1025, 1038, 1075, 1089, 1130, 1302, 1367, 1461, 1495, 1566, 1596, 1657, 2850, 2919, 2954, 3082, 3190 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.52 (1H, d, J = 5.1 Hz, H6), 8.11 (1H, t, J = 5.7 Hz, NH), 7.86 (1H, d, J = 1.4 Hz, H3), 7.75 (1H, dd, J = 5.2, 1.5 Hz, H5), 3.25 (2H, q, J = 6.6 Hz, NH-CH2), 1.56 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.33–1.16 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.52, 154.84, 151.76, 142.28, 134.17, 122.64, 118.59, 42.71, 31.47, 29.21, 29.18, 29.13, 29.12, 28.88, 28.86, 28.79, 26.28, 22.26, 14.12. Elemental analysis for C19H29BrN4O (409.37); calculated C, 55.75; H, 7.14; N, 13.69, found: C, 55.89; H, 7.22; N, 13.80. Rf: 0.78.
N-Dodecyl-5-(pyridazin-4-yl)-1,3,4-oxadiazol-2-amine 3r. Brown solid, yield 41%; mp 142–143 °C. IR (ATR): 665, 719, 727, 882, 975, 1058, 1340, 1380, 1395, 1471, 1480, 1527, 1568, 1617, 1662, 2850, 2917, 2953, 3267 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 9.55 (1H, dd, J = 2.3, 1.3 Hz, H3), 9.37 (1H, dd, J = 5.5, 1.3 Hz, H6), 8.19 (1H, t, J = 5.7 Hz, NH-CH2), 7.91 (1H, dd, J = 5.4, 2.3 Hz, H5), 3.26 (2H, q, J = 6.6 Hz, NH-CH2), 1.56 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.34–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.67, 154.11, 152.28, 146.46, 122.72, 121.04, 42.75, 31.46, 29.20, 29.17, 29.13, 29.10, 28.87, 28.84, 28.81, 26.30, 22.25, 14.10. Elemental analysis for C18H29N5O (331.46); calculated C, 65.23; H, 8.82; N, 21.13, found: C, 65.31; H, 8.91; N, 21.24. Rf: 0.42.
5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-thiol 2s (2 mmol, 358 mg) was suspended together with 2 equivalents of potassium carbonate (4 mmol, 552 mg) in 30 mL of N,N-dimethylformamide. Then, 1-bromododecane was added in one portion (2 mmol, 498 mg). Reaction was allowed to react for 5 h. The reaction mixture was evaporated to dryness on a rotary evaporator and transferred to a separatory funnel with 30 mL of EtOAc and 30 mL of demineralized water. The organic phase was washed with 30 mL of a saturated brine. The organic phase was dried over anhydrous sodium sulfate for 45 min, then the desiccant was separated, and the mixture was evaporated to dryness. The crude product was overlaid with dry DCM and stored at −20 °C. The solid product was filtered off and washed twice with 5 mL of ice-cold DCM.
2-(Dodecylthio)-5-(pyridin-4-yl)-1,3,4-oxadiazole 3s. Yellow solid, yield 100%; mp 63–64 °C. IR (ATR): 707, 723, 835, 961, 989, 1063, 1083, 1182, 1226, 1415, 1460, 1539, 1608, 2850, 2919, 2955 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.82–8.79 (2H, m, H2, H6), 7.90–7.87 (2H, m, H3, H5), 3.32 (2H, t, J = 7.2 Hz, S-CH2), 1.76 (2H, p, J = 7.4 Hz, S-CH2-CH2), 1.38 (2H, p, J = 7.3 Hz, C3H2), 1.31–1.16 (16H, m, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 165.63, 163.72, 151.13, 130.33, 120.14, 32.28, 31.48, 29.21, 29.19, 29.12, 29.08, 29.03, 28.89, 28.55, 27.96, 22.28, 14.14. Elemental analysis for C19H29N3OS (347.52); calculated C, 65.67; H, 8.41; N, 12.09, found: C, 65.79; H, 8.52; N, 12.09. Rf: 0.72.
