Synthesis of 2-Aryloyl-N-Dodecylhydrazine-1-(thio)Carboxamides (2a–2r) and Other Precursors (2s–2u)
Appropriate aryl hydrazide (1.0 mmol) was suspended in 10 mL of acetonitrile and heated to initiate boiling of the mixture, then dodecyl isocyanate (or dodecyl isothiocyanate for precursor of 3t; 1.05 of equivalents, 1.05 mmol) was added quickly in one portion. The reaction mixture was refluxed continuously for 2 h, then left to cool down at RT and stored for 1 h at −20 °C. Solid material was filtered off and recrystallized from methanol to provide pure hydrazides 2. The progress of the reaction was monitored by TLC.
2-Benzoyl-N-dodecylhydrazine-1-carboxamide 2a. White solid; yield 98%; mp 137–137 °C. IR (ATR): 608, 661, 689, 749, 902, 1026, 1065, 1246, 1275, 1465, 1489, 1540, 1563, 1662, 1682, 2850, 2917, 2954, 3296 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.05 (1H, s, NH-CO-NH-CH2), 7.85 (2H, dd, J = 7.1, 1.7 Hz, H2, H6), 7.73 (1H, s, NH-CO-Ar), 7.53–7.49 (1H, m, H4), 7.43 (2H, t, J = 7.7 Hz, H3, H5), 6.40 (1H, t, J = 5.8 Hz, NH-CH2), 2.97 (2H, q, J = 6.6 Hz, NH-CH2), 1.34 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.82, 158.87, 133.32, 132.12, 128.79, 128.07, 31.84, 30.41, 29.61, 29.56, 29.38, 29.36, 29.34, 29.32, 29.26, 26.85, 22.63, 14.48. Elemental analysis for C20H33N3O2 (347.50); calculated: C, 69.13; H, 9.57; N, 12.09, found: C, 68.19; H, 9.44; N, 12.30. Rf: 0.66.
N-Dodecyl-2-(4-methylbenzoyl)hydrazine-1-carboxamide 2b. White solid; yield 95%; mp 121–122 °C. IR (ATR): 608, 661, 721, 746, 838, 911, 1191, 1252, 1265, 1338, 1469, 1495, 1542, 1583, 1613, 1642, 1665, 2849, 2917, 2954, 3294 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.96 (1H, s, NH-CO-NH-CH2), 7.75 (2H, d, J = 8.2 Hz, H2, H6), 7.69 (1H, s, NH-CO-Ar), 7.24 (2H, d, J = 8.1 Hz, H3, H5), 6.37 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.6 Hz, NH-CH2), 2.32 (3H, s, Ph-CH3), 1.38–1.29 (2H, m, NH-CH2-CH2), 1.21–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.73, 158.92, 142.05, 130.53, 129.31, 128.09, 31.84, 30.41, 29.61, 29.56, 29.42, 29.40, 29.38, 29.36, 29.26, 26.85, 22.63, 21.52, 14.48. Elemental analysis for C21H35N3O2 (361.53); calculated: C, 69.77; H, 9.76; N, 11.62, found: C, 69.89; H, 9.70; N, 11.73. Rf: 0.60.
N-Dodecyl-2-(4-methoxybenzoyl)hydrazine-1-carboxamide 2c. White solid; yield 95%; mp 158–159 °C. IR (ATR): 605, 726, 756, 841, 912, 1038, 1127, 1181, 1254, 1335, 1463, 1504, 1540, 1564, 1609, 1642, 2850, 2921, 2956, 3310 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.90 (1H, s, NH-CO-NH-CH2), 7.83 (2H, d, J = 9.1 Hz, H2, H6), 7.66 (1H, s, NH-CO-Ar), 6.96 (2H, d, J = 8.9 Hz, H3, H5), 6.37 (1H, t, J = 5.9 Hz, NH-CH2), 3.77 (3H, s, OCH3), 2.96 (2H, q, J = 6.7 Hz, NH-CH2), 1.37–1.30 (2H, m, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.36, 162.40, 159.00, 129.94, 125.49, 114.02, 55.90, 31.84, 30.41, 29.65, 29.63, 29.61, 29.59, 29.57, 29.38, 29.26, 26.85, 22.63, 14.48. Elemental analysis for C21H35N3O3 (377.53); calculated: C, 66.81; H, 9.34; N, 11.13, found: C, 66.85; H, 9.38; N, 11.14. Rf: 0.51.
2-[4-(tert-Butyl)benzoyl]-N-dodecylhydrazine-1-carboxamide 2d. White solid; yield 91%; mp 119–120 °C. IR (ATR): 621, 632, 718, 897, 1018, 1083, 1113, 1200, 1290, 1305, 1364, 1464, 1506, 1537, 1614, 1668, 2853, 2924, 2958, 3250 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.98 (1H, s, NH-CO-NH-CH2), 7.79 (2H, d, J = 8.7 Hz, H2, H6), 7.71 (1H, s, NH-CO-Ar), 7.44 (2H, d, J = 8.7 Hz, H3, H5), 6.37 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.7 Hz, NH-CH2), 1.34 (2H, q, J = 7.0 Hz, NH-CH2-CH2), 1.26 (9H, s, C(CH3)3), 1.23–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.68, 158.92, 154.96, 130.56, 127.94, 125.53, 35.17, 31.85, 31.45, 30.40, 29.68. 29.66, 29.64, 29.62, 29.57, 29.39, 29.27, 26.85, 22.64, 14.48. Elemental analysis for C24H41N3O2 (403.61); calculated: C, 71.42; H, 10.24; N, 10.41, found: C, 71.55; H, 10.29; N, 10.40. Rf: 0.62.
N-Dodecyl-2-(4-nitrobenzoyl)hydrazine-1-carboxamide 2e. White solid; yield 88%; mp 220–221 °C. IR (ATR): 625, 679, 713, 852, 868, 911, 1011, 1056, 1250, 1263, 1299, 1316, 1344, 1468, 1484, 1534, 1551, 1590, 1605, 1641, 1671, 2852, 2920, 3275 cm−1.1H NMR (600 MHz, DMSO-D6) δ 10.26 (1H, s, NH-CO-NH-CH2), 8.27 (2H, d, J = 8.8 Hz, H3, H5), 8.07 (2H, d, J = 8.8 Hz, H2, H6), 7.76 (1H, s, NH-CO-Ar), 6.35 (1H, t, J = 6.1 Hz, NH-CH2), 3.00 (2H, q, J = 6.6 Hz, NH-CH2), 1.37 (2H, q, J = 7.0 Hz, NH-CH2-CH2), 1.23–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.28, 158.52, 149.91, 139.22, 129.58, 123.91, 31.77, 30.33, 29.53, 29.50, 29.48, 29.46, 29.44, 29.30, 29.16, 26.83, 22.53, 14.33. Elemental analysis for C20H32N4O4 (392.50); calculated: C, 61.20; H, 8.22; N, 14.27, found: C, 61.29; H, 8.11; N, 14.36. Rf: 0.51.
