
Liposome-Derived Nanosystems for the Treatment of Behavioral and Neurodegenerative Diseases: The Promise of Niosomes, Transfersomes, and Ethosomes for Increased Brain Drug Bioavailability



Abstract
: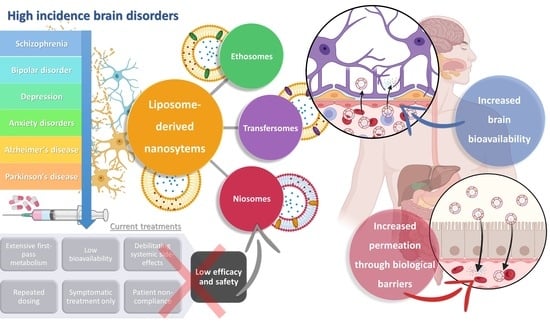
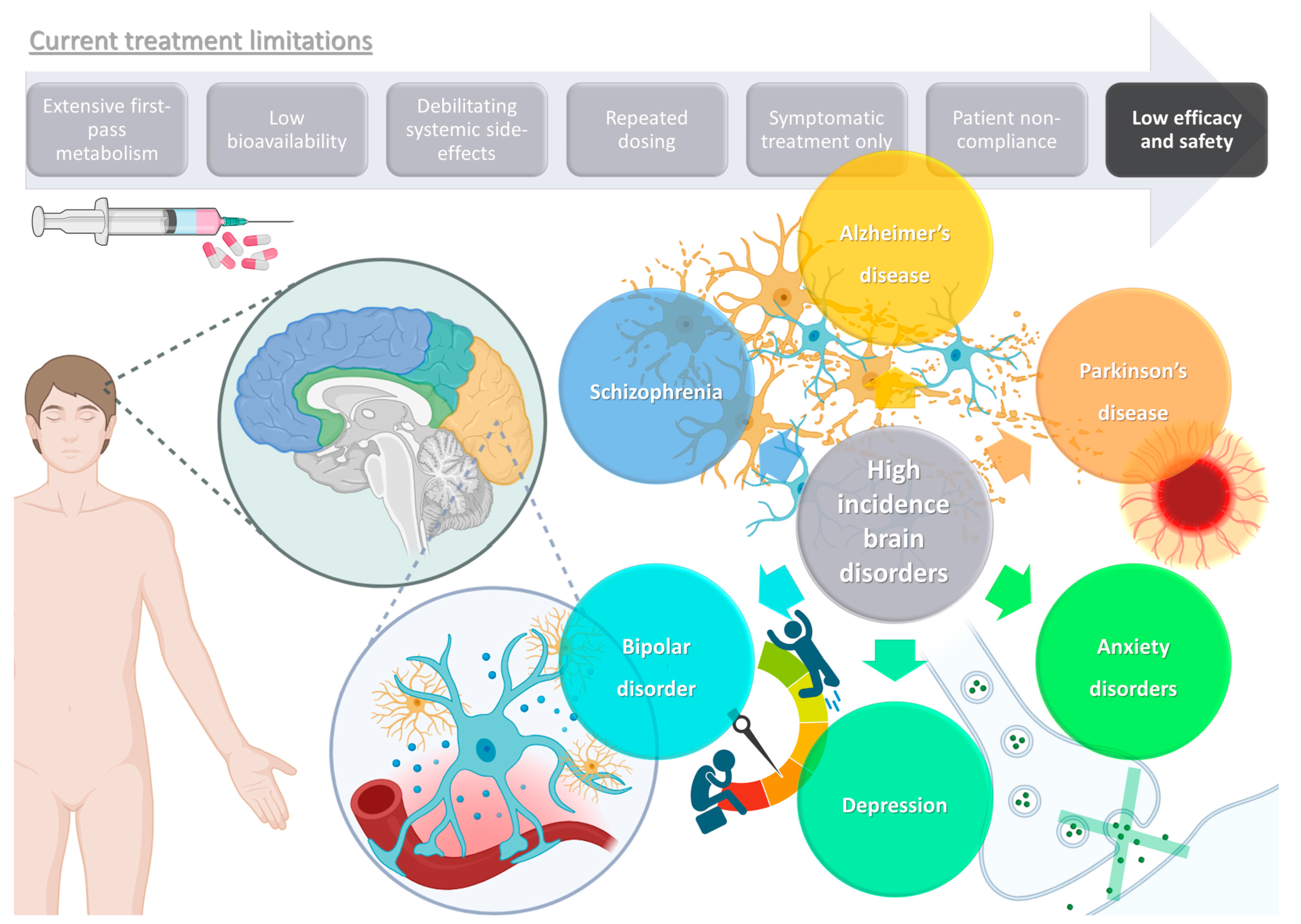
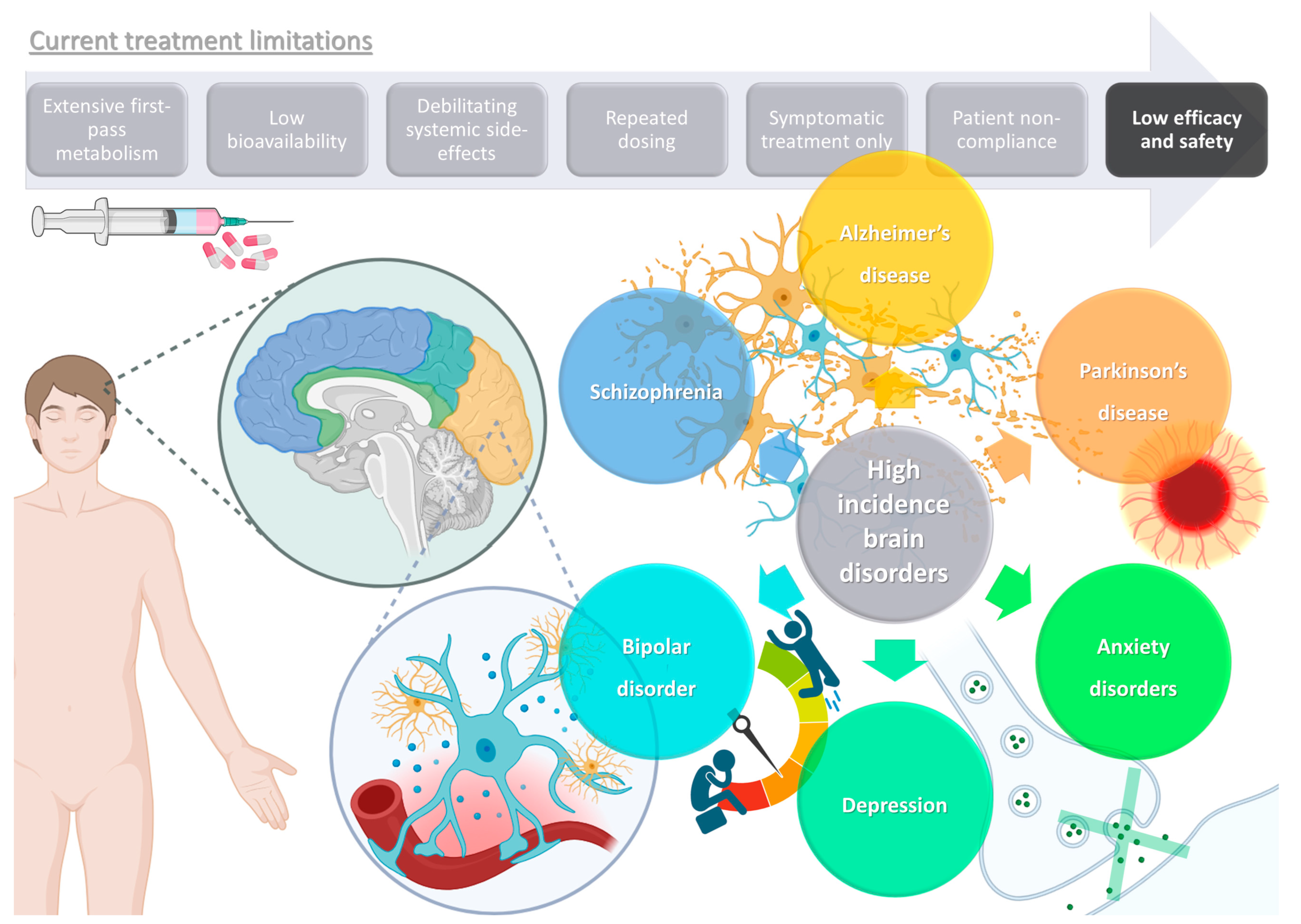
1. High Prevalence Brain Disorders: Current Treatments and Their Limitations
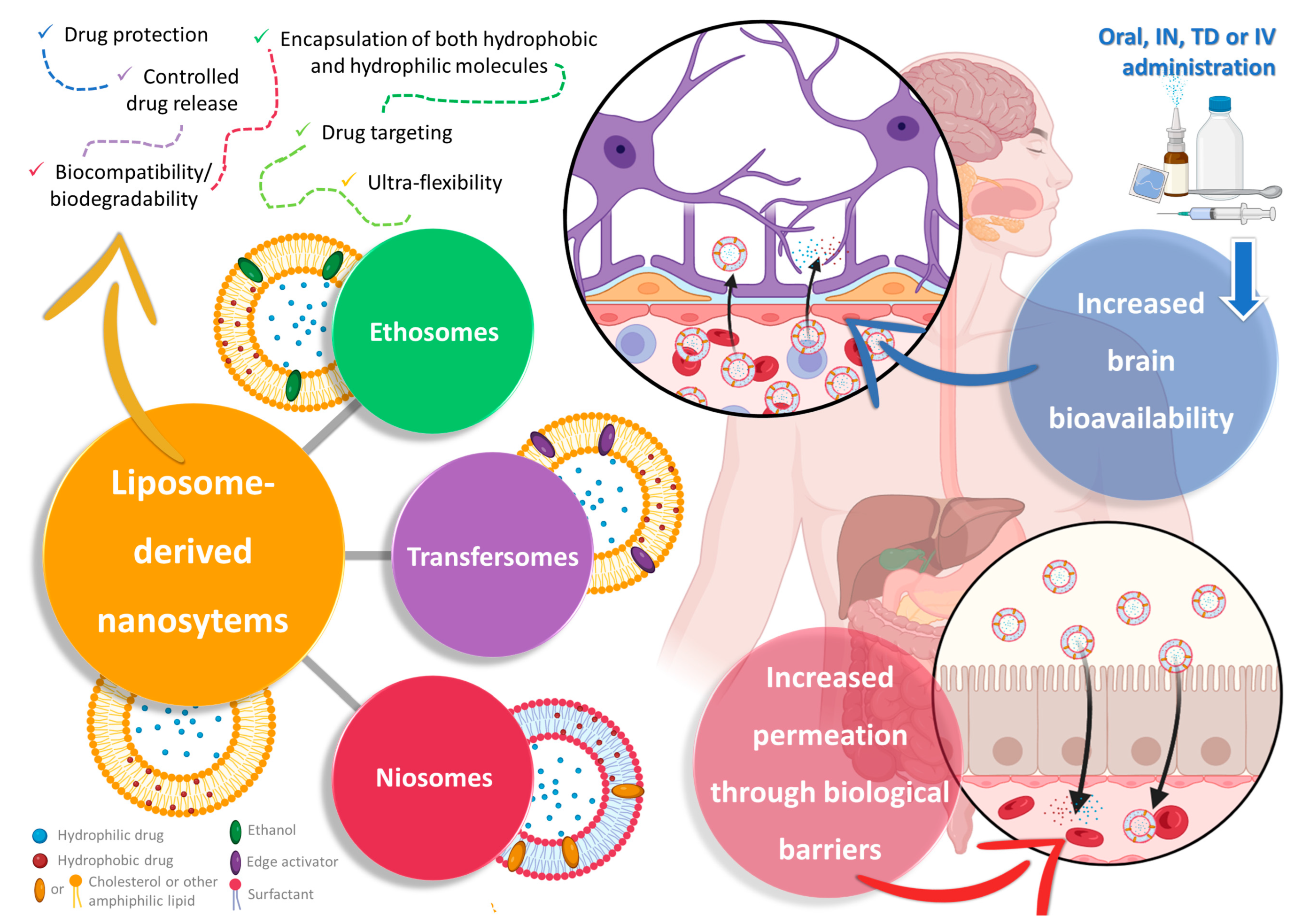
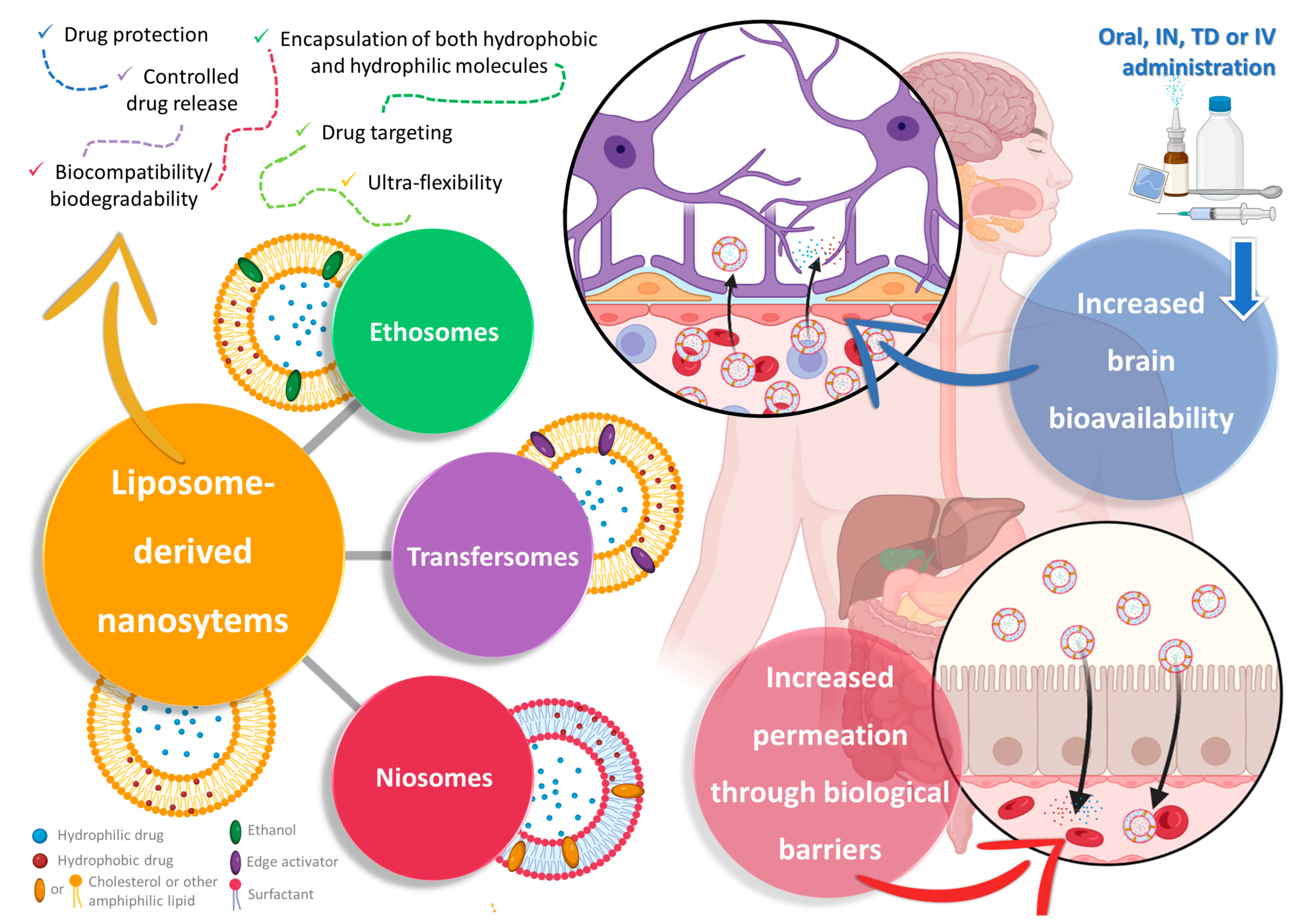
2. Nanosystems as Non-Conventional Forms of Treatment for Increased Efficacy and Safety
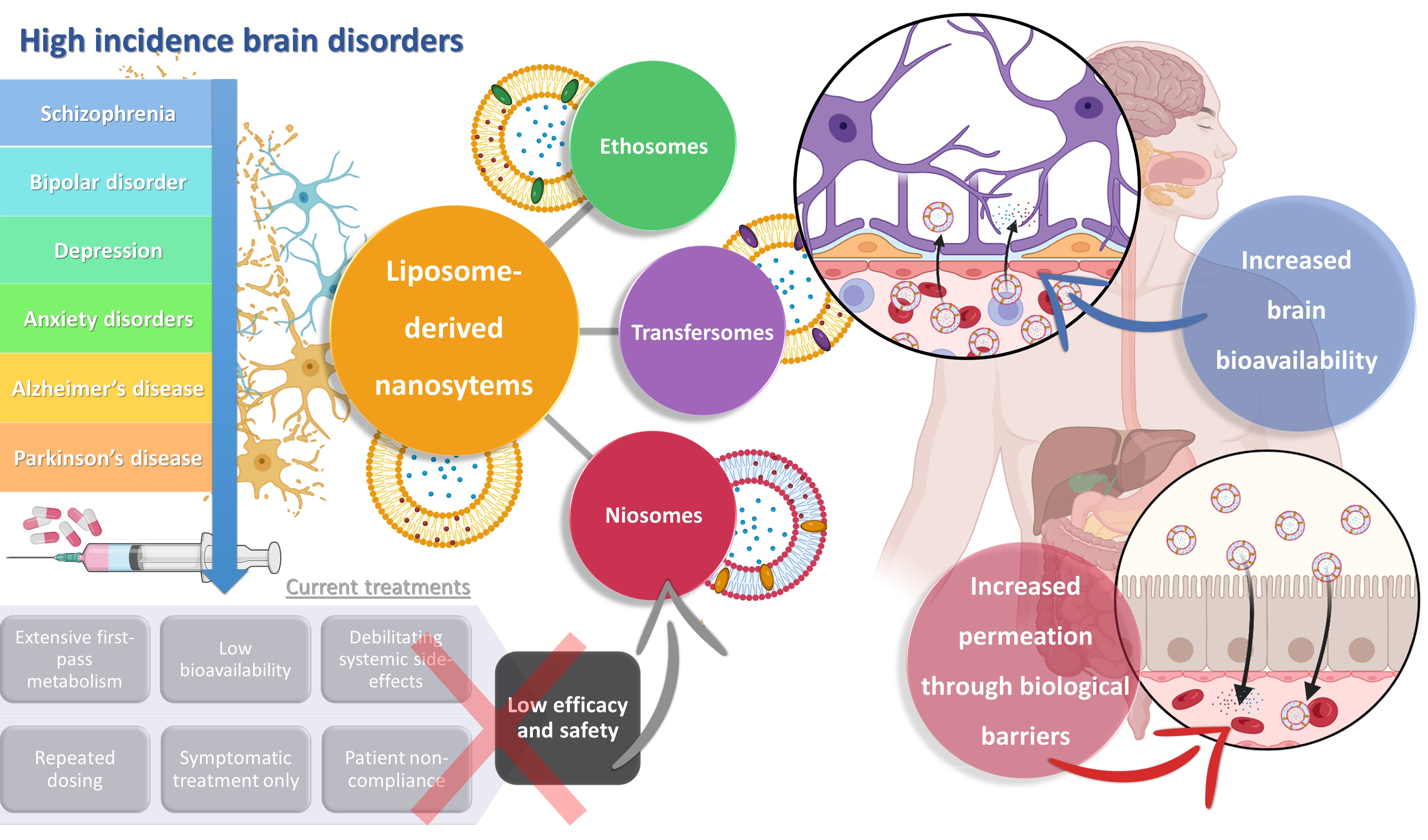
3. Liposome-Derived Nanosystems: Ethosomes, Transfersomes, and Niosomes for Brain Drug Delivery
3.1. Ethosomes
3.2. Transfersomes
3.3. Niosomes
4. Liposome-Derived Vesicles: The Future for Brain Drug Delivery?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Rehm, J.; Shield, K.D. Global Burden of Disease and the Impact of Mental and Addictive Disorders. Curr. Psychiatry Rep. 2019, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.R.; McGee, R.E.; Druss, B.G. Mortality in Mental Disorders and Global Disease Burden Implications. JAMA Psychiatry 2015, 72, 334. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Mental Disorders Collaborators Global, Regional, and National Burden of 12 Mental Disorders in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Psychiatry 2022, 9, 137–150. [CrossRef] [PubMed]
- Prince, M. The Global Burden of Mental Disorder. In Oxford Textbook of Community Mental Health; Oxford University Press: Oxford, UK, 2010; Chapter 7. [Google Scholar]
- Vigo, D.; Thornicroft, G.; Atun, R. Estimating the True Global Burden of Mental Illness. Lancet Psychiatry 2016, 3, 171–178. [Google Scholar] [CrossRef]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Reis Marques, T.; Howes, O.D. Schizophrenia—An Overview. JAMA Psychiatry 2020, 77, 201. [Google Scholar] [CrossRef]
- Karlsgodt, K.H.; Sun, D.; Cannon, T.D. Structural and Functional Brain Abnormalities in Schizophrenia. Curr. Dir. Psychol. Sci. 2010, 19, 226–231. [Google Scholar] [CrossRef]
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Primers 2015, 1, 15067. [Google Scholar] [CrossRef]
- Wong, A.H.C.; Van Tol, H.H.M. Schizophrenia: From Phenomenology to Neurobiology. Neurosci. Biobehav. Rev. 2003, 27, 269–306. [Google Scholar] [CrossRef]
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and Treatment Options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar]
- Vieta, E.; Berk, M.; Schulze, T.G.; Carvalho, A.F.; Suppes, T.; Calabrese, J.R.; Gao, K.; Miskowiak, K.W.; Grande, I. Bipolar Disorders. Nat. Rev. Dis. Primers 2018, 4, 18008. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S.; Berk, M.; Brietzke, E.; Goldstein, B.I.; López-Jaramillo, C.; Kessing, L.V.; Malhi, G.S.; Nierenberg, A.A.; Rosenblat, J.D.; Majeed, A.; et al. Bipolar Disorders. Lancet 2020, 396, 1841–1856. [Google Scholar] [CrossRef]
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar Disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Carvalho, A.F.; Firth, J.; Vieta, E. Bipolar Disorder. N. Engl. J. Med. 2020, 383, 58–66. [Google Scholar] [CrossRef]
- Johnson, W.M.; Fields, S.A.; Bluett, E. Bipolar Disorder: Managing the Peaks and Valleys. Int. J. Psychiatry Med. 2020, 55, 349–356. [Google Scholar] [CrossRef]
- Gitlin, M.J. Antidepressants in Bipolar Depression: An Enduring Controversy. Int. J. Bipolar Disord. 2018, 6, 25. [Google Scholar] [CrossRef]
- Shah, N.; Grover, S.; Rao, G.P. Clinical Practice Guidelines for Management of Bipolar Disorder. Indian J. Psychiatry 2017, 59, 51. [Google Scholar] [CrossRef]
- Malhi, G.S.; Tanious, M.; Das, P.; Coulston, C.M.; Berk, M. Potential Mechanisms of Action of Lithium in Bipolar Disorder. CNS Drugs 2013, 27, 135–153. [Google Scholar] [CrossRef]
- Marwaha, S.; Palmer, E.; Suppes, T.; Cons, E.; Young, A.H.; Upthegrove, R. Novel and Emerging Treatments for Major Depression. Lancet 2023, 401, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative Efficacy and Acceptability of 21 Antidepressant Drugs for the Acute Treatment of Adults with Major Depressive Disorder: A Systematic Review and Network Meta-Analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Teng, T.; Zhang, Y.; Del Giovane, C.; Furukawa, T.A.; Weisz, J.R.; Li, X.; Cuijpers, P.; Coghill, D.; Xiang, Y.; et al. Comparative Efficacy and Acceptability of Antidepressants, Psychotherapies, and Their Combination for Acute Treatment of Children and Adolescents with Depressive Disorder: A Systematic Review and Network Meta-Analysis. Lancet Psychiatry 2020, 7, 581–601. [Google Scholar] [CrossRef] [PubMed]