N-Dodecyl-5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine 3t. Yellow solid, yield 68%; mp 152–154 °C. IR (ATR): 632, 683, 719, 802, 816, 992, 1010, 1035, 1121, 1195, 1375, 1420, 1468, 1494, 1523, 1549, 1592, 1635, 2854, 2920, 2955, 3194 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.76 (2H, d, J = 6.1 Hz, H2, H6), 8.01 (2H, d, J = 6.0 Hz, H3), 3.40 (2H, t, J = 7.0 Hz, NH-CH2), 1.65 (2H, p, J = 7.2 Hz, NH-CH2-CH2), 1.41–1.21 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.86 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 170.82, 159.97, 145.49, 142.65, 121.18, 45.14, 30.86, 28.59, 28.56, 28.54, 28.52, 28.26, 28.22, 28.12, 25.97, 21.58, 13.34. Elemental analysis for C19H30N4S (346.54); calculated C, 65.85; H, 8.73; N, 16.17, found: C, 65.99; H, 8.70; N, 16.25. Rf: 0.45.
N′-Tridecylideneisonicotinohydrazide 2u (0.5 mmol, 159 mg) was dissolved in 30 mL of DMSO, iodine (1.2 eq., 0.6 mmol, 152 mg) and potassium carbonate (3 eq., 1.5 mmol, 207 mg) was added to the solution, and heated to 100 °C. After 3 h, the mixture was evaporated to dryness on a rotary evaporator and transferred to a separatory funnel with 30 mL of EtOAc acetate and 30 mL of demineralized water. The organic phase was washed with 30 mL of a saturated Na2S2O3 solution and finally 30 mL of a saturated brine. The organic phase was dried over anhydrous sodium sulfate for 45 min, then the desiccant was separated, and the mixture was evaporated to dryness. The crude product was overlaid with dry DCM and stored at −20 °C. The solid product was filtered off on and washed twice with 5 mL of ice-cold DCM [36].
2-Dodecyl-5-(pyridin-4-yl)-1,3,4-oxadiazole 3u. Yellow solid, yield 76%; mp 75–76 °C. IR (ATR): 636, 704, 724, 730, 833, 989, 1023, 1099, 1182, 1232, 1417, 1464, 1473, 1541, 1565, 1608, 2849, 2872, 2917, 2955 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.82–8.80 (2H, m, H2, H6), 7.90–7.88 (2H, m, H3, H5), 2.95 (2H, t, J = 7.4 Hz, C1H2), 1.78 (2H, p, J = 7.4 Hz, C2H2), 1.38 (2H, p, J = 7.3 Hz, C3H2), 1.34–1.20 (16H, m, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 167.84, 162.32, 150.84, 130.58, 119.96, 31.15, 28.86, 28.85, 28.80, 28.66, 28.52, 28.35, 28.15, 25.63, 24.54, 21.92, 13.72. Elemental analysis for C19H29N3O (315.46); calculated C, 72.34; H, 9.27; N, 13.32, found: C, 72.41; H, 9.30; N, 13.22. Rf: 0.64.
3.2. Acetyl- and Butyrylcholinesterase Inhibition
Enzyme inhibition activity of 3 quantified as IC50 values was determined spectrophotometrically using modified Ellman’s method [38]. It has widely been used for in vitro screening of potential AChE and BChE inhibitors. The enzyme activity is measured indirectly by determination of 5-mercapto-2-nitrobenzoic acid ion formed after reaction of the thiol reagent 5,5′-disulfanediyl-bis(2-nitrobenzoic acid) (DTNB) and thiocholine, a product of acetylthiocholine (ATCh) and butyrylthiocholine (BTCh) as substrates for hydrolysis by AChE and BChE, respectively [39].