2-[4-(Dimethylamino)benzoyl]-N-dodecylhydrazine-1-carboxamide 2f. White solid; yield 88%; mp 160–161 °C. IR (ATR): 609, 721, 763, 836, 950, 1068, 1173, 1211, 1265, 1288, 1340, 1375, 1468, 1515, 1540, 1556, 1607, 1637, 1685, 2850, 2918, 2952, 3292 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 9.72 (1H, s, NH-CO-NH-CH2), 7.75 (2H, d, J = 9.0 Hz, H2, H6), 7.59 (1H, s, NH-CO-Ar), 6.70 (2H, d, J = 8.9 Hz, H3, H5), 6.33 (1H, t, J = 5.8 Hz, NH-CH2), 3.02–2.95 (8H, m, N-CH3, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.42, 158.81, 152.51, 129.08, 119.38, 110.85, 39.85, 31.48, 30.06, 29.26, 29.25, 29.24, 29.21, 29.20, 29.02, 28.90, 26.49, 22.27, 14.12. Elemental analysis for C22H38N4O2 (390.57); calculated: C, 67.66; H, 9.81; N, 14.35, found: C, 67.75; H, 9.90; N, 14.34. Rf: 0.46.
N-Dodecyl-2-(4-fluorobenzoyl)hydrazine-1-carboxamide 2g. White solid; yield 88%; mp 202–203 °C. IR (ATR): 610, 645, 726, 849, 914, 1011, 1164, 1232, 1262, 1339, 1466, 1502, 1544, 1569, 1605, 1638, 1661, 2851, 2921, 3301 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.08 (1H, s, NH-CO-NH-CH2), 7.95–7.89 (2H, m, H2, H6), 7.74 (1H, s, NH-CO-Ar), 7.31–7.24 (2H, m, H3, H5), 6.43 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.6 Hz, NH-CH2), 1.35–1.32 (2H, m, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.64 (d, J = 54.1 Hz), 163.81, 158.84, 130.77 (d, J = 8.9 Hz), 129.81 (d, J = 2.9 Hz), 115.76 (d, J = 21.8 Hz), 31.84, 30.40, 29.61, 29.58, 29.56, 29.38, 29.36, 29.34, 29.26, 26.85, 22.63, 14.47. Elemental analysis for C20H32FN3O2 (365.49); calculated: C, 65.72; H, 8.83; N, 11.50, found: C, 65.80; H, 8.92; N, 11.51. Rf: 0.48.
2-(4-Chlorobenzoyl)-N-dodecylhydrazine-1-carboxamide 2h. White solid; yield 91%; mp 215–216 °C. IR (ATR): 602, 680, 719, 754, 845, 911, 1011, 1092, 1252, 1266, 1339, 1470, 1485, 1587, 1599, 1639, 1664, 2850, 2918, 2954, 3299 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.13 (1H, s, NH-CO-NH-CH2), 7.86 (2H, d, J = 8.2 Hz, H2, H6), 7.76 (1H, s, NH-CO-Ar), 7.52 (2H, d, J = 8.3 Hz, H3, H5), 6.44 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (2H, q, J = 6.6 Hz, NH-CH2), 1.36–1.32 (2H, m, NH-CH2-CH2), 1.21–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.85, 158.77, 136.98, 132.11, 130.01, 128.92, 31.84, 30.39, 29.67, 29.65, 29.63, 29.61, 29.56, 29.37, 29.26, 26.84, 22.63, 14.48. Elemental analysis for C20H32ClN3O2 (381.95); calculated: C, 62.89; H, 8.45; N, 11.00, found: C, 62.81; H, 8.56; N, 11.02. Rf: 0.48.
2-(4-Bromobenzoyl)-N-dodecylhydrazine-1-carboxamide 2i. White solid; yield 90%; mp 214–215 °C. IR (ATR): 607, 681, 719, 752, 845, 855, 911, 1015, 1033, 1254, 1260, 1266, 1339, 1474, 1499, 1587, 1599, 1639, 1664, 2854, 2928, 2954, 3250 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.19 (1H, s, NH-CO-NH-CH2), 7.85 (2H, d, J = 8.3 Hz, H2, H6), 7.79 (1H, s, NH-CO-Ar), 7.70 (2H, d, J = 8.3 Hz, H3, H5), 6.46 (1H, t, J = 5.9 Hz, NH-CH2), 2.95 (2H, q, J = 6.6 Hz, NH-CH2), 1.37–1.33 (2H, m, NH-CH2-CH2), 1.22–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.62, 158.40, 132.10, 131.49, 129.83, 125.54, 31.48, 30.03, 29.27, 29.25, 29.23, 29.21, 29.17, 29.02, 28.90, 26.48, 22.28, 14.13. Elemental analysis for C20H32BrN3O2 (426.40); calculated: C, 56.34; H, 7.56; N, 9.85, found: C, 56.42; H, 7.66; N, 9.79. Rf: 0.51.
N-Dodecyl-2-(4-iodobenzoyl)hydrazine-1-carboxamide 2j. White solid; yield 94%; mp 216–217 °C. IR (ATR): 623, 635, 718, 750, 840, 908, 1005, 1066, 1252, 1268, 1339, 1470, 1479, 1507, 1542, 1590, 1636, 1663, 2850, 2917, 2951, 3303 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.11 (1H, s, NH-CO-NH-CH2), 7.84–7.82 (2H, m, H3, H5), 7.74 (1H, s, NH-CO-Ar), 7.63–7.61 (2H, m, H2, H6), 6.42 (1H, t, J = 5.8 Hz, NH-CH2), 2.96 (q, J = 6.8 Hz, NH-CH2), 1.36–1.31 (2H, m, NH-CH2-CH2), 1.21–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 166.25, 158.76, 137.71, 132.78, 130.01, 99.82, 31.84, 30.38, 29.65, 29.63, 29.61, 29.59, 29.56, 29.37, 29.26, 26.84, 22.64, 14.49. Elemental analysis for C20H32IN3O2 (473.40); calculated: C, 50.74; H, 6.81; N, 8.88, found: C, 50.86; H, 6.90; N, 8.95. Rf: 0.51.