- Ménard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of Depression: Insights from Human and Rodent Studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef] [PubMed]
- Penn, E.; Tracy, D.K. The Drugs Don’t Work? Antidepressants and the Current and Future Pharmacological Management of Depression. Ther. Adv. Psychopharmacol. 2012, 2, 179–188. [Google Scholar] [CrossRef]
- Ramic, E.; Prasko, S.; Gavran, L.; Spahic, E. Assessment of the Antidepressant Side Effects Occurrence in Patients Treated in Primary Care. Mater. Socio-Medica 2020, 32, 131. [Google Scholar] [CrossRef]
- Santarsieri, D.; Schwartz, T. Antidepressant Efficacy and Side-Effect Burden: A Quick Guide for Clinicians. Drugs Context 2015, 4, 1–12. [Google Scholar] [CrossRef]
- Penninx, B.W.; Pine, D.S.; Holmes, E.A.; Reif, A. Anxiety Disorders. Lancet 2021, 397, 914–927. [Google Scholar] [CrossRef]
- Strawn, J.R.; Geracioti, L.; Rajdev, N.; Clemenza, K.; Levine, A. Pharmacotherapy for Generalized Anxiety Disorder in Adult and Pediatric Patients: An Evidence-Based Treatment Review. Expert Opin. Pharmacother. 2018, 19, 1057–1070. [Google Scholar] [CrossRef]
- Craske, M.G.; Stein, M.B. Anxiety. Lancet 2016, 388, 3048–3059. [Google Scholar] [CrossRef] [PubMed]
- Bandelow, B. Current and Novel Psychopharmacological Drugs for Anxiety Disorders. In Anxiety Disorders: Rethinking and Understanding Recent Discoveries; Springer: Berlin/Heidelberg, Germany, 2020; pp. 347–365. [Google Scholar]
- Slee, A.; Nazareth, I.; Bondaronek, P.; Liu, Y.; Cheng, Z.; Freemantle, N. Pharmacological Treatments for Generalised Anxiety Disorder: A Systematic Review and Network Meta-Analysis. Lancet 2019, 393, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B. Anxiolytics: Past, Present, and Future Agents. J. Clin. Psychiatry 2003, 64 (Suppl. 3), 3–6. [Google Scholar]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Abubakar, M.B.; Sanusi, K.O.; Ugusman, A.; Mohamed, W.; Kamal, H.; Ibrahim, N.H.; Khoo, C.S.; Kumar, J. Alzheimer’s Disease: An Update and Insights Into Pathophysiology. Front. Aging Neurosci. 2022, 14, 742408. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Piaceri, I. Genetics of Familial and Sporadic Alzheimer’s Disease. Front. Biosci. 2013, 5, 167–177. [Google Scholar] [CrossRef]
- Kim, D.H.; Yeo, S.H.; Park, J.-M.; Choi, J.Y.; Lee, T.-H.; Park, S.Y.; Ock, M.S.; Eo, J.; Kim, H.-S.; Cha, H.-J. Genetic Markers for Diagnosis and Pathogenesis of Alzheimer’s Disease. Gene 2014, 545, 185–193. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer’s Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.H.; Sawhney, G.; Pottoo, F.H.; Mohi-ud-din, R.; Madishetti, S.; Jachak, S.M.; Ahmed, Z.; Masoodi, M.H. Role of Environmental Pollutants in Alzheimer’s Disease: A Review. Environ. Sci. Pollut. Res. 2020, 27, 44724–44742. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug Treatments in Alzheimer’s Disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. Anti-Amyloid Monoclonal Antibodies Are Transformative Treatments That Redefine Alzheimer’s Disease Therapeutics. Drugs 2023, 83, 569–576. [Google Scholar] [CrossRef]
- Gauthier, S.; Boxer, A.; Knopman, D.; Sims, J.; Doody, R.; Aisen, P.; Iwatsubo, T.; Bateman, R.; Vellas, B. Therapeutic Targets for Alzheimer’s Disease: Amyloid Vs. Non-Amyloid. Where Does Consensus Lie Today? An CTAD Task Force Report. J. Prev. Alzheimer’s Dis. 2022, 9, 231–235. [Google Scholar] [CrossRef]
- Lew, M. Overview of Parkinson’s Disease. Pharmacotherapy 2007, 27, 155S–160S. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson Disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Sivanandy, P.; Leey, T.C.; Xiang, T.C.; Ling, T.C.; Wey Han, S.A.; Semilan, S.L.A.; Hong, P.K. Systematic Review on Parkinson’s Disease Medications, Emphasizing on Three Recently Approved Drugs to Control Parkinson’s Symptoms. Int. J. Environ. Res. Public Health 2021, 19, 364. [Google Scholar] [CrossRef] [PubMed]