The enzyme activity in final reaction mixture (2000 µL) was 0.2 U/mL, concentration of ATCh (or BTCh) 40 µM and concentration of DTNB 100 µM for all reactions. The compounds 3 were dissolved in DMSO and then diluted in demineralized water (conductivity 3 μS, equipment supplier BKG Water Treatment, Hradec Králové, Czech Republic) to the concentration of 1000 µM. For all the tested compounds and rivastigmine, five different concentrations of inhibitor in final reaction mixture were used. The final concentration of DMSO was 0.2%. All experiments were carried in triplicates. The average values of reaction rate (v0-uninhibited reaction, vi-inhibited reaction) were used for construction of the dependence v0/vi vs. concentration of inhibitor. From obtained equation of regression curve, IC50 values were calculated.
Then, we assessed reversibility of AChE inhibition caused by the most potent inhibitor 3t. Three different inhibitor concentrations (1, 10, and 100 µM) were chosen. The aim was to observe the effect of the inhibitor on enzyme activity (A) over time. Based on this, it is possible to distinguish reversible and irreversible inhibition. The procedure was analogous to the determination of enzyme activity. The reaction mixture containing phosphate buffer, AChE and 3t (at one of the chosen concentrations) was prepared and stirred vigorously. At given times (5, 10, 15, 20, 30, 40, 50, 60, 260, and 1450 min), DTNB and ATCh were added to the sample taken from the reaction mixture, mixed rapidly, and the absorbance was measured to quantify the enzyme activity. Based on knowledge of the enzyme activity in the absence of the inhibitor (i.e., 100% activity), the percentages of residual enzyme activity in presence of inhibitor were calculated. Then, the dependence of percentage of residual enzyme activity (% A) on time was constructed. Based on these kinetic data, it is possible to distinguish between reversible, pseudo-irreversible and irreversible inhibition.
For the determination of the type of inhibition of the most active inhibitor 3t, Lineweaver–Burk plot [34] was used. The procedure was analogous to the determination of IC50. The enzyme activity in the final reaction mixture (2000 µL) was 0.2 U/mL, concentration of ATCh 20–80 µM and concentration of DTNB was 100 µM. For each of the substrate concentrations, four different concentrations of the inhibitor were used (10, 25, 35 and 50 µM). The dependence of absorbance vs. time was observed and the reaction rate was calculated. All experiments were carried in duplicates, and the average values of reaction rate were used for the construction of the Lineweaver–Burk plot. From obtained equations of regression curves in Lineweaver–Burk plot, the values of Michaelis constant (KM) and maximum velocity (Vm) were calculated. Based on these results, the type of inhibition was identified.
Acetylcholinesterase was used from electric eel (Electrophorus electricus L.; EeAChE) and butyrylcholinesterase originated from equine serum (EqBChE). A drug rivastigmine was used as a reference dual enzyme inhibitor. All the enzymes, substrates and rivastigmine were purchased from Merck (Prague, Czech Republic).
3.3. Molecular Docking
Crystallographic structures of human AChE and human BChE were obtained from protein data bank (www.rcsb.org accessed on 30 January 2022; pdb codes 4PQE and 1POI, respectively). The 3D structures of ligands were prepared in Chem3D Pro 19.1 (ChemBioOffice 2019 Package, CambridgeSoft, Cambridge, MA, USA). In the preparation process, water molecules were removed from the enzymes and the structures of enzymes and ligands were optimized using UCSF Chimera software package (Amber force field) [40]. Docking was performed using Autodock Vina [41] and Autodock 4.2 [42]; a Lamarckian genetic algorithm was used. The 3D affinity grid box was designed to include the full active and peripheral site of AChE and BChE. The number of grid points in the X-, Y- and Z-axes was 20, 20 and 20, respectively, with grid points separated by 1 Å (Autodock Vina), and 40, 40 and 40, respectively, with grid points separated by 0.4 Å (Autodock 4). The graphic visualizations of the ligand-enzyme interactions were prepared in PyMOL (The PyMOL Molecular Graphics System, Version 1.5 Schrödinger, LLC, New York, NY, USA).