2-(3,5-Dinitrobenzoyl)-N-dodecylhydrazine-1-carboxamide 2k. Greyish solid, yield 76%; mp 157–159 °C. IR (ATR): 717, 732, 920, 1084, 1241, 1350, 1466, 1476, 1538, 1569, 1615, 1653, 2848, 2924, 3099, 3216, 3357 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.81 (1H, s, NH-CO-NH-CH2), 9.06 (2H, d, J = 2.0 Hz, H2, H6), 8.99 (1H, t, J = 2.1 Hz, H4), 8.04 (1H, s, NH-CO-Ar), 6.67 (1H, s, NH-CH2), 3.02 (2H, q, J = 6.6 Hz, NH-CH2), 1.41–1.36 (2H, m, NH-CH2-CH2), 1.27–1.21 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 162.70, 158.10, 148.31, 135.70, 128.01, 121.41, 39.20, 31.49, 30.06, 29.27, 29.25, 29.22, 29.20, 29.03, 28.91, 26.49, 22.29, 14.14. Elemental analysis for C20H31N5O6 (437.50); calculated C, 54.91; H, 7.14; N, 16.01, found: C, 55.01; H, 7.21; N, 16.00. Rf: 0.50.
N-Dodecyl-2-isonicotinoylhydrazine-1-carboxamide
2l. The synthesis and characterization of the compound was published previously by our group [
21].
N-Dodecyl-2-nicotinoylhydrazine-1-carboxamide 2m. White solid; yield 95%; mp 143–144 °C. IR (ATR): 632, 721, 826, 903, 1028, 1124, 1194, 1253, 1266, 1317, 1379, 1470, 1525, 1589, 1638, 1695, 2848, 2916, 2979, 3197, 3311 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.25 (1H, s, NH-CO-NH-CH2), 9.00 (1H, dd, J = 2.3, 0.9 Hz, H2), 8.69 (1H, dd, J = 4.8, 1.7 Hz, H4), 8.21–8.15 (1H, m, H6), 7.82 (1H, s, NH-CO-Ar), 7.48 (1H, ddd, J = 7.9, 4.8, 0.9 Hz, H5), 6.50 (t, J = 5.9 Hz, NH-CH2), 3.00–2.94 (2H, m, NH-CH2), 1.37–1.32 (2H, m, NH-CH2-CH2), 1.21–1.18 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.81 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 165.48, 158.70, 152.74, 149.17, 135.79, 128.98, 123.97, 31.84, 30.39, 29.61, 29.59, 29.57, 29.56, 29.55, 29.37, 29.25, 26.85, 22.63, 14.48. Elemental analysis for C19H32N4O2 (348.49); calculated: C, 65.48; H, 9.26; N, 16.08, found: C, 65.42; H, 9.15; N, 16.01. Rf: 0.24.
N-Dodecyl-2-picolinoylhydrazine-1-carboxamide 2n. White solid; yield 89%; mp 101–103 °C. IR (ATR): 613, 727, 747, 823, 1017, 1038, 1066, 1240, 1271, 1435, 1465, 1481, 1497, 1565, 1573, 1634, 1715, 2849, 2926, 2956, 3316, 3377 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 10.13 (1H, s, NH-CO-NH-CH2), 8.65 (1H, dd, J = 4.6, 1.4 Hz, H6), 8.03–7.97 (2H, m, H3, H4), 7.85 (1H, s, NH-CO-Ar), 7.62 (1H, ddd, J = 6.9, 4.7, 2.5 Hz, H5), 6.37 (1H, t, J = 5.7 Hz, NH-CH2), 2.99 (2H, q, J = 6.6 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 163.71, 158.08, 149.65, 148.67, 137.84, 126.95, 122.41, 39.40, 31.47, 29.99, 29.24, 29.22, 29.21, 29.19, 29.00, 28.89, 26.48, 22.26, 14.11. Elemental analysis for C19H32N4O2 (348.49); calculated: C, 65.48; H, 9.26; N, 16.08, found: C, 65.59; H, 9.19; N, 16.19. Rf: 0.49.
N-Dodecyl-2-(pyrimidine-4-carbonyl)hydrazine-1-carboxamide 2o. White solid, yield 89%; mp 133–134 °C. IR (ATR): 605, 639, 664, 765, 870, 997, 1096, 1157, 1231, 1289, 1342, 1389, 1466, 1506, 1556, 1582, 1659, 1696, 2852, 2922, 3090, 2922, 3090, 3214, 3292 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.46 (1H, s, NH-CO-NH-CH2), 9.33 (1H, d, J = 1.5 Hz, H2), 9.07 (1H, d, J = 5.0 Hz, H5), 8.00 (1H, dd, J = 5.0, 1.4 Hz, H6), 7.95 (1H, s, NH-CO-Ar), 6.43 (1H, t, J = 5.7 Hz, NH-CH2), 2.99 (2H, q, J = 6.8 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 162.63, 159.69, 158.06, 157.82, 156.37, 119.01, 39.40, 31.46, 29.97, 29.23, 29.22, 29.19, 29.00, 28.88, 26.48, 22.26, 14.12. Elemental analysis for C18H31N5O2 (349.48); calculated C, 61.86; H, 8.94; N, 20.04, found: C, 61.95; H, 9.01; N, 20.15. Rf: 0.32.
N-Dodecyl-2-(pyrazine-2-carbonyl)hydrazine-1-carboxamide 2p. White solid, yield 79%; mp 128–132 °C. IR (ATR): 632, 725, 760, 915, 1021, 1072, 1117, 1170, 1224, 1263, 1268, 1408, 1466, 1490, 1567, 1644, 1698, 1717, 2849, 2923, 2955, 3310 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.35 (1H, s, NH-CO-NH-CH2), 9.16 (1H, d, J = 1.5 Hz, H3), 8.88 (1H, d, J = 2.5 Hz, H6), 8.74 (1H, dd, J = 2.4, 1.5 Hz, H5), 7.91 (1H, s, NH-CO-Ar), 6.42 (1H, t, J = 5.7 Hz, NH-CH2), 3.00 (2H, q, J = 6.8 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 162.98, 157.99, 147.84, 144.81, 143.86, 143.61, 39.41, 31.48, 30.00, 29.27, 29.25, 29.24, 29.20, 29.01, 28.90, 26.49, 22.27, 14.11. Elemental analysis for C18H31N5O2 (349.48); calculated C, 61.86; H, 8.94; N, 20.04, found: C, 62.00; H, 9.14; N, 20.07. Rf: 0.46.