- Katzenschlager, R.; Sampaio, C.; Costa, J.; Lees, A. Anticholinergics for Symptomatic Management of Parkinson´s Disease. Cochrane Database Syst. Rev. 2002, 2010, CD003735. [Google Scholar] [CrossRef]
- Müller, T. Catechol-O-Methyltransferase Inhibitors in Parkinson’s Disease. Drugs 2015, 75, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Bogosian, A.; Rixon, L.; Hurt, C.S. Prioritising Target Non-Pharmacological Interventions for Research in Parkinson’s Disease: Achieving Consensus from Key Stakeholders. Res. Involv. Engagem. 2020, 6, 35. [Google Scholar] [CrossRef]
- Ahn, S.; Chen, Y.; Bredow, T.; Cheung, C.; Yu, F. Effects of Non-PharmacologicalTreatments on Quality of Life in Parkinson’s Disease: A Review. J. Park. Dis. Alzheimer’s Dis. 2017, 4, 1–10. [Google Scholar] [CrossRef]
- Romba, C.; Perez-Reisler, M. Management of Adverse Effects of Psychotropic Medications. Pediatr. Ann. 2020, 49, e431–e435. [Google Scholar] [CrossRef]
- Schwartz, T.L.; Nihalani, N.; Jindal, S.; Virk, S.; Jones, N. Psychiatric Medication-Induced Obesity: A Review. Obes. Rev. 2004, 5, 115–121. [Google Scholar] [CrossRef]
- Smith, F.A.; Wittmann, C.W.; Stern, T.A. Medical Complications of Psychiatric Treatment. Crit. Care Clin. 2008, 24, 635–656. [Google Scholar] [CrossRef]
- Hilt, R.J.; Chaudhari, M.; Bell, J.F.; Wolf, C.; Koprowicz, K.; King, B.H. Side Effects from Use of One or More Psychiatric Medications in a Population-Based Sample of Children and Adolescents. J. Child. Adolesc. Psychopharmacol. 2014, 24, 83–89. [Google Scholar] [CrossRef]
- Ballard, C.; Grace, J.; McKeith, L.; Holmes, C. Neuroleptic Sensitivity in Dementia with Lewy Bodies and Alzheimer’s Disease. Lancet 1998, 351, 1032–1033. [Google Scholar] [CrossRef] [PubMed]
- Muller, C. Prodrug Approaches for Enhancing the Bioavailability of Drugs with Low Solubility. Chem. Biodivers. 2009, 6, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Hoffman, A. Rationalizing the Selection of Oral Lipid Based Drug Delivery Systems by an in Vitro Dynamic Lipolysis Model for Improved Oral Bioavailability of Poorly Water Soluble Drugs. J. Control Release 2008, 129, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Pires, P.C.; Rodrigues, M.; Alves, G.; Santos, A.O. Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs. Pharmaceutics 2022, 14, 588. [Google Scholar] [CrossRef]
- Zhang, F.; Xu, C.; Liu, C. Drug Delivery Strategies to Enhance the Permeability of the Blood–Brain Barrier for Treatment of Glioma. Drug Des. Devel Ther. 2015, 9, 2089–2100. [Google Scholar] [CrossRef]
- He, X.; Deng, H.; Hwang, H. The Current Application of Nanotechnology in Food and Agriculture. J. Food Drug Anal. 2019, 27, 1–21. [Google Scholar] [CrossRef]
- Yan, H.; Zhai, B.; Yang, F.; Chen, Z.; Zhou, Q.; Paiva-Santos, A.C.; Yuan, Z.; Zhou, Y. Nanotechnology-Based Diagnostic and Therapeutic Strategies for Neuroblastoma. Front. Pharmacol. 2022, 13, 908713. [Google Scholar] [CrossRef]
- Mazayen, Z.M.; Ghoneim, A.M.; Elbatanony, R.S.; Basalious, E.B.; Bendas, E.R. Pharmaceutical Nanotechnology: From the Bench to the Market. Futur. J. Pharm. Sci. 2022, 8, 12. [Google Scholar] [CrossRef]
- Katz, L.M.; Dewan, K.; Bronaugh, R.L. Nanotechnology in Cosmetics. Food Chem. Toxicol. 2015, 85, 127–137. [Google Scholar] [CrossRef]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The History of Nanoscience and Nanotechnology: From Chemical –Physical Applications to Nanomedicine. Molecules 2019, 25, 112. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liang, W.; Xiao, X.; Qian, Y. Nanotechnology, an Alternative with Promising Prospects and Advantages for the Treatment of Cardiovascular Diseases. Int. J. Nanomed. 2018, 13, 7349–7362. [Google Scholar] [CrossRef] [PubMed]
- Sethi, B.; Kumar, V.; Mahato, K.; Coulter, D.W.; Mahato, R.I. Recent Advances in Drug Delivery and Targeting to the Brain. J. Control Release 2022, 350, 668–687. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.-G.; Grumezescu, A.M. Polymer-Based Nanosystems—A Versatile Delivery Approach. Materials 2021, 14, 6812. [Google Scholar] [CrossRef]