4. Conclusions
A series of twenty 1,3,4-oxadiazoles and one 1,3,4-thiadiazole were designed, prepared, and evaluated as potential inhibitors of acetylcholinesterase and butyrylcholinesterase. Almost all of them showed significant inhibition of both cholinesterases with IC50 values in the micromolar range. Their activity toward AChE was found to be comparatively higher than that toward BChE. Many of them showed higher inhibitory effect against AChE than the drug rivastigmine. We described structure–activity relationships. The most active compound, thiadiazole decorated with 4-pyridyl and dodecylamine, was the best inhibitor of both enzymes. It represents a reversible, mixed-type AchE inhibitor. The interactions of the most potent inhibitors with target structures were also investigated using molecular docking, which showed that the binding is different for particular acetylcholine hydrolysing enzymes.
Other types of biological activities, especially antimicrobial, will be investigated in the future.
Author Contributions
Conceptualization, M.K.; methodology, V.P., Š.Š., M.Š. and M.K.; investigation, V.P., Š.Š., K.S., M.Š. and M.K.; writing—original draft preparation, V.P., Š.Š., M.Š., J.V. and M.K.; writing—review and editing, Š.Š., K.S., J.V. and M.K.; supervision, Š.Š., J.V. and M.K.; funding acquisition, J.V. and M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Czech Science Foundation (grant number 20-19638Y); the EFSA-CDN (grant No. CZ.02.1.01/0.0/0.0/16_019/0000841) co-funded by ERDF and Charles University (SVV 260 547).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bennion, B.J.; Law, R.J.; Fattebert, J.L.; Schwegler, E.; Lightstone, F.C. Modeling the binding of CWAs to human AChE and BuChE. Mil. Med. Sci. Lett. 2013, 8, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.; Radic, Z.; Hosea, N.A.; Camp, S.; Marchot, P.; Berman, H.A. Structural bases for the specificity of cholinesterase catalysis and inhibition. Toxicol. Lett. 1995, 82, 453–458. [Google Scholar] [CrossRef]
- Čolović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stedman, E.; Easson, L.H. Choline-esterase. An enzyme present in the blood-serum of the horse. Biochem. J. 1932, 26, 2056–2066. [Google Scholar] [CrossRef] [Green Version]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef]
- Pohanka, M. Cholinesterases, a target of pharmacology and toxicology. Biomed. Pap. 2011, 155, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Price, D.L.; Koliatsos, V.E.; Clatterbuck, R.C. Cholinergic systems: Human diseases, animal models, and prospects for therapy. Prog. Brain Res. 1993, 98, 51–60. [Google Scholar]
- Ohno, K.; Engel, A.G.; Brengman, J.M.; Shen, X.M.; Heidenreich, F.; Vincent, A.; Milone, M.; Tan, E.; Demirci, M.; Walsh, P.; et al. The spectrum of mutations causing end-plate acetylcholinesterase deficiency. Ann. Neurol. 2000, 47, 162–170. [Google Scholar] [CrossRef]
- Vincent, A. Immunology of the neuromuscular junction and presynaptic nerve terminal. Curr. Opin. Neurol. 1999, 12, 545–551. [Google Scholar] [CrossRef]
- Masson, P.; Lockridge, O. Butyrylcholinesterase for protection from organophosphorus poisons: Catalytic complexities and hysteretic behavior. Arch. Biochem. Biophys. 2010, 494, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jbilo, O.; Bartels, C.F.; Chatonnet, A.; Toutant, J.P.; Lockridge, O. Tissue distribution of human acetylcholinesterase and butyrylcholinesterase messenger-RNA. Toxicon 1994, 32, 1445–1457. [Google Scholar] [CrossRef]
