
Dehydrocrenatidine Induces Liver Cancer Cell Apoptosis by Suppressing JNK-Mediated Signaling

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
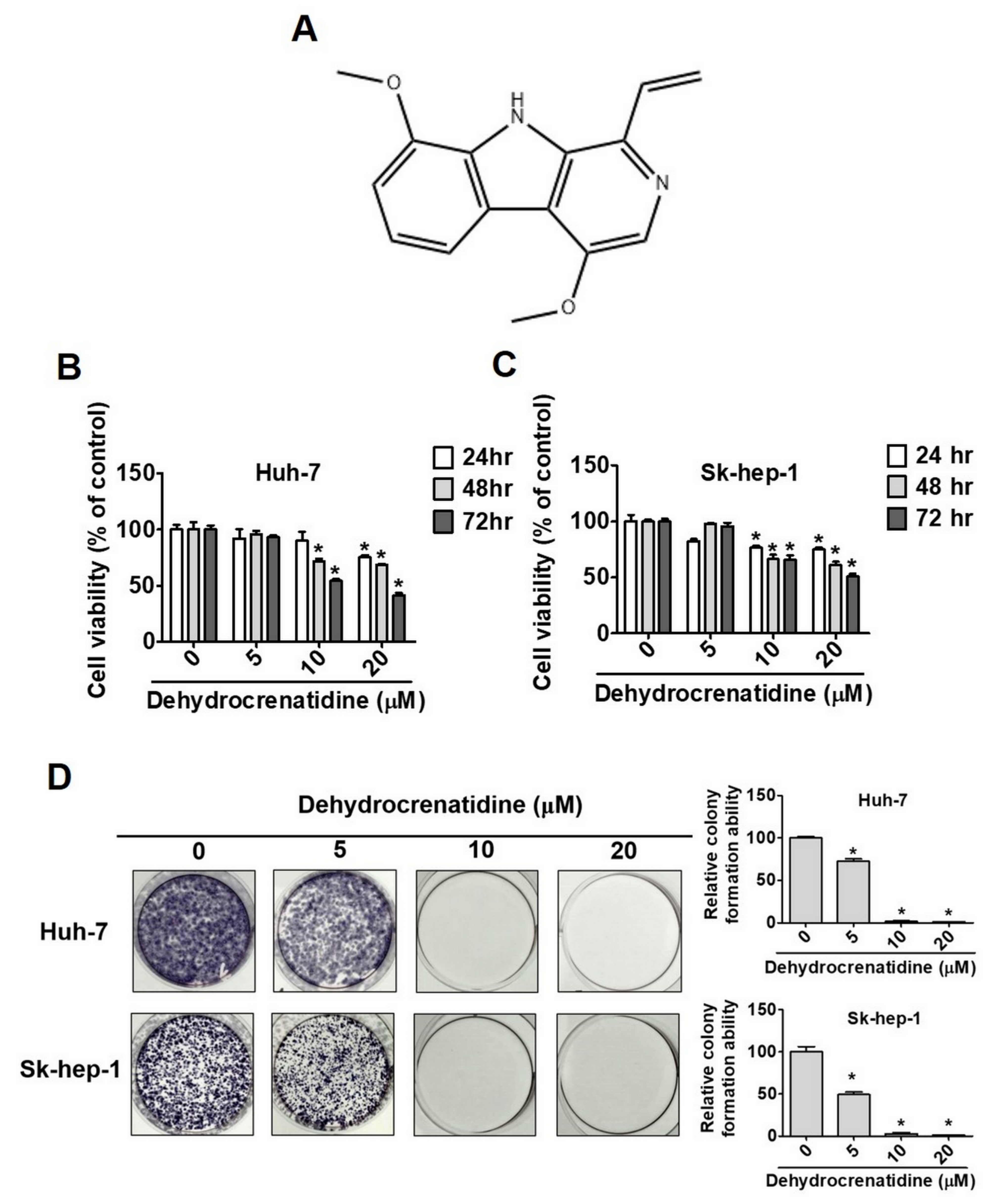
2.1. Effect of Dehydrocrenatidine on Cell Viability in Liver Cancer Cells
2.2. Effect of Dehydrocrenatidine on Cell Cycle Progression in Liver Cancer Cells
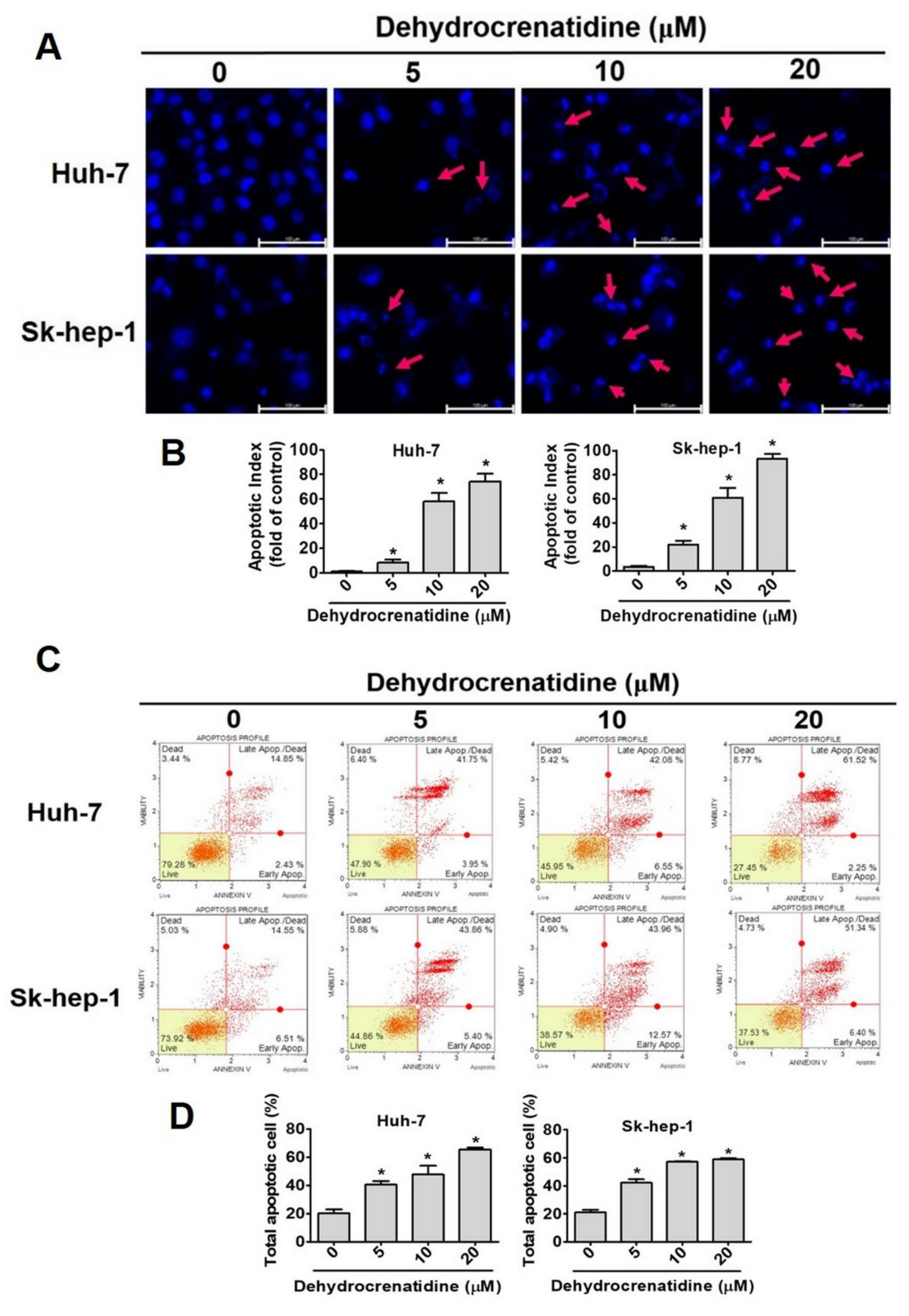
2.3. Effect of Dehydrocrenatidine on Apoptosis of Liver Cancer Cells
2.4. Pro-Apoptotic Mode of Action of Dehydrocrenatidine in Liver Cancer Cells
2.5. Pro-Apoptotic Mechanism of Dehydrocrenatidine in Liver Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Culture
4.3. MTT Assay
4.4. Colony-Formation Assay
4.5. Cell Cycle Analysis
4.6. DAPI Staining
4.7. Annexin V/PI Double Staining Assay
4.8. Mitochondrial Membrane Potential Assay
4.9. Protein Extraction and Western Blotting Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sagnelli, E.; Macera, M.; Russo, A.; Coppola, N.; Sagnelli, C. Epidemiological and etiological variations in hepatocellular carcinoma. Infection 2020, 48, 7–17. [Google Scholar] [CrossRef]
- Phan, D.V.; Chan, C.L.; Li, A.A.; Chien, T.Y.; Nguyen, V.C. Liver cancer prediction in a viral hepatitis cohort: A deep learning approach. Int. J. Cancer 2020, 147, 2871–2878. [Google Scholar] [CrossRef]
- Li, I.H.; Shih, J.H.; Tsai, C.S.; Chien, W.C.; Kao, H.H.; Pan, K.T.; Cheng, Y.D.; Kao, L.T. Inverse Association of Fibrates and Liver Cancer: A Population-Based Case-Control Study in Taiwan. J. Clin. Pharmacol. 2019, 59, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Yeh, C.T.; Lin, K.H. Cancer Stem Cell Functions in Hepatocellular Carcinoma and Comprehensive Therapeutic Strategies. Cells 2020, 9, 1331. [Google Scholar] [CrossRef]