- Tapeinos, C.; Battaglini, M.; Ciofani, G. Advances in the Design of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Targeting Brain Diseases. J. Control Release 2017, 264, 306–332. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in Nose-to-Brain Drug Delivery: A Review of Non-Clinical Brain Targeting Studies. J. Control Release 2018, 270, 89–100. [Google Scholar] [CrossRef]
- Nehra, M.; Uthappa, U.T.; Kumar, V.; Kumar, R.; Dixit, C.; Dilbaghi, N.; Mishra, Y.K.; Kumar, S.; Kaushik, A. Nanobiotechnology-Assisted Therapies to Manage Brain Cancer in Personalized Manner. J. Control Release 2021, 338, 224–243. [Google Scholar] [CrossRef]
- Alenzi, A.M.; Albalawi, S.A.; Alghamdi, S.G.; Albalawi, R.F.; Albalawi, H.S.; Qushawy, M. Review on Different Vesicular Drug Delivery Systems (VDDSs) and Their Applications. Recent Pat. Nanotechnol. 2023, 17, 18–32. [Google Scholar] [CrossRef]
- Dymek, M.; Sikora, E. Liposomes as Biocompatible and Smart Delivery Systems—The Current State. Adv. Colloid. Interface Sci. 2022, 309, 102757. [Google Scholar] [CrossRef]
- Shah, S.; Dhawan, V.; Holm, R.; Nagarsenker, M.S.; Perrie, Y. Liposomes: Advancements and Innovation in the Manufacturing Process. Adv. Drug Deliv. Rev. 2020, 154–155, 102–122. [Google Scholar] [CrossRef]
- Adnan, M.; Akhter, M.H.; Afzal, O.; Altamimi, A.S.A.; Ahmad, I.; Alossaimi, M.A.; Jaremko, M.; Emwas, A.-H.; Haider, T.; Haider, M.F. Exploring Nanocarriers as Treatment Modalities for Skin Cancer. Molecules 2023, 28, 5905. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Nie, L.; Zhu, S.; Zhang, X. Nanovesicles-Mediated Drug Delivery for Oral Bioavailability Enhancement. Int. J. Nanomed. 2022, 17, 4861–4877. [Google Scholar] [CrossRef]
- Jafari, A.; Daneshamouz, S.; Ghasemiyeh, P.; Mohammadi-Samani, S. Ethosomes as Dermal/Transdermal Drug Delivery Systems: Applications, Preparation and Characterization. J. Liposome Res. 2023, 33, 34–52. [Google Scholar] [CrossRef]
- Paiva-Santos, A.C.; Silva, A.L.; Guerra, C.; Peixoto, D.; Pereira-Silva, M.; Zeinali, M.; Mascarenhas-Melo, F.; Castro, R.; Veiga, F. Ethosomes as Nanocarriers for the Development of Skin Delivery Formulations. Pharm. Res. 2021, 38, 947–970. [Google Scholar] [CrossRef]
- Lu, J.; Guo, T.; Fan, Y.; Li, Z.; He, Z.; Yin, S.; Feng, N. Recent Developments in the Principles, Modification and Application Prospects of Functionalized Ethosomes for Topical Delivery. Curr. Drug Deliv. 2021, 18, 570–582. [Google Scholar] [CrossRef]
- Pilch, E.; Musiał, W. Liposomes with an Ethanol Fraction as an Application for Drug Delivery. Int. J. Mol. Sci. 2018, 19, 3806. [Google Scholar] [CrossRef]
- Cascione, M.; De Matteis, V.; Leporatti, S.; Rinaldi, R. The New Frontiers in Neurodegenerative Diseases Treatment: Liposomal-Based Strategies. Front. Bioeng. Biotechnol. 2020, 8, 566767. [Google Scholar] [CrossRef]
- Niu, X.-Q.; Zhang, D.-P.; Bian, Q.; Feng, X.-F.; Li, H.; Rao, Y.-F.; Shen, Y.-M.; Geng, F.-N.; Yuan, A.-R.; Ying, X.-Y.; et al. Mechanism Investigation of Ethosomes Transdermal Permeation. Int. J. Pharm. X 2019, 1, 100027. [Google Scholar] [CrossRef]
- Matharoo, N.; Mohd, H.; Michniak-Kohn, B. Transferosomes as a Transdermal Drug Delivery System: Dermal Kinetics and Recent Developments. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, e1918. [Google Scholar] [CrossRef]
- Opatha, S.A.T.; Titapiwatanakun, V.; Chutoprapat, R. Transfersomes: A Promising Nanoencapsulation Technique for Transdermal Drug Delivery. Pharmaceutics 2020, 12, 855. [Google Scholar] [CrossRef]
- Gupta, R.; Kumar, A. Transfersomes: The Ultra-Deformable Carrier System for Non-Invasive Delivery of Drug. Curr. Drug Deliv. 2021, 18, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Fernández-García, R.; Lalatsa, A.; Statts, L.; Bolás-Fernández, F.; Ballesteros, M.P.; Serrano, D.R. Transferosomes as Nanocarriers for Drugs across the Skin: Quality by Design from Lab to Industrial Scale. Int. J. Pharm. 2020, 573, 118817. [Google Scholar] [CrossRef] [PubMed]
- Yasamineh, S.; Yasamineh, P.; Ghafouri Kalajahi, H.; Gholizadeh, O.; Yekanipour, Z.; Afkhami, H.; Eslami, M.; Hossein Kheirkhah, A.; Taghizadeh, M.; Yazdani, Y.; et al. A State-of-the-Art Review on the Recent Advances of Niosomes as a Targeted Drug Delivery System. Int. J. Pharm. 2022, 624, 121878. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Tripathi, P.; Gupta, R.; Pandey, S. Niosomes: A Review on Niosomal Research in the Last Decade. J. Drug Deliv. Sci. Technol. 2020, 56, 101581. [Google Scholar] [CrossRef]