- Kumar, V.; Saha, A.; Roy, K. In silico modeling for dual inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) enzymes in Alzheimer’s disease. Comput. Biol. Chem. 2020, 88, 107355. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sharma, P.K.; Kumar, N.; Dudhe, R.A. Review: Oxadiazole Their Chemistry and Pharmacological Potentials. Pharma Chem. 2010, 2, 253–263. [Google Scholar]
- Patel, K.D.; Prajapati, S.M.; Panchal, S.N.; Patel, H.D. Review of synthesis of 1,3,4-oxadiazole derivatives. Synth. Commun. 2014, 44, 1859–1875. [Google Scholar] [CrossRef]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in medicinal chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef]
- Nayak, S.G.; Poojary, B. A Review on the Preparation of 1,3,4-Oxadiazoles from the Dehydration of Hydrazines and Study of Their Biological Roles. Chem. Afr. 2019, 2, 551–571. [Google Scholar] [CrossRef] [Green Version]
- Khalilullah, H.; Ahsan, M.J.; Hedaitullah, M.; Khan, S.; Ahmed, B. 1,3,4-Oxadiazole: A biologically active scaffold. Mini Rev. Med. Chem. 2012, 12, 789–801. [Google Scholar] [CrossRef]
- Chawla, G. 1,2,4-Oxadiazole as a Privileged Scaffold for Anti-inflammatory and Analgesic Activities: A Review. Mini Rev. Med. Chem. 2018, 18, 1536–1547. [Google Scholar] [CrossRef]
- Verma, G.; Khan, M.F.; Akhtar, W.; Alam, M.M.; Akhter, M.; Shaquiquzzaman, M. A Review Exploring Therapeutic Worth of 1,3,4-Oxadiazole Tailored Compounds. Mini Rev. Med. Chem. 2019, 19, 477–509. [Google Scholar] [CrossRef]
- Vosátka, R.; Krátký, M.; Švarcová, M.; Janoušek, J.; Stolaříková, J.; Madacki, J.; Huszár, S.; Mikušová, K.; Korduláková, J.; Trejtnar, F.; et al. New lipophilic isoniazid derivatives and their 1,3,4-oxadiazole analogues: Synthesis, antimycobacterial activity and investigation of their mechanism of action. Eur. J. Med. Chem. 2018, 151, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.R.W.; Boger, D.L.; Jorgensen, W.L. Elucidation of Fatty Acid Amide Hydrolase Inhibition by Potent α-Ketoheterocycle Derivatives from Monte Carlo Simulations. J. Am. Chem. Soc. 2005, 127, 17377. [Google Scholar] [CrossRef] [PubMed]
- Kareem, R.T.; Abedinifar, F.; Mahmood, E.A.; Ebadi, A.G.; Rajabi, F.; Vessally, E. The recent development of donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s agents: Highlights from 2010 to 2020. RSC Adv. 2021, 11, 30781. [Google Scholar] [CrossRef]
- Tripathi, A.; Choubey, P.K.; Sharma, P.; Seth, A.; Tripathi, P.N.; Tripathi, M.K.; Prajapati, S.K.; Krishnamurthy, S.; Shrivastava, S.K. Design and development of molecular hybrids of 2-pyridylpiperazine and 5-phenyl-1,3,4-oxadiazoles as potential multifunctional agents to treat Alzheimer’s disease. Eur. J. Med. Chem. 2019, 183, 111707. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Sharma, P.; Tripathi, P.N.; Gupta, S.K.; Srivastava, P.; Seth, A.; Tripathi, A.; Krishnamurthy, S.; Shrivastava, S.K. Design and development of 1,3,4-oxadiazole derivatives as potential inhibitors of acetylcholinesterase to ameliorate scopolamine-induced cognitive dysfunctions. Bioorg. Chem. 2019, 89, 103025. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.-R.; Ren, S.-T.; Yin, L.; Liu, X.-J.; Cheng, F.-C.; Liu, W.-W.; Shi, D.-H.; Cao, Z.-L.; Sun, H.-M. Synthesis and bioactivity of novel C2-glycosyl oxadiazole derivatives as acetylcholinesterase inhibitors. Heterocycl. Comm. 2018, 24, 333–338. [Google Scholar] [CrossRef]