2-(2-Bromoisonicotinoyl)-N-dodecylhydrazine-1-carboxamide 2q. White solid; yield 67%; mp 169–171 °C. IR (ATR): 634, 725, 739, 929, 1078, 1245, 1261, 1337, 1370, 1462, 1539, 1567, 1650, 2850, 2923, 2953, 3040, 3281 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.46 (1H, s, NH-CO-NH-CH2), 8.56 (1H, d, J = 5.1 Hz, H6), 8.03 (1H, d, J = 1.4 Hz, H3), 7.94 (1H, s, NH-CO-Ar), 7.81 (1H, dd, J = 5.1, 1.4 Hz, H5), 6.58 (1H, t, J = 5.9 Hz, NH-CH2), 3.00 (2H, q, J = 6.6 Hz, NH-CH2), 1.36 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 163.48, 158.06, 151.37, 143.17, 141.92, 126.05, 121.50, 39.36, 30.00, 29.31, 29.24, 29.20, 29.19, 19.11, 29.00, 28.89, 26.46, 22.27, 14.12. Elemental analysis for C19H31BrN4O2 (427.39); calculated: C, 53.40; H, 7.31; N, 13.11, found: C, 53.51; H, 7.22; N, 13.18. Rf: 0.33.
N-Dodecyl-2-(pyridazine-4-carbonyl)hydrazine-1-carboxamide 2r. White solid; yield 91%; mp 165–166 °C. IR (ATR): 644, 675, 716, 724, 753, 895, 979, 1052, 1247, 1265, 1288, 1351, 1464, 1481, 1545, 1569, 1620, 1662, 2849, 2922, 2954, 3080, 3177, 3349 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.60 (1H, s, NH-CO-NH-CH2), 9.56 (1H, dd, J = 2.3, 1.3 Hz, H3), 9.45 (1H, dd, J = 5.2, 1.3 Hz, H6), 8.02 (1H, dd, J = 5.3, 2.3 Hz, H5), 7.98 (1H, s, NH-CO-Ar), 6.60 (1H, t, J = 5.7 Hz, NH-CH2), 3.01 (2H, q, J = 6.6 Hz, NH-CH2), 1.37 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.28–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 163.44, 157.99, 152.29, 148.91, 130.31, 124.53, 39.87, 31.45, 29.98, 29.94, 29.21, 29.19, 29.17, 28.97, 28.86, 26.45, 22.24, 14.02. Elemental analysis for C18H31N5O2 (349.48); calculated: C, 61.86; H, 8.94; N, 20.04, found: C, 61.90; H, 9.02; N, 20.14. Rf: 0.13.
Isoniazid (10 mmol, 1.37 g) was dissolved in 50 mL of 5% aqueous solution of potassium hydroxide, then carbon disulfide (1.5 eq., 15 mmol, 1.14 g) was added in one portion. Reaction mixture was heated to 90 °C for 10 h. The mixture was cooled to room temperature and acidified to pH = 6 with 1M HCl. Residue precipitate was filtered off, washed with 10 mL of cold water and recrystallized from ethanol, gave pure 5-(pyridin-4-yl)-1,3,4-oxadiazol-2-thiol
2s [
35].
5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-thiol
2s [
35]. Yellow solid, yield 73%; mp 264–266 °C. IR (ATR): 693, 722, 734, 828, 887, 1010, 1051, 1076, 1147, 1218, 1236, 1311, 1331, 1368, 1421, 1496, 1553, 1597, 1846, 2385 cm
−1.
1H NMR (600 MHz, DMSO-
D6) δ 8.82–8.79 (2H, m, H2, H6), 7.82–7.79 (2H, m, H3, H5).
13C NMR (151 MHz, DMSO-
D6) δ 178.02, 158.94, 151.02, 129.95, 119.79. Elemental analysis for C
7H
5N
3OS (179.20); calculated C, 46.92; H, 2.81; N, 23.45, found: C, 47.02; H, 2.95; N, 23.51.
Rf: 0.46.
N-Dodecyl-2-isonicotinoylhydrazine-1-carbothioamide 2t. White solid, yield 76%; mp 199–201 °C. IR (ATR): 656, 692, 720, 752, 850, 908, 999, 1062, 1115, 1229, 1249, 1269, 1360, 1404, 1472, 1481, 1502, 1530, 1556, 1653, 2852, 2923, 3136, 3287 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 10.59 (1H, s, NH-CS-NH-CH2), 9.32 (1H, s, NH-CS-Ar), 8.77–8.74 (2H, m, H2, H6), 8.14 (1H, t, J = 5.7 Hz, NH-CH2), 7.82–7.79 (2H, m, H3, H5), 3.41 (2H, q, J = 6.8 Hz, NH-CH2), 1.47 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.29–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 201.01, 164.58, 150.32, 139.78, 121.82, 43.91, 31.47, 29.24, 29.19, 29.17, 28.99, 28.89, 28.88, 28.86, 26.42, 22.27, 14.12. Elemental analysis for C19H32N4OS (364.55); calculated C, 62.60; H, 8.85; N, 15.37, found: C, 62.69; H, 8.92; N, 15.48. Rf: 0.32.
Tridecanal (1 mmol, 198 mg) was dissolved in 20 mL of ethanol, 1 eq. of isoniazid (1 mmol, 137 mg) was added to the solution and reaction mixture was heated to reflux for 5 h. The mixture was reduced the volume in a rotary evaporator, and then stored in the freezer to crystalize (−20 °C). The solid product was filtered off and washed with methanol to provide pure hydrazone
2u [
36].
(
E)-
N′-Tridecylideneisonicotinohydrazide
2u [
37]. White solid, yield 90%; mp 90–92 °C. IR (ATR): 655, 670, 728, 754, 846, 877, 1043, 1295, 1367, 1412, 1460, 1548, 1598, 1624, 1655, 2848, 2923, 2955, 3070, 3257 cm
−1.
1H NMR (600 MHz, DMSO-
D6) δ 11.62 (1H, s, NH), 8.75–8.73 (2H, m, H2, H6), 7.76–7.73 (3H, m, H3, H5, N=CH), 2.26 (2H, td,
J = 7.4, 5.5 Hz, =CH-C
H2), 1.47 (2H, p,
J = 7.1 Hz, =CH-CH
2-C
H2), 1.32–1.19 (18H, m, C
4H
2, C
5H
2, C
6H
2, C
7H
2, C
8H
2, C
9H
2, C
10H
2, C
11H
2, C
12H
2), 0.84 (3H, t,
J = 6.9 Hz, CH
3).