- Witika, B.A.; Bassey, K.E.; Demana, P.H.; Siwe-Noundou, X.; Poka, M.S. Current Advances in Specialised Niosomal Drug Delivery: Manufacture, Characterization and Drug Delivery Applications. Int. J. Mol. Sci. 2022, 23, 9668. [Google Scholar] [CrossRef]
- Chen, S.; Hanning, S.; Falconer, J.; Locke, M.; Wen, J. Recent Advances in Non-Ionic Surfactant Vesicles (Niosomes): Fabrication, Characterization, Pharmaceutical and Cosmetic Applications. Eur. J. Pharm. Biopharm. 2019, 144, 18–39. [Google Scholar] [CrossRef]
- Kheilnezhad, B.; Hadjizadeh, A. Factors Affecting the Penetration of Niosome into the Skin, Their Laboratory Measurements and Dependency to the Niosome Composition: A Review. Curr. Drug Deliv. 2021, 18, 555–569. [Google Scholar] [CrossRef]
- Hamishehkar, H.; Rahimpour, Y.; Kouhsoltani, M. Niosomes as a Propitious Carrier for Topical Drug Delivery. Expert Opin. Drug Deliv. 2013, 10, 261–272. [Google Scholar] [CrossRef]
- Choi, M.J.; Maibach, H.I. Liposomes and Niosomes as Topical Drug Delivery Systems. Skin. Pharmacol. Physiol. 2005, 18, 209–219. [Google Scholar] [CrossRef]
- Moghtaderi, M.; Sedaghatnia, K.; Bourbour, M.; Fatemizadeh, M.; Salehi Moghaddam, Z.; Hejabi, F.; Heidari, F.; Quazi, S.; Farasati Far, B. Niosomes: A Novel Targeted Drug Delivery System for Cancer. Med. Oncol. 2022, 39, 240. [Google Scholar] [CrossRef] [PubMed]
- Osanloo, M.; Assadpour, S.; Mehravaran, A.; Abastabar, M.; Akhtari, J. Niosome-Loaded Antifungal Drugs as an Effective Nanocarrier System: A Mini Review. Curr. Med. Mycol. 2019, 4, 31. [Google Scholar] [CrossRef]
- Aparajay, P.; Dev, A. Functionalized Niosomes as a Smart Delivery Device in Cancer and Fungal Infection. Eur. J. Pharm. Sci. 2022, 168, 106052. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, Y.; Luo, G. Ligustrazine Phosphate Ethosomes for Treatment of Alzheimer’s Disease, in Vitro and in Animal Model Studies. AAPS PharmSciTech 2012, 13, 485–492. [Google Scholar] [CrossRef]
- Mishra, G.; Awasthi, R.; Singh, A.K.; Singh, S.; Mishra, S.K.; Singh, S.K.; Nandi, M.K. Intranasally Co-Administered Berberine and Curcumin Loaded in Transfersomal Vesicles Improved Inhibition of Amyloid Formation and BACE-1. ACS Omega 2022, 7, 43290–43305. [Google Scholar] [CrossRef]
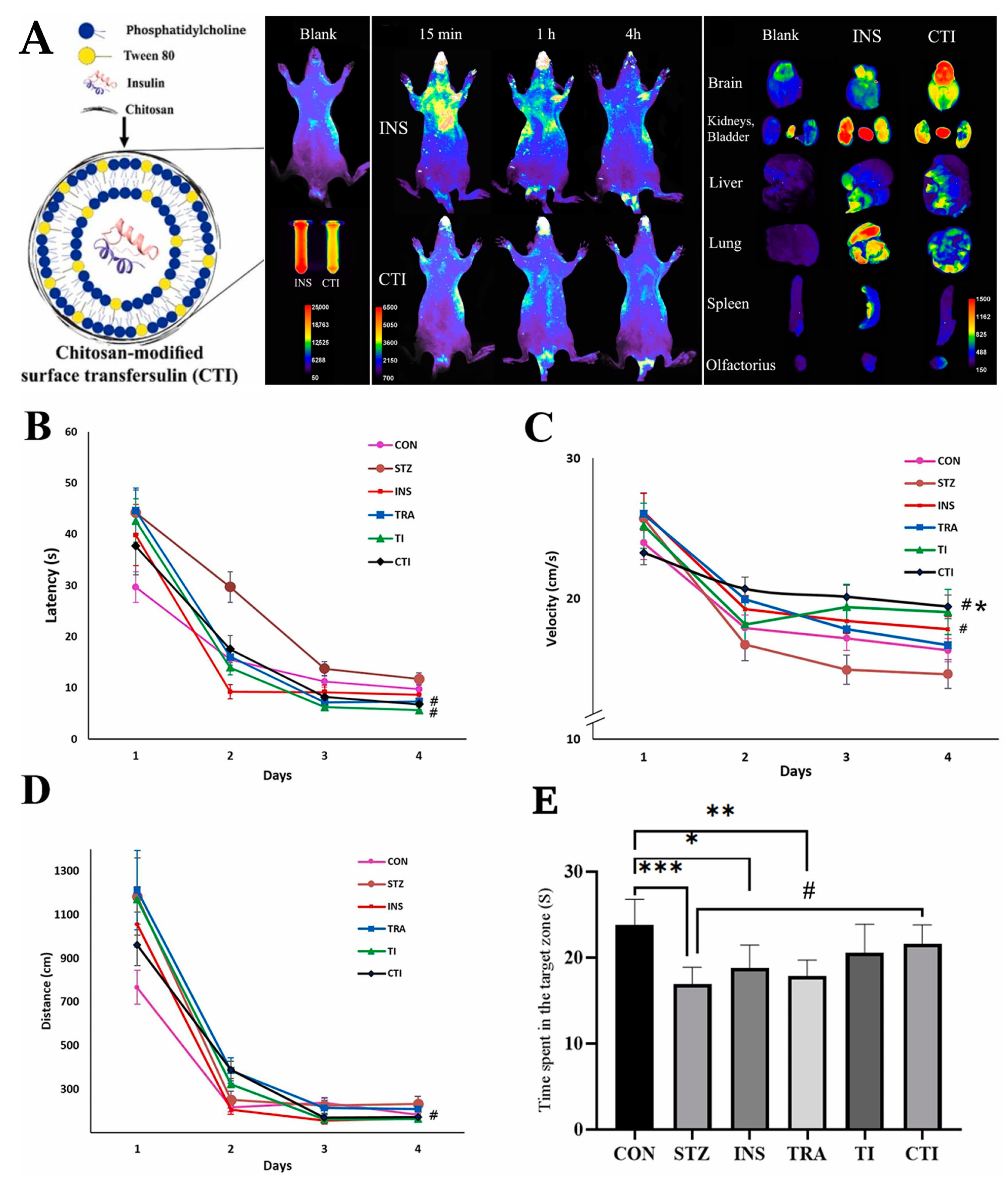
- Nojoki, F.; Ebrahimi-Hosseinzadeh, B.; Hatamian-Zarmi, A.; Khodagholi, F.; Khezri, K. Design and Development of Chitosan-Insulin-Transfersomes (Transfersulin) as Effective Intranasal Nanovesicles for the Treatment of Alzheimer’s Disease: In Vitro, in Vivo, and Ex Vivo Evaluations. Biomed. Pharmacother. 2022, 153, 113450. [Google Scholar] [CrossRef]
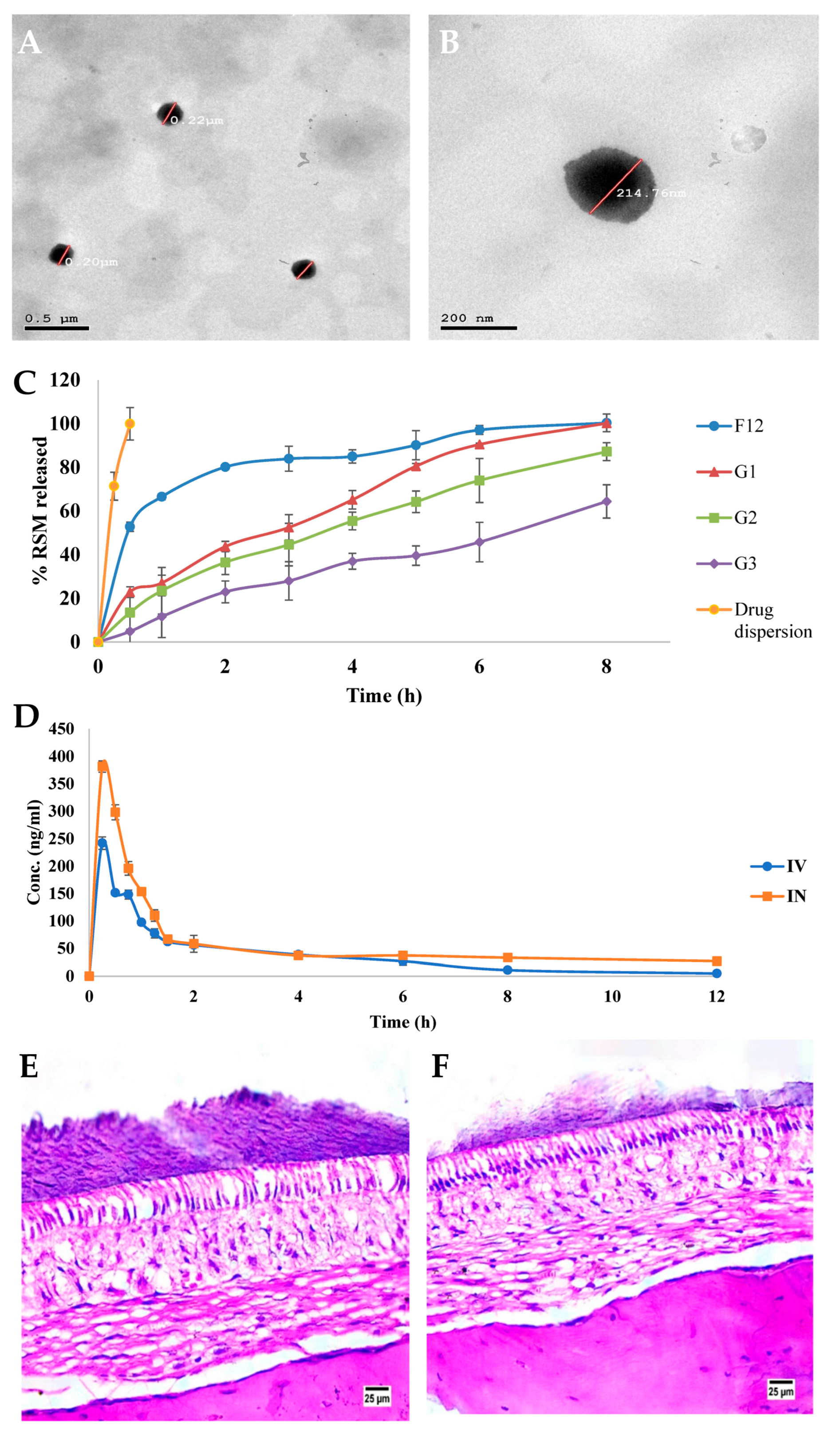
- ElShagea, H.N.; Makar, R.R.; Salama, A.H.; Elkasabgy, N.A.; Basalious, E.B. Investigating the Targeting Power to Brain Tissues of Intranasal Rasagiline Mesylate-Loaded Transferosomal In Situ Gel for Efficient Treatment of Parkinson’s Disease. Pharmaceutics 2023, 15, 533. [Google Scholar] [CrossRef]
- Shreya, A.B.; Managuli, R.S.; Menon, J.; Kondapalli, L.; Hegde, A.R.; Avadhani, K.; Shetty, P.K.; Amirthalingam, M.; Kalthur, G.; Mutalik, S. Nano-Transfersomal Formulations for Transdermal Delivery of Asenapine Maleate: In Vitro and in Vivo Performance Evaluations. J. Liposome Res. 2016, 26, 221–232. [Google Scholar] [CrossRef]
- Singh, R.P.; Narke, R.M.; Jadhav, P.V. Formulation and Evaluation of Asenapine Maleate Loaded Niosomes for the Treatment of Schizophrenia. Indian J. Pharm. Educ. Res. 2020, 54, S128–S139. [Google Scholar] [CrossRef]
- Khallaf, R.A.; Aboud, H.M.; Sayed, O.M. Surface Modified Niosomes of Olanzapine for Brain Targeting via Nasal Route; Preparation, Optimization, and in Vivo Evaluation. J. Liposome Res. 2020, 30, 163–173. [Google Scholar] [CrossRef]
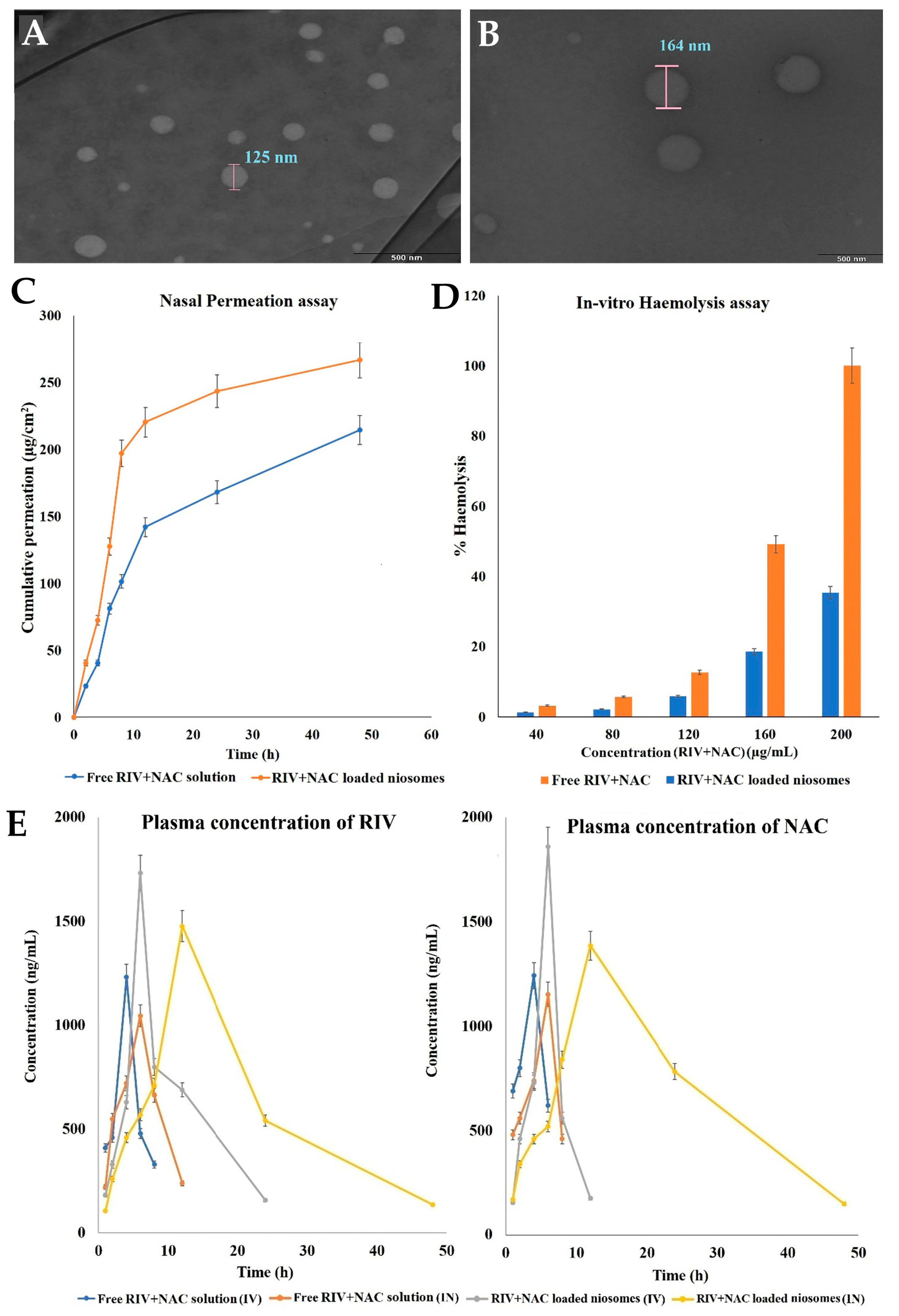
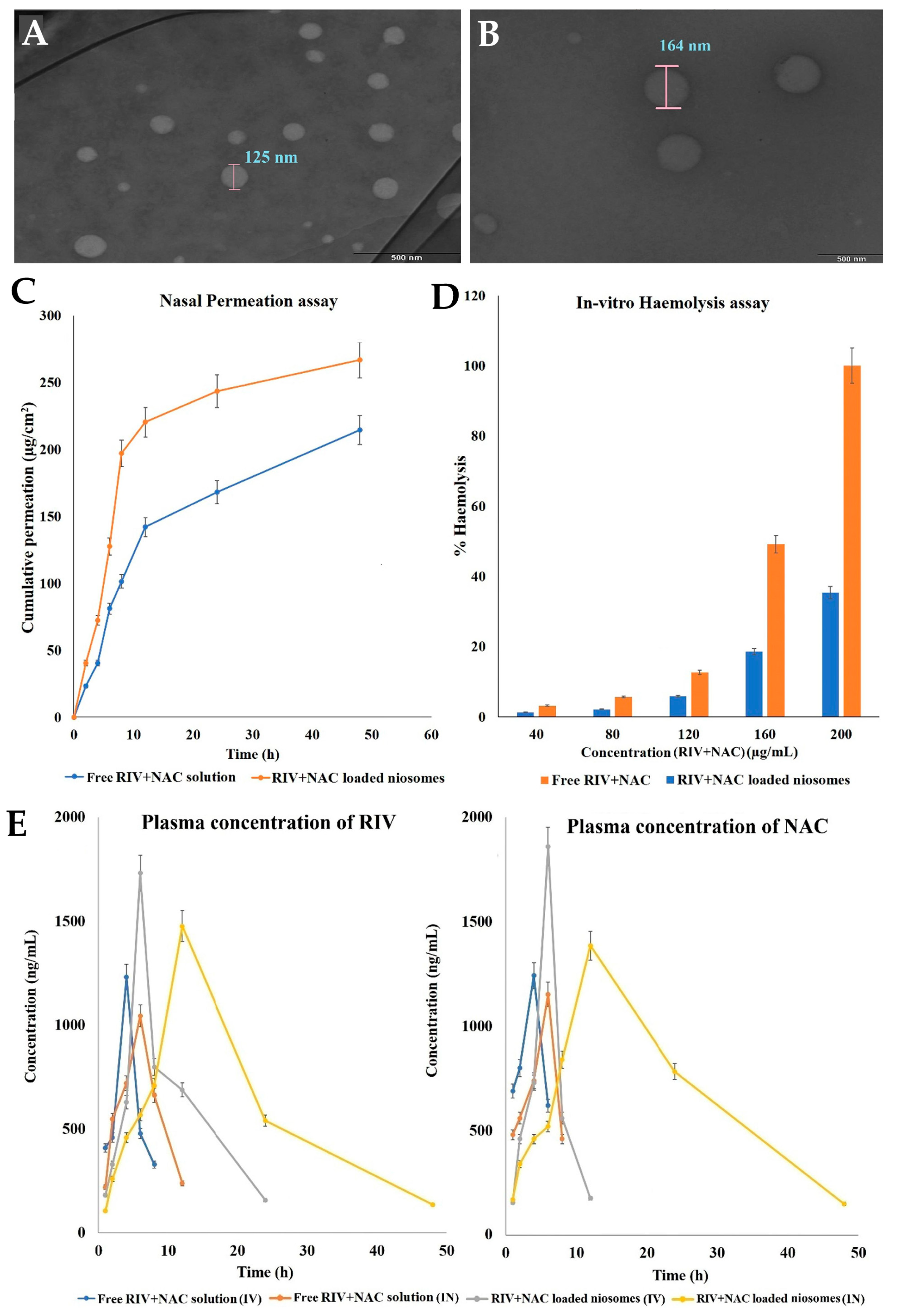
- Kulkarni, P.; Rawtani, D.; Barot, T. Design, Development and in-Vitro/in-Vivo Evaluation of Intranasally Delivered Rivastigmine and N-Acetyl Cysteine Loaded Bifunctional Niosomes for Applications in Combinative Treatment of Alzheimer’s Disease. Eur. J. Pharm. Biopharm. 2021, 163, 1–15. [Google Scholar] [CrossRef] [PubMed]