- Bingul, M.; Saglam, M.F.; Kandemir, H.; Boga, M.; Sengul, I.F. Synthesis of indole-2-carbohydrazides and 2-(indol-2-yl)-1,3,4-oxadiazoles as antioxidants and their acetylcholinesterase inhibition properties. Monatsh. Chem. 2019, 150, 1553–1560. [Google Scholar] [CrossRef]
- Krátký, M.; Baranyai, Z.; Štěpánková, Š.; Svrčková, K.; Švarcová, M.; Stolaříková, J.; Horváth, L.; Bősze, S.; Vinšová, J. N-Alkyl-2-[4-(trifluoromethyl)benzoyl]hydrazine-1-carboxamides and Their Analogues: Synthesis and Multitarget Biological Activity. Molecules 2020, 25, 2268. [Google Scholar] [CrossRef]
- Krátký, M.; Štěpánková, Š.; Brablíková, M.; Svrčková, K.; Švarcová, M.; Vinšová, J. Novel Iodinated Hydrazide-hydrazones and their Analogues as Acetyl- and Butyrylcholinesterase Inhibitors. Curr. Top. Med. Chem. 2020, 20, 2106–2117. [Google Scholar] [CrossRef]
- Yusufzai, S.K.; Khan, M.S.; Sulaiman, O.; Osman, H.; Lamjin, D.N. Molecular docking studies of coumarin hybrids as potential acetylcholinesterase, butyrylcholinesterase, monoamine oxidase A/B and β-amyloid inhibitors for Alzheimer’s disease. Chem. Cent. J. 2018, 12, 128. [Google Scholar] [CrossRef]
- Ahmad, R.; Iqbal, R.; Akhtar, H.; Zia-ul-Haq; Duddeck, H.; Stefaniak, L.; Sitkowski, J. Synthesis and structure determination of some oxadiazole-2-thione and triazole-3-thione galactosides. Nucleosides Nucleotides Nucleic Acids 2001, 20, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Ujan, R.; Saeed, A.; Channar, P.A.; Larik, F.A.; Abbas, Q.; Alajmi, M.F.; El-Seedi, H.R.; Rind, M.A.; Hassan, M.; Raza, H.; et al. Drug-1,3,4-Thiadiazole Conjugates as Novel Mixed-Type Inhibitors of Acetylcholinesterase: Synthesis, Molecular Docking, Pharmacokinetics, and ADMET Evaluation. Molecules 2019, 24, 860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, M.; Cavrini, V.; Andrisano, V. Characterization of reversible and pseudo-irreversible acetylcholinesterase inhibitors by means of an immobilized enzyme reactor. J. Chromatogr. A 2007, 1144, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Karabanovich, G.; Zemanová, J.; Smutný, T.; Székely, R.; Šarkan, M.; Centárová, I.; Vocat, A.; Pávková, I.; Čonka, P.; Němeček, J.; et al. Development of 3,5-Dinitrobenzylsulfanyl-1,3,4-oxadiazoles and Thiadiazoles as Selective Antitubercular Agents Active Against Replicating and Nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 2362–2380. [Google Scholar] [CrossRef]
- Yu, W.; Huang, G.; Zhang, Y.; Liu, H.; Dong, L.; Yu, X.; Li, Y.; Chang, J. I2-Mediated Oxidative C–O Bond Formation for the Synthesis of 1,3,4-Oxadiazoles from Aldehydes and Hydrazides. J. Org. Chem. 2013, 78, 10337–10343. [Google Scholar] [CrossRef]
- Hearn, M.J.; Cynamon, M.H.; Chen, M.F.; Coppins, R.; Davis, J.; Kang, H.J.O.; Noble, A.; Tu-Sekine, B.; Terrot, M.S.; Trombino, D.; et al. Preparation and antitubercular activities in vitro and in vivo of novel Schiff bases of isoniazid. Eur. J. Med. Chem. 2009, 44, 4169–4178. [Google Scholar] [CrossRef] [Green Version]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Nat. C 2004, 59, 293–296. [Google Scholar]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).