13C NMR (151 MHz, DMSO-
D6) δ 161.36, 153.98, 150.42, 140.82, 121.63, 32.18, 31.48, 29.24, 29.21, 29.14, 28.99, 28.90, 28.83, 26.12, 22.28, 14.12. Elemental analysis for C
19H
31N
3O (317.48); calculated C, 71.88; H, 9.84; N, 13.24, found: C, 71.95; H, 9.99; N, 13.15.
Rf: 0.31.
Procedure for Synthesis of 2-Aryloyl-N-Dodecyl-1,3,4-Oxadiazol-2-Amines (3a–3r) and Their Analogues (3s–3u)
An amount of 0.5 mmol of the appropriate 2-aryl-N-dodecylhydrazine-1-carboxamide (or N-dodecyl-2-isonicotinoylhydrazine-1-carbothioamide) 2 was suspended in 30 mL of DCM. Thereafter, 1.5 mmol (3 eq., 286 mg) of p-toluenesulfonyl chloride was added to the solution. After a complete dissolution of p-toluenesulfonyl chloride, 2.5 mmol of triethylamine (5 eq., 348 µL) was added dropwise. The mixture was allowed to react at room temperature for 12 h. The reaction mixture was evaporated to dryness on a rotary evaporator and transferred to a separatory funnel with 30 mL of ethyl acetate (EtOAc) and 30 mL of demineralized water. The organic phase was washed with 30 mL of a saturated NaHCO3 solution followed by 30 mL of a saturated brine. The organic phase was dried over anhydrous sodium sulfate for 45 min, then the desiccant was separated, and the mixture was evaporated to dryness. The crude product was overlaid with dry DCM and stored at −20 °C overnight. The solid product was filtered off and washed twice with 5 mL of ice-cold DCM. The progress of the reaction was monitored by TLC using a mixture of DCM with MeOH (93:7 v/v) as a mobile phase.
N-Dodecyl-5-phenyl-1,3,4-oxadiazol-2-amine 3a. White solid, yield 100%; mp 114–115 °C. IR (ATR): 631, 635, 722, 750, 829, 833, 838, 1036, 1050, 1111, 1184, 1270, 1301, 1400, 1484, 1505, 1588, 1674, 2851, 2924, 3240 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.87–7.67 (2H, m, H2, H6), 7.55–7.38 (3H, m, H3, H5, NH), 3.22 (2H, q, J = 6.6 Hz, NH-CH2), 1.56 (2H, p, J = 7.2 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.35, 158.05, 130.77, 129.62, 125.65, 125.13, 43.30, 31.74, 29.48, 29.46, 29.44, 29.41, 29.39, 29.14, 29.11, 26.71, 22.48, 14.26. Elemental analysis for C20H31N3O (329.49); calculated C, 72.91; H, 9.48; N, 12.75, found: C, 73.00; H, 9.59; N, 12.64. Rf: 0.77.
N-Dodecyl-5-(p-tolyl)-1,3,4-oxadiazol-2-amine 3b. White solid, yield 91%; mp 121–122 °C. IR (ATR): 630, 724, 741, 829, 838, 1030, 1052, 1113, 1173, 1266, 1300, 1441, 1469, 1506, 1587, 1614, 2850, 2920, 3268 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.66 (2H, d, J = 7.8 Hz, H2, H6), 7.41–7.39 (1H, m, NH), 7.29 (2H, d, J = 7.9 Hz, H3, H5), 3.20 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 2.33 (3H, s, Ph-CH3), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.24–1.17 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.17, 158.16, 140.72, 130.19, 125.65, 122.41, 43.28, 31.74, 29.47, 29.44, 29.42, 29.40, 29.38, 29.13, 29.11, 26.70, 22.48, 21.45, 14.27. Elemental analysis for C21H33N3O (343.52); calculated C, 73.43; H, 9.68; N, 12.23, found: C, 73.52; H, 9.60; N, 12.33. Rf: 0.75.
N-Dodecyl-5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-amine 3c. White solid, yield 90%; mp 116–117 °C. IR (ATR): 606, 634, 724, 739, 903, 828, 838, 956, 1029, 1049, 1173, 1266, 1299, 1469, 1506, 1587, 1614, 2850, 2921, 3266 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.70 (2H, d, J = 8.4 Hz, H2, H6), 7.35–7.32 (1H, m, NH), 7.03 (2H, d, J = 8.4 Hz, H3, H5), 3.80 (3H, s, OCH3), 3.19 (2H, q, J = 6.6 Hz, NH-CH2), 1.55 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.29–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.7 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.03, 161.56, 158.01, 127.41, 117.72, 115.28, 55.99, 43.30, 31.74, 29.48, 29.46, 29.44, 29.43, 29.41, 29.14, 29.11, 26.71, 22.48, 14.27. Elemental analysis for C21H33N3O2 (359.51); calculated C, 70.16; H, 9.25; N, 11.69, found: C, 70.11; H, 9.15; N, 11.81. Rf: 0.69.
5-[4-(tert-Butyl)phenyl]-N-dodecyl-1,3,4-oxadiazol-2-amine 3d. White solid, yield 87%; mp 97–98 °C. IR (ATR): 614, 636, 725, 836, 852, 902, 1016, 1049, 1114, 1269, 1363, 1468, 1500, 1544, 1613, 2851, 2921, 3261 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.70 (2H, d, J = 8.5 Hz, H3, H5), 7.50 (2H, d, J = 8.5 Hz, H2, H6), 7.41 (1H, s, NH), 3.20 (2H, td, J = 7.1, 5.7 Hz, NH-CH2), 1.56 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.28 (9H, s, C(CH3)3), 1.25–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.46, 157.36, 132.75, 127.53, 124.27, 124.12, 43.28, 31.74, 29.48, 29.44, 29.43, 29.41, 29.39, 29.37, 29.36, 29.34, 29.12, 26.68, 22.49, 14.27. Rf: 0.77.
N-Dodecyl-5-(4-nitrophenyl)-1,3,4-oxadiazol-2-amine 3e. White solid, yield 90%; mp 127–128 °C. IR (ATR): 680, 719, 733, 857, 953, 1013, 1047, 1072, 1296, 1350, 1479, 1535, 1507, 1562, 1605, 2849, 2916, 3250 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 8.32–8.29 (2H, m, H2, H6), 8.01–7.99 (2H, m, H3, H5), 7.76 (1H, t, J = 5.8 Hz, NH), 3.25 (2H, q, J = 6.1 Hz, NH-CH2), 1.57 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.26–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.98, 156.77, 148.72, 130.52, 126.61, 124.97, 43.30, 31.74, 29.48, 29.46, 29.44, 29.43, 29.41, 29.33, 29.12, 26.68, 22.48, 14.25. Elemental analysis for C20H30N4O3 (374.49); calculated C, 64.15; H, 8.08; N, 14.96, found: C, 64.26; H, 8.00; N, 15.08. Rf: 0.85.