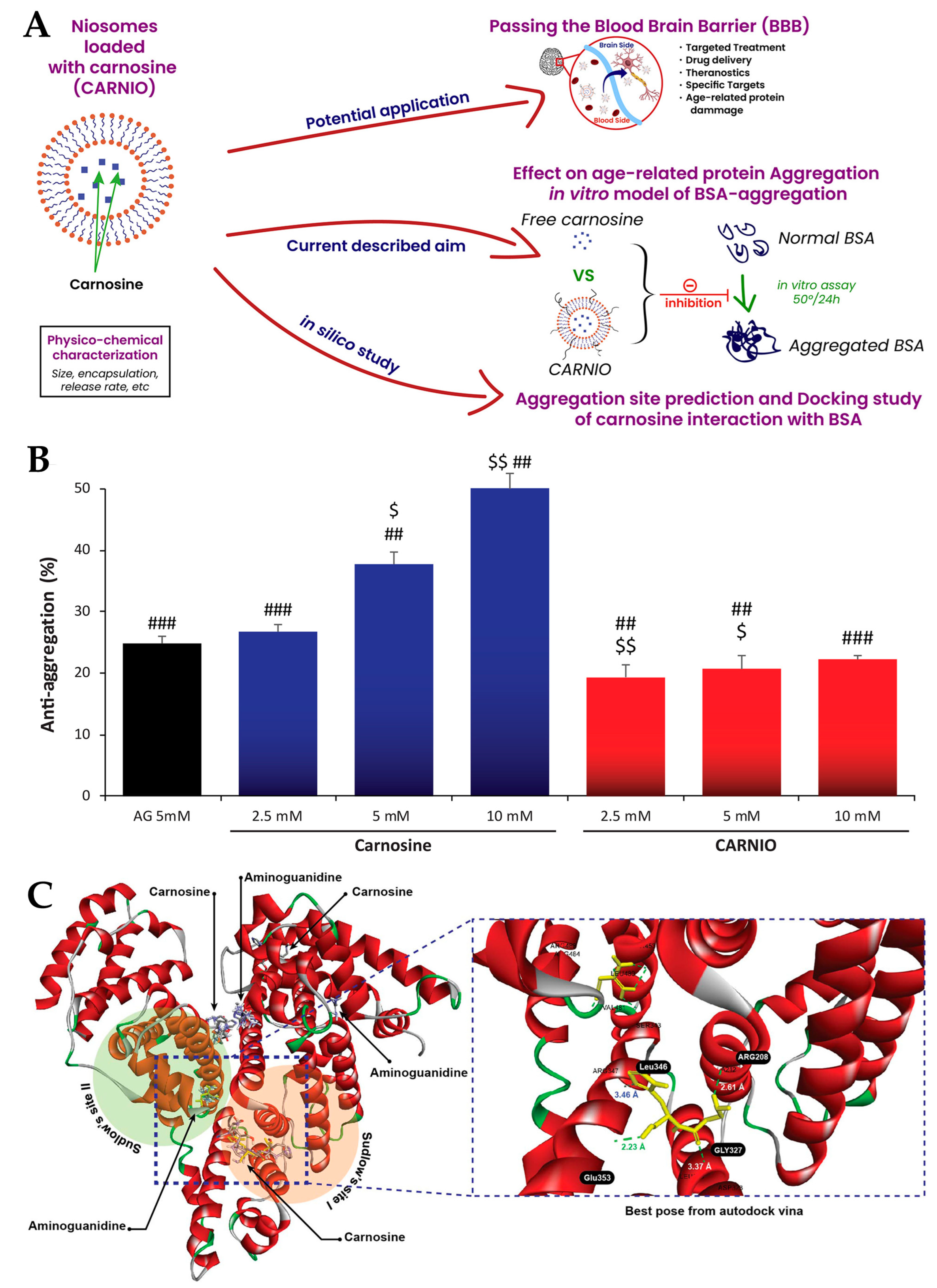
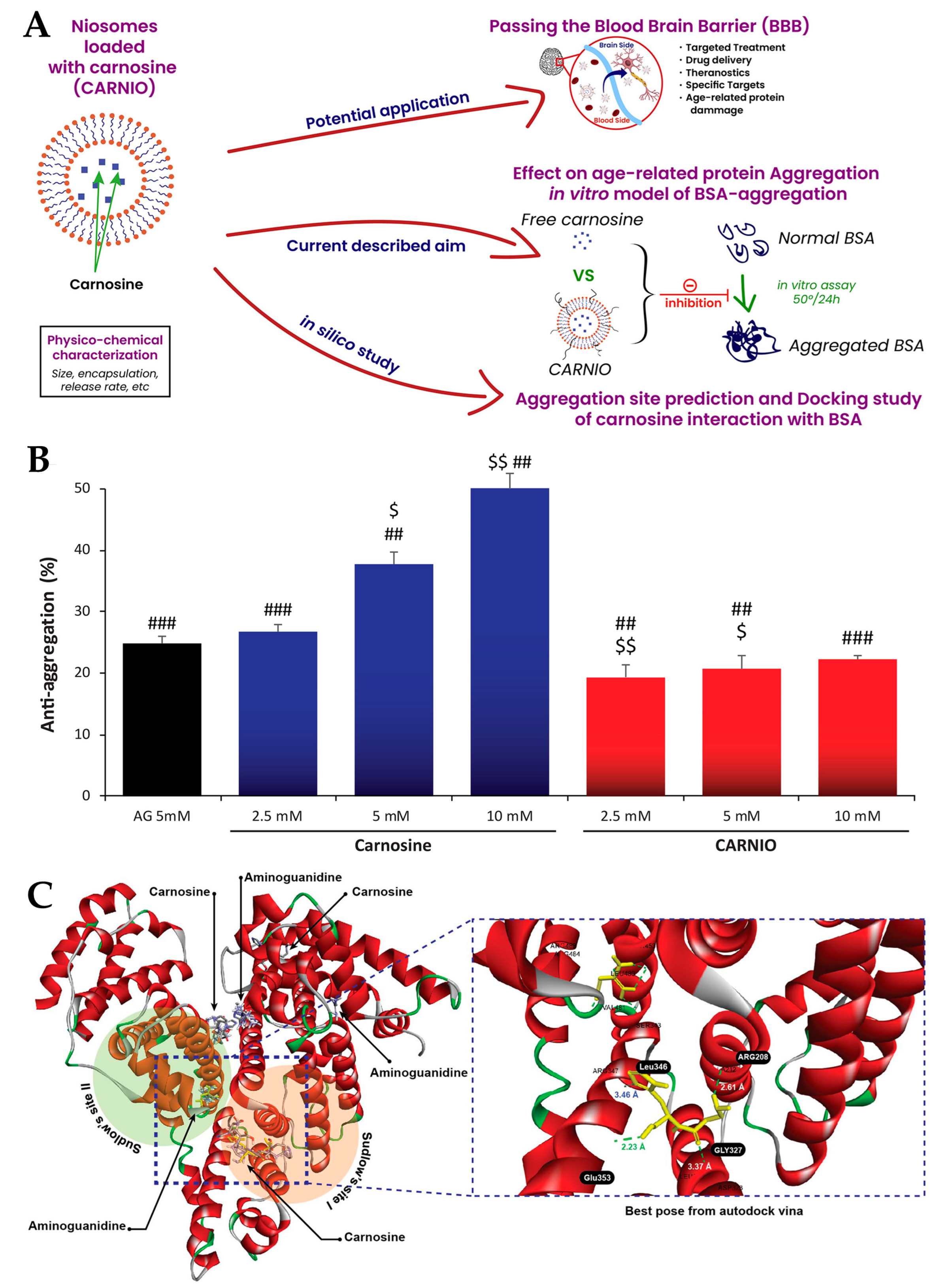
- Moulahoum, H.; Sanli, S.; Timur, S.; Zihnioglu, F. Potential Effect of Carnosine Encapsulated Niosomes in Bovine Serum Albumin Modifications. Int. J. Biol. Macromol. 2019, 137, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, X.; Zhao, Z.; Wang, Z.; Li, S.; Chen, C.; Yu, S.; Qu, X.; Li, K.; Tian, Y.; et al. Preparation of a Novel Ginkgolide B Niosomal Composite Drug. Open Chem. 2020, 18, 1064–1074. [Google Scholar] [CrossRef]
- Mathure, D.; Madan, J.R.; Gujar, K.N.; Tupsamundre, A.; Ranpise, H.A.; Dua, K. Formulation and Evaluation of Niosomal In-Situ Nasal Gel of Buspirone Hydrochloride for Brain Delivery. Pharm. Nanotechnol. 2018, 6, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, S.; Bai, Y.; Zhao, Z.; Bai, Y.; Gong, G.; He, X.; Zheng, X. Design and Synthesis of Ligustrazine Derivatives as Potential Anti-Alzheimer’s Agents. Nat. Prod. Res. 2023, 1–11. [Google Scholar] [CrossRef]
- Li, Z.; Meng, X.; Ma, G.; Liu, W.; Li, W.; Cai, Q.; Wang, S.; Huang, G.; Zhang, Y. Increasing Brain Glucose Metabolism by Ligustrazine Piperazine Ameliorates Cognitive Deficits through PPARγ-Dependent Enhancement of Mitophagy in APP/PS1 Mice. Alzheimer’s Res. Ther. 2022, 14, 150. [Google Scholar] [CrossRef]
- Deng, C.; Meng, Z.; Chen, H.; Meng, S. Tetramethylpyrazine Ameliorates Systemic Streptozotocin-Induced Alzheimer-like Pathology. J. Chem. Neuroanat. 2023, 127, 102207. [Google Scholar] [CrossRef]
- Alkilani, A.Z.; McCrudden, M.T.C.; Donnelly, R.F. Transdermal Drug Delivery: Innovative Pharmaceutical Developments Based on Disruption of the Barrier Properties of the Stratum Corneum. Pharmaceutics 2015, 7, 438–470. [Google Scholar] [CrossRef]
- Ramadon, D.; McCrudden, M.T.C.; Courtenay, A.J.; Donnelly, R.F. Enhancement Strategies for Transdermal Drug Delivery Systems: Current Trends and Applications. Drug Deliv. Transl. Res. 2022, 12, 758–791. [Google Scholar] [CrossRef]
- Jeong, W.Y.; Kwon, M.; Choi, H.E.; Kim, K.S. Recent Advances in Transdermal Drug Delivery Systems: A Review. Biomater. Res. 2021, 25, 24. [Google Scholar] [CrossRef]
- Naik, A.; Kalia, Y.N.; Guy, R.H. Transdermal Drug Delivery: Overcoming the Skin’s Barrier Function. Pharm. Sci. Technol. Today 2000, 3, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Bird, D.; Ravindra, N.M. Transdermal Drug Delivery and Patches—An Overview. Med. Devices Sens. 2020, 3, e10069. [Google Scholar] [CrossRef]
- Peña-Juárez, M.C.; Guadarrama-Escobar, O.R.; Escobar-Chávez, J.J. Transdermal Delivery Systems for Biomolecules. J. Pharm. Innov. 2022, 17, 319–332. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; El Rayess, Y.; Rizk, A.A.; Sadaka, C.; Zgheib, R.; Zam, W.; Sestito, S.; Rapposelli, S.; Neffe-Skocińska, K.; Zielińska, D.; et al. Turmeric and Its Major Compound Curcumin on Health: Bioactive Effects and Safety Profiles for Food, Pharmaceutical, Biotechnological and Medicinal Applications. Front. Pharmacol. 2020, 11, 1021. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.; He, L.; Liu, L.; Cheng, B.; Zhou, F.; Cao, D.; He, Y. A Comprehensive Review on the Benefits and Problems of Curcumin with Respect to Human Health. Molecules 2022, 27, 4400. [Google Scholar] [CrossRef] [PubMed]
- Razavi, B.M.; Ghasemzadeh Rahbardar, M.; Hosseinzadeh, H. A Review of Therapeutic Potentials of Turmeric (Curcuma Longa) and Its Active Constituent, Curcumin, on Inflammatory Disorders, Pain, and Their Related Patents. Phytother. Res. 2021, 35, 6489–6513. [Google Scholar] [CrossRef]
- Wei, W.; Yao, J.; Zhang, T.; Wen, J.; Zhang, Z.; Luo, Y.; Cao, Y.; Li, H. Network Pharmacology Reveals That Berberine May Function against Alzheimer’s Disease via the AKT Signaling Pathway. Front. Neurosci. 2023, 17, 1059496. [Google Scholar] [CrossRef]
- Senol Deniz, F.S.; Salmas, R.E.; Emerce, E.; Sener, B.; Erdogan Orhan, I. Cholinesterase Inhibitory and In Silico Toxicity Assessment of Thirty-Four Isoquinoline Alkaloids—Berberine as the Lead Compound. CNS Neurol. Disord. Drug Targets 2023, 22, 1. [Google Scholar] [CrossRef]