- Su, S.Y.; Lee, W.C. Mortality trends of liver diseases from 1981 to 2016 and the projection to 2035 in Taiwan: An age-period-cohort analysis. Liver Int. 2019, 39, 770–776. [Google Scholar] [CrossRef]
- Chen, L.J.; Chang, Y.J.; Chang, Y.J. Survival Predictability Between the American Joint Committee on Cancer 8th Edition Staging System and the Barcelona Clinic Liver Cancer Classification in Patients with Hepatocellular Carcinoma. Oncologist 2021, 26, e445–e453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, W.; Zhang, H.; Qiao, L. Combination of anti-angiogenesis agents and transarterial embolization: Is it a promising approach for the treatment of liver cancer? Discov. Med. 2015, 20, 51–55. [Google Scholar]
- Halberstein, R.A. Medicinal plants: Historical and cross-cultural usage patterns. Ann. Epidemiol. 2005, 15, 686–699. [Google Scholar] [CrossRef]
- Ashraf, M.A. Phytochemicals as Potential Anticancer Drugs: Time to Ponder Nature’s Bounty. BioMed Res. Int. 2020, 2020, 8602879. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, B.; Kanwar, S.S.; Kumar, A. Lead Phytochemicals for Anticancer Drug Development. Front. Plant. Sci. 2016, 7, 1667. [Google Scholar] [CrossRef] [Green Version]
- Mondal, A.; Gandhi, A.; Fimognari, C.; Atanasov, A.G.; Bishayee, A. Alkaloids for cancer prevention and therapy: Current progress and future perspectives. Eur. J. Pharmacol. 2019, 858, 172472. [Google Scholar] [CrossRef]