5-[4-(Dimethylamino)phenyl]-N-dodecyl-1,3,4-oxadiazol-2-amine 3f. White solid, yield 60%; mp 118–121 °C. IR (ATR): 612, 664, 721, 739, 815, 952, 1047, 1064, 1164, 1196, 1227, 1270, 1369, 1468, 1515, 1578, 1614, 1687, 2848, 2916, 3237 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 7.77 (2H, d, J = 8.8 Hz, H3, H5), 7.59 (2H, d, J = 8.9 Hz, H2, H6), 6.80 (1H, t, J = 5.7 Hz, NH), 3.19 (2H, q, J = 6.6 Hz, NH-CH2), 2.97 (6H, s, N-CH3), 1.54 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.33–1.16 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.84 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 161.84, 160.94, 152.33, 127.63, 112.08, 111.90, 42.28, 31.49, 29.23, 29.19, 29.13, 29.05, 28.91, 28.70, 27.07, 26.12, 22.29, 14.15. Elemental analysis for C22H36N4O (372.56); calculated C, 70.93; H, 9.74; N, 15.04, found: C, 80.02; H, 9.81; N, 14.95. Rf: 0.74.
N-Dodecyl-5-(4-fluorophenyl)-1,3,4-oxadiazol-2-amine 3g. White solid, yield 81%; mp 104–105 °C. IR (ATR): 609, 660, 737, 820, 841, 1029, 1050, 1158, 1249, 1268, 1286, 1412, 1471, 1505, 1609, 1623, 2849, 2917, 2954, 3271 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.83–7.79 (2H, m, H2, H6), 7.45 (1H, s, NH), 7.31–7.27 (2H, m, H3, H5), 3.21 (2H, td, J = 7.1, 5.7 Hz, NH-CH2), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.27–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.47 (d, J = 32.7 Hz), 162.93, 157.32, 128.10 (d, J = 8.8 Hz), 121.78 (d, J = 3.2 Hz), 116.77 (d, J = 22.4 Hz), 43.29, 31.74, 29.48, 29.44, 29.43, 29.41, 29.37, 29.14, 29.11, 26.70, 22.48, 14.24. Elemental analysis for C20H30FN3O (347.48); calculated C, 69.13; H, 8.70; N, 12.09, found: C, 69.24; H, 8.69; N, 11.55. Rf: 0.71.
5-(4-Chlorophenyl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3h. White solid, yield 92%; mp 120–121 °C. IR (ATR): 621, 719, 736, 827, 836, 955, 1014, 1051, 1074, 1099, 1287, 1406, 1487, 1521, 1607, 1621, 2849, 2918, 2953, 3246 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.78–7.75 (2H, m, H2, H6), 7.55–7.51 (3H, m, H3, H5, NH), 3.21 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 1.59–1.52 (2H, m, NH-CH2-CH2), 1.25–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.45, 157.27, 135.53, 129.81, 127.35, 123.93, 43.28, 31.74, 29.48, 29.44, 29.43, 29.41, 29.36, 29.13, 29.11, 26.69, 22.48, 14.26. Elemental analysis for C20H30ClN3O (363.93); calculated C, 66.01; H, 8.31; N, 11.55, found: C, 66.12; H, 8.30; N, 11.67. Rf: 0.75.
5-(4-Bromophenyl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3i. White solid, yield 96%; mp 129–130 °C. IR (ATR): 620, 720, 832, 1011, 1030, 1050, 1070, 1156, 1269, 1287, 1402, 1471, 1483, 1600, 1618, 2848, 2917, 2951, 3253 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.71–7.66 (4H, m, H2, H3, H5, H6), 7.54–7.51 (1H, m, NH), 3.21 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.29–1.19 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.46, 157.36, 132.75, 127.53, 124.27, 124.12, 43.28, 31.74, 29.48, 29.46, 29.44, 29.43, 29.41, 29.36, 29.12, 26.68, 22.49, 14.27. Elemental analysis for C20H30BrN3O (408.38); calculated C, 58.82; H, 7.40; N, 10.29, found: C, 58.97; H, 7.49; N, 10.37. Rf: 0.75.
N-Dodecyl-5-(4-iodophenyl)-1,3,4-oxadiazol-2-amine 3j. White solid, yield 74%; mp 150–152 °C. IR (ATR): 625, 711, 720, 736, 829, 1007, 1049, 1062, 1156, 1269, 1287, 1339, 1472, 1480, 1524, 1598, 1621, 2849, 2920, 2955, 3240 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 7.87–7.84 (2H, m, H3, H5), 7.55–7.51 (3H, m, H2, H6, NH), 3.21 (2H, q, J = 6.6 Hz, NH-CH2), 1.55 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.24–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 7.0 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.44, 157.53, 138.60, 127.39, 124.56, 97.15, 43.28, 31.74, 29.47, 29.46, 29.44, 29.42, 29.40, 29.35, 29.11, 26.68, 22.49, 14.28. Elemental analysis for C20H30IN3O (455.38); calculated C, 52.75; H, 6.64; N, 9.23, found: C, 52.89; H, 6.71; N, 9.17. Rf: 0.77.
5-(3,5-Dinitrophenyl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3k. Yellow solid, yield 92%; mp 131–133 °C. IR (ATR): 685, 728, 911, 1027, 1072, 1135, 1343, 1352, 1469, 1545, 1558, 1568, 1671, 2850, 2921, 3100 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.86 (2H, d, J = 2.1 Hz, H2, H6), 8.76 (1H, t, J = 2.1 Hz, H4), 8.13 (1H, t, J = 5.7 Hz, NH-CH2), 3.27 (2H, q, J = 6.7 Hz, NH-CH2), 1.58 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.37–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.47, 154.91, 148.89, 127.01, 124.66, 119.33, 42.75, 31.48, 29.23, 29.20, 29.17, 29.16, 28.90, 28.87, 28.85, 26.33, 22.28, 14.12. Elemental analysis for C20H29N5O5 (419.48); calculated C, 57.27; H, 6.97; N, 16.70, found: C, 57.34; H, 7.00; N, 16.78. Rf: 0.62.