- Zhang, R.; Lei, B.; Wu, G.; Wang, Y.; Huang, Q. Protective Effects of Berberine against β-Amyloid-Induced Neurotoxicity in HT22 Cells via the Nrf2/HO-1 Pathway. Bioorg Chem. 2023, 133, 106210. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of Intranasal Drug Delivery Directly to the Brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Keller, L.-A.; Merkel, O.; Popp, A. Intranasal Drug Delivery: Opportunities and Toxicologic Challenges during Drug Development. Drug Deliv. Transl. Res. 2022, 12, 735–757. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.R.; Shekara, H.H.; Sahoo, A.K.; Harsha Vardhan, P.V.; Thiruppathi, N.; Venkatesh, M.P. Intranasal Nanoparticulate Delivery Systems for Neurodegenerative Disorders: A Review. Ther. Deliv. 2023. [Google Scholar] [CrossRef]
- Fonseca, L.C.; Lopes, J.A.; Vieira, J.; Viegas, C.; Oliveira, C.S.; Hartmann, R.P.; Fonte, P. Intranasal Drug Delivery for Treatment of Alzheimer’s Disease. Drug Deliv. Transl. Res. 2021, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Zha, S.; Wong, K.; All, A.H. Intranasal Delivery of Functionalized Polymeric Nanomaterials to the Brain. Adv. Healthc. Mater. 2022, 11, 2102610. [Google Scholar] [CrossRef]
- Patel, D.; Patel, B.; Wairkar, S. Intranasal Delivery of Biotechnology-Based Therapeutics. Drug Discov. Today 2022, 27, 103371. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Kellar, D.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2021, 8, 240–248. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-Acting Intranasal Insulin Detemir Improves Cognition for Adults with Mild Cognitive Impairment or Early-Stage Alzheimer’s Disease Dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef]
- Stocchi, F.; Fossati, C.; Torti, M. Rasagiline for the Treatment of Parkinson’s Disease: An Update. Expert Opin. Pharmacother. 2015, 16, 2231–2241. [Google Scholar] [CrossRef]
- Chen, J.; Swope, D.; DASHTIPOUR, K. Comprehensive Review of Rasagiline, a Second-Generation Monoamine Oxidase Inhibitor, for the Treatment of Parkinson’s Disease. Clin. Ther. 2007, 29, 1825–1849. [Google Scholar] [CrossRef]
- Mínguez-Mínguez, S.; Solís-García del Pozo, J.; Jordán, J. Rasagiline in Parkinson’s Disease: A Review Based on Meta-Analysis of Clinical Data. Pharmacol. Res. 2013, 74, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Taymouri, S.; Shahnamnia, S.; Mesripour, A.; Varshosaz, J. In Vitro and in Vivo Evaluation of an Ionic Sensitive in Situ Gel Containing Nanotransfersomes for Aripiprazole Nasal Delivery. Pharm. Dev. Technol. 2021, 26, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Pae, C.-U. A Review of the Safety and Tolerability of Aripiprazole. Expert Opin. Drug Saf. 2009, 8, 373–386. [Google Scholar] [CrossRef] [PubMed]
- DeLeon, A.; Patel, N.C.; Lynn Crismon, M. Aripiprazole: A Comprehensive Review of Its Pharmacology, Clinical Efficacy, and Tolerability. Clin. Ther. 2004, 26, 649–666. [Google Scholar] [CrossRef]
- Kinghorn, W.A.; McEvoy, J.P. Aripiprazole: Pharmacology, Efficacy, Safety and Tolerability. Expert Rev. Neurother. 2005, 5, 297–307. [Google Scholar] [CrossRef]
- Musselman, M.; Faden, J.; Citrome, L. Asenapine: An Atypical Antipsychotic with Atypical Formulations. Ther. Adv. Psychopharmacol. 2021, 11, 20451253211035269. [Google Scholar] [CrossRef]
- Weber, J.; McCormack, P.L. Asenapine. CNS Drugs 2009, 23, 781–792. [Google Scholar] [CrossRef]
- Plosker, G.L.; Deeks, E.D. Asenapine: A Review in Schizophrenia. CNS Drugs 2016, 30, 655–666. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral Delivery of Proteins and Peptides: Challenges, Status Quo and Future Perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Verma, S.; Goand, U.K.; Husain, A.; Katekar, R.A.; Garg, R.; Gayen, J.R. Challenges of Peptide and Protein Drug Delivery by Oral Route: Current Strategies to Improve the Bioavailability. Drug Dev. Res. 2021, 82, 927–944. [Google Scholar] [CrossRef]
- Bianchera, A.; Bettini, R. Polysaccharide Nanoparticles for Oral Controlled Drug Delivery: The Role of Drug–Polymer and Interpolymer Interactions. Expert Opin. Drug Deliv. 2020, 17, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Moodley, K.; Pillay, V.; Choonara, Y.E.; du Toit, L.C.; Ndesendo, V.M.K.; Kumar, P.; Cooppan, S.; Bawa, P. Oral Drug Delivery Systems Comprising Altered Geometric Configurations for Controlled Drug Delivery. Int. J. Mol. Sci. 2011, 13, 18–43. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Islam, T.; Nurunnabi, M. Mucoadhesive Carriers for Oral Drug Delivery. J. Control Release 2022, 351, 504–559. [Google Scholar] [CrossRef] [PubMed]