- Lu, J.J.; Bao, J.L.; Chen, X.P.; Huang, M.; Wang, Y.T. Alkaloids isolated from natural herbs as the anticancer agents. Evid.-Based Complement. Altern. Med. 2012, 2012, 485042. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Tang, Q.; Xu, J.; Wang, S.; Li, S.; Zou, X.; Cao, Z. Dehydrocrenatidine Inhibits Voltage-Gated Sodium Channels and Ameliorates Mechanic Allodia in a Rat Model of Neuropathic Pain. Toxins 2019, 11, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhu, N.; Du, Y.; Bai, Q.; Chen, X.; Nan, J.; Qin, X.; Zhang, X.; Hou, J.; Wang, Q.; et al. Dehydrocrenatidine is a novel janus kinase inhibitor. Mol. Pharmacol. 2015, 87, 572–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.Y.; Lin, C.C.; Chuang, Y.C.; Lo, Y.S.; Hsieh, M.J.; Chen, M.K. Apoptotic effects of dehydrocrenatidine via JNK and ERK pathway regulation in oral squamous cell carcinoma. Biomed. Pharmacother. 2021, 137, 111362. [Google Scholar] [CrossRef]
- Zhang, J.H.; Xu, M. DNA fragmentation in apoptosis. Cell Res. 2000, 10, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Gradinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.L.; Han, F.Y.; Lou, L.L.; Zhao, W.Y.; Huang, X.X.; Yao, G.D.; Song, S.J. The nature compound dehydrocrenatidine exerts potent antihepatocellular carcinoma by destroying mitochondrial complexes in vitro and in vivo. Phytother. Res. 2022, 36, 1353–1371. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.C.; Lo, Y.S.; Chuang, Y.C.; Lin, C.C.; Ho, H.Y.; Hsieh, M.J.; Lin, J.T. Dehydrocrenatidine extracted from Picrasma quassioides induces the apoptosis of nasopharyngeal carcinoma cells through the JNK and ERK signaling pathways. Oncol. Rep. 2021, 46, 166. [Google Scholar] [CrossRef]