N-Dodecyl-5-(pyridin-4-yl)-1,3,4-oxadiazol-2-amine
3l. The synthesis and characterization of the compound was published previously by our group [
21].
N-Dodecyl-5-(pyridin-3-yl)-1,3,4-oxadiazol-2-amine 3m. White solid, yield 87%; mp 121–122 °C. IR (ATR): 625, 706, 718, 733, 820, 961, 1016, 1056, 1079, 1390, 1430, 1471, 1479, 1551, 1619, 2851, 2917, 2953, 3269 cm−1. 1H NMR (600 MHz, DMSO-D6) δ 8.95 (1H, d, J = 2.2 Hz, H2), 8.64 (1H, dd, J = 4.8, 1.6 Hz, H6), 8.11 (1H, dt, J = 8.0, 2.0 Hz, H4), 7.62–7.58 (1H, m, NH), 7.50 (1H, ddd, J = 8.0, 4.8, 0.9 Hz, H5), 3.23 (2H, td, J = 7.0, 5.7 Hz, NH-CH2), 1.56 (2H, p, J = 7.1 Hz, NH-CH2-CH2), 1.27–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.82 (3H, t, J = 6.9 Hz, CH3). 13C NMR (151 MHz, DMSO-D6) δ 164.64, 156.07, 151.45, 146.49, 132.99, 124.58, 121.46, 43.31, 31.74, 29.47, 29.45, 29.43, 29.40, 29.35, 29.13, 29.10, 26.69, 22.48, 14.26. Elemental analysis for C19H30N4O (330.48); calculated C, 69.05; H, 9.15; N, 16.95, found: C, 69.14; H, 9.14; N, 17.08. Rf: 0.47.
N-Dodecyl-5-(pyridin-2-yl)-1,3,4-oxadiazol-2-amine 3n. White solid, yield 47%; mp 114–116 °C. IR (ATR): 622, 685, 717, 741, 786, 961,1096, 1149, 1288, 1392, 1439, 1460, 1479, 1559, 1591, 1632, 2852, 2918, 3266 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 10.37 (1H, s, NH-CH2), 8.65 (1H, dd, J = 4.4, 1.3 Hz, H6), 7.98–7.91 (2H, m, H3, H4), 7.49 (1H, ddd, J = 6.8, 4.6, 1.8 Hz, H5), 3.23 (2H, q, J = 6.7 Hz, NH-CH2), 1.56 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.33–1.17 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.36, 157.51, 150.01, 143.71, 137.64, 125.08, 121.35, 45.50, 42.70, 31.46, 29.21, 29.17, 29.13, 28.87, 28.82, 26.32, 22.25, 14.11, 8.59. Elemental analysis for C19H30N4O (330.48); calculated C, 69.05; H, 9.15; N, 16.95, found: C, 69.15; H, 9.26; N, 16.81. Rf: 0.75.
N-Dodecyl-5-(pyrimidin-4-yl)-1,3,4-oxadiazol-2-amine 3o. Pinkish solid, yield 76%; mp 154–155 °C. IR (ATR): 665, 683, 723, 845, 971, 989, 1060, 1074, 1156, 1292, 1397, 1470, 1481, 1582, 1625, 2850, 2916, 2954, 3032, 3269 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 9.26 (1H, s, H2), 8.93 (1H, d, J = 5.2 Hz, H5), 8.03 (1H, s, NH-CH2), 7.96 (1H, d, J = 5.2 Hz, H6), 3.28 (2H, t, J = 7.0 Hz, NH-CH2), 1.60 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.38–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.71, 158.76, 158.19, 155.89, 149.85, 117.16, 42.54, 31.05, 28.77, 28.74, 28.73, 28.71, 28.70, 28.57, 28.41, 25.95, 21.80, 13.60. Elemental analysis for C18H29N5O (331.46); calculated C, 65.23; H, 8.82; N, 21.13, found: C, 65.31; H, 8.90; N, 21.11. Rf: 0.58.
N-Dodecyl-5-(pyrazin-2-yl)-1,3,4-oxadiazol-2-amine 3p. White solid, yield 61%; mp 130–131 °C. IR (ATR): 716, 737, 756, 850, 967, 1015, 1041, 1058, 1098, 1175, 1288, 1389, 1423, 1472, 1523, 1627, 2852, 2918, 3294 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 9.16 (1H, d, J = 1.5 Hz, H3), 8.73–8.70 (2H, m, H5, H6), 7.87 (1H, s, NH-CH2), 3.28 (2H, q, J = 6.6 Hz, NH-CH2), 1.60 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.38–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.45, 155.48, 145.02, 144.33, 142.09, 139.64, 42.58, 31.04, 28.77, 28.74, 28.72, 28.70, 28.68, 28.59, 28.41, 25.96, 21.79, 13.58. Elemental analysis for C18H29N5O (331.46); calculated C, 65.23; H, 8.82; N, 21.13, found: C, 65.20; H, 8.74; N, 21.00. Rf: 0.60.
5-(2-Bromopyridin-4-yl)-N-dodecyl-1,3,4-oxadiazol-2-amine 3q. White solid, yield 50%; mp 92–93 °C. IR (ATR): 672, 689, 724, 736, 747, 850, 872, 1005, 1025, 1038, 1075, 1089, 1130, 1302, 1367, 1461, 1495, 1566, 1596, 1657, 2850, 2919, 2954, 3082, 3190 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.52 (1H, d, J = 5.1 Hz, H6), 8.11 (1H, t, J = 5.7 Hz, NH), 7.86 (1H, d, J = 1.4 Hz, H3), 7.75 (1H, dd, J = 5.2, 1.5 Hz, H5), 3.25 (2H, q, J = 6.6 Hz, NH-CH2), 1.56 (2H, p, J = 6.9 Hz, NH-CH2-CH2), 1.33–1.16 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.52, 154.84, 151.76, 142.28, 134.17, 122.64, 118.59, 42.71, 31.47, 29.21, 29.18, 29.13, 29.12, 28.88, 28.86, 28.79, 26.28, 22.26, 14.12. Elemental analysis for C19H29BrN4O (409.37); calculated C, 55.75; H, 7.14; N, 13.69, found: C, 55.89; H, 7.22; N, 13.80. Rf: 0.78.
N-Dodecyl-5-(pyridazin-4-yl)-1,3,4-oxadiazol-2-amine 3r. Brown solid, yield 41%; mp 142–143 °C. IR (ATR): 665, 719, 727, 882, 975, 1058, 1340, 1380, 1395, 1471, 1480, 1527, 1568, 1617, 1662, 2850, 2917, 2953, 3267 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 9.55 (1H, dd, J = 2.3, 1.3 Hz, H3), 9.37 (1H, dd, J = 5.5, 1.3 Hz, H6), 8.19 (1H, t, J = 5.7 Hz, NH-CH2), 7.91 (1H, dd, J = 5.4, 2.3 Hz, H5), 3.26 (2H, q, J = 6.6 Hz, NH-CH2), 1.56 (2H, p, J = 7.0 Hz, NH-CH2-CH2), 1.34–1.20 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 164.67, 154.11, 152.28, 146.46, 122.72, 121.04, 42.75, 31.46, 29.20, 29.17, 29.13, 29.10, 28.87, 28.84, 28.81, 26.30, 22.25, 14.10. Elemental analysis for C18H29N5O (331.46); calculated C, 65.23; H, 8.82; N, 21.13, found: C, 65.31; H, 8.91; N, 21.24. Rf: 0.42.
5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-thiol 2s (2 mmol, 358 mg) was suspended together with 2 equivalents of potassium carbonate (4 mmol, 552 mg) in 30 mL of N,N-dimethylformamide. Then, 1-bromododecane was added in one portion (2 mmol, 498 mg). Reaction was allowed to react for 5 h. The reaction mixture was evaporated to dryness on a rotary evaporator and transferred to a separatory funnel with 30 mL of EtOAc and 30 mL of demineralized water. The organic phase was washed with 30 mL of a saturated brine. The organic phase was dried over anhydrous sodium sulfate for 45 min, then the desiccant was separated, and the mixture was evaporated to dryness. The crude product was overlaid with dry DCM and stored at −20 °C. The solid product was filtered off and washed twice with 5 mL of ice-cold DCM.
2-(Dodecylthio)-5-(pyridin-4-yl)-1,3,4-oxadiazole 3s. Yellow solid, yield 100%; mp 63–64 °C. IR (ATR): 707, 723, 835, 961, 989, 1063, 1083, 1182, 1226, 1415, 1460, 1539, 1608, 2850, 2919, 2955 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.82–8.79 (2H, m, H2, H6), 7.90–7.87 (2H, m, H3, H5), 3.32 (2H, t, J = 7.2 Hz, S-CH2), 1.76 (2H, p, J = 7.4 Hz, S-CH2-CH2), 1.38 (2H, p, J = 7.3 Hz, C3H2), 1.31–1.16 (16H, m, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.83 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 165.63, 163.72, 151.13, 130.33, 120.14, 32.28, 31.48, 29.21, 29.19, 29.12, 29.08, 29.03, 28.89, 28.55, 27.96, 22.28, 14.14. Elemental analysis for C19H29N3OS (347.52); calculated C, 65.67; H, 8.41; N, 12.09, found: C, 65.79; H, 8.52; N, 12.09. Rf: 0.72.
N-Dodecyl-5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine 3t. Yellow solid, yield 68%; mp 152–154 °C. IR (ATR): 632, 683, 719, 802, 816, 992, 1010, 1035, 1121, 1195, 1375, 1420, 1468, 1494, 1523, 1549, 1592, 1635, 2854, 2920, 2955, 3194 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.76 (2H, d, J = 6.1 Hz, H2, H6), 8.01 (2H, d, J = 6.0 Hz, H3), 3.40 (2H, t, J = 7.0 Hz, NH-CH2), 1.65 (2H, p, J = 7.2 Hz, NH-CH2-CH2), 1.41–1.21 (18H, m, C3H2, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.86 (3H, t, J = 6.7 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 170.82, 159.97, 145.49, 142.65, 121.18, 45.14, 30.86, 28.59, 28.56, 28.54, 28.52, 28.26, 28.22, 28.12, 25.97, 21.58, 13.34. Elemental analysis for C19H30N4S (346.54); calculated C, 65.85; H, 8.73; N, 16.17, found: C, 65.99; H, 8.70; N, 16.25. Rf: 0.45.
N′-Tridecylideneisonicotinohydrazide
2u (0.5 mmol, 159 mg) was dissolved in 30 mL of DMSO, iodine (1.2 eq., 0.6 mmol, 152 mg) and potassium carbonate (3 eq., 1.5 mmol, 207 mg) was added to the solution, and heated to 100 °C. After 3 h, the mixture was evaporated to dryness on a rotary evaporator and transferred to a separatory funnel with 30 mL of EtOAc acetate and 30 mL of demineralized water. The organic phase was washed with 30 mL of a saturated Na
2S
2O
3 solution and finally 30 mL of a saturated brine. The organic phase was dried over anhydrous sodium sulfate for 45 min, then the desiccant was separated, and the mixture was evaporated to dryness. The crude product was overlaid with dry DCM and stored at −20 °C. The solid product was filtered off on and washed twice with 5 mL of ice-cold DCM [
36].
2-Dodecyl-5-(pyridin-4-yl)-1,3,4-oxadiazole 3u. Yellow solid, yield 76%; mp 75–76 °C. IR (ATR): 636, 704, 724, 730, 833, 989, 1023, 1099, 1182, 1232, 1417, 1464, 1473, 1541, 1565, 1608, 2849, 2872, 2917, 2955 cm−1. 1H NMR (500 MHz, DMSO-D6) δ 8.82–8.80 (2H, m, H2, H6), 7.90–7.88 (2H, m, H3, H5), 2.95 (2H, t, J = 7.4 Hz, C1H2), 1.78 (2H, p, J = 7.4 Hz, C2H2), 1.38 (2H, p, J = 7.3 Hz, C3H2), 1.34–1.20 (16H, m, C4H2, C5H2, C6H2, C7H2, C8H2, C9H2, C10H2, C11H2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (126 MHz, DMSO-D6) δ 167.84, 162.32, 150.84, 130.58, 119.96, 31.15, 28.86, 28.85, 28.80, 28.66, 28.52, 28.35, 28.15, 25.63, 24.54, 21.92, 13.72. Elemental analysis for C19H29N3O (315.46); calculated C, 72.34; H, 9.27; N, 13.32, found: C, 72.41; H, 9.30; N, 13.22. Rf: 0.64.


 ,
, 
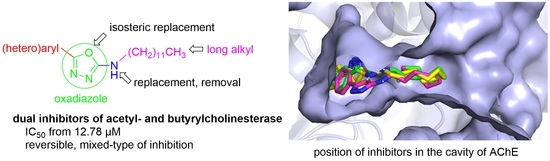









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