- Brookes, A.; Ji, L.; Bradshaw, T.D.; Stocks, M.; Gray, D.; Butler, J.; Gershkovich, P. Is Oral Lipid-Based Delivery for Drug Targeting to the Brain Feasible? Eur. J. Pharm. Biopharm. 2022, 172, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Komossa, K.; Rummel-Kluge, C.; Hunger, H.; Schmid, F.; Schwarz, S.; Duggan, L.; Kissling, W.; Leucht, S. Olanzapine versus Other Atypical Antipsychotics for Schizophrenia. Cochrane Database Syst. Rev. 2010, 3, CD006654. [Google Scholar] [CrossRef]
- Lund, B.C.; Perry, P.J. Olanzapine: An Atypical Antipsychotic for Schizophrenia. Expert Opin. Pharmacother. 2000, 1, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Bhana, N.; Foster, R.H.; Olney, R.; Plosker, G.L. Olanzapine. Drugs 2001, 61, 111–161. [Google Scholar] [CrossRef]
- Nguyen, K.; Hoffman, H.; Chakkamparambil, B.; Grossberg, G.T. Evaluation of Rivastigmine in Alzheimer’s Disease. Neurodegener. Dis. Manag. 2021, 11, 35–48. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The Advantages of Dual Inhibition of Acetylcholinesterase and Butyrylcholinesterase and Its Role in Subcortical Vascular Dementia and Parkinson’s Disease Dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef]
- Birks, J.S.; Grimley Evans, J. Rivastigmine for Alzheimer’s Disease. In Cochrane Database of Systematic Reviews; Birks, J.S., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2015. [Google Scholar]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef]
- More, J.; Galusso, N.; Veloso, P.; Montecinos, L.; Finkelstein, J.P.; Sanchez, G.; Bull, R.; Valdés, J.L.; Hidalgo, C.; Paula-Lima, A. N-Acetylcysteine Prevents the Spatial Memory Deficits and the Redox-Dependent RyR2 Decrease Displayed by an Alzheimer’s Disease Rat Model. Front. Aging Neurosci. 2018, 10, 399. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, S.; Zargooshnia, S.; Asl, S.S.; Komaki, A.; Sarihi, A. Influence of N -Acetyl Cysteine on Beta-Amyloid-Induced Alzheimer’s Disease in a Rat Model: A Behavioral and Electrophysiological Study. Brain Res. Bull. 2017, 131, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Di Pietro, L.; Cardaci, V.; Maugeri, S.; Caraci, F. The Therapeutic Potential of Carnosine: Focus on Cellular and Molecular Mechanisms. Curr. Res. Pharmacol. Drug Discov. 2023, 4, 100153. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.; Mousa, A.; Berk, M.; Chia, W.L.; Ukropec, J.; Majid, A.; Ukropcová, B.; de Courten, B. The Potential of Carnosine in Brain-Related Disorders: A Comprehensive Review of Current Evidence. Nutrients 2019, 11, 1196. [Google Scholar] [CrossRef]
- Dai, Z.; Lu, X.-Y.; Zhu, W.-L.; Liu, X.-Q.; Li, B.-Y.; Song, L.; Liu, H.-F.; Cai, W.-W.; Deng, Y.-X.; Xu, T.-T.; et al. Carnosine Ameliorates Age-Related Dementia via Improving Mitochondrial Dysfunction in SAMP8 Mice. Food Funct. 2020, 11, 2489–2497. [Google Scholar] [CrossRef]
- Distefano, A.; Caruso, G.; Oliveri, V.; Bellia, F.; Sbardella, D.; Zingale, G.A.; Caraci, F.; Grasso, G. Neuroprotective Effect of Carnosine Is Mediated by Insulin-Degrading Enzyme. ACS Chem. Neurosci. 2022, 13, 1588–1593. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B Inactivates the NLRP3 Inflammasome by Promoting Autophagic Degradation to Improve Learning and Memory Impairment in Alzheimer’s Disease. Metab. Brain Dis. 2022, 37, 329–341. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B Protects against Cognitive Impairment in Senescence-Accelerated P8 Mice by Mitigating Oxidative Stress, Inflammation and Ferroptosis. Biochem. Biophys. Res. Commun. 2021, 572, 7–14. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, C.; Li, J.; Hou, Q.; Li, J.; Ma, J.; Wang, W.; Wang, Z. Ginkgolide B Protects Hippocampal Neurons from Apoptosis Induced by Beta-Amyloid 25–35 Partly via up-Regulation of Brain-Derived Neurotrophic Factor. Eur. J. Pharmacol. 2010, 647, 48–54. [Google Scholar] [CrossRef]
- Miao, Q.; Chai, Z.; Song, L.-J.; Wang, Q.; Song, G.-B.; Wang, J.; Yu, J.-Z.; Xiao