- Velmurugan, B.K.; Lin, J.T.; Mahalakshmi, B.; Lin, C.C.; Chuang, Y.C.; Lo, Y.S.; Ho, H.Y.; Hsieh, M.J.; Chen, M.K. Dehydrocrenatidine inhibits head and neck cancer cells invasion and migration by modulating JNK1/2 and ERK1/2 pathway and decreases MMP-2 expression. Environ. Toxicol. 2021, 36, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Wang, C.C.; Pai, H.T.; Tu, S.L.; Hou, P.Y.; Huang, C.Y.; Huang, M.T.; Chang, Y.J. The Natural Compound Dehydrocrenatidine Attenuates Nicotine-Induced Stemness and Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma by Regulating a7nAChR-Jak2 Signaling Pathways. Dis. Markers 2022, 2022, 8316335. [Google Scholar] [CrossRef] [PubMed]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.T.; Ho, H.Y.; Lin, C.C.; Chuang, Y.C.; Lo, Y.S.; Hsieh, M.J.; Chen, M.K. Platyphyllenone Induces Autophagy and Apoptosis by Modulating the AKT and JNK Mitogen-Activated Protein Kinase Pathways in Oral Cancer Cells. Int. J. Mol. Sci. 2021, 22, 4211. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velmurugan, B.K.; Hsieh, M.-J.; Lin, C.-C.; Ho, H.-Y.; Hsieh, M.-C. Dehydrocrenatidine Induces Liver Cancer Cell Apoptosis by Suppressing JNK-Mediated Signaling. Pharmaceuticals 2022, 15, 402. https://doi.org/10.3390/ph15040402
Velmurugan BK, Hsieh M-J, Lin C-C, Ho H-Y, Hsieh M-C. Dehydrocrenatidine Induces Liver Cancer Cell Apoptosis by Suppressing JNK-Mediated Signaling. Pharmaceuticals. 2022; 15(4):402. https://doi.org/10.3390/ph15040402
Chicago/Turabian StyleVelmurugan, Bharath Kumar, Ming-Ju Hsieh, Chia-Chieh Lin, Hsin-Yu Ho, and Ming-Chang Hsieh. 2022. "Dehydrocrenatidine Induces Liver Cancer Cell Apoptosis by Suppressing JNK-Mediated Signaling" Pharmaceuticals 15, no. 4: 402. https://doi.org/10.3390/ph15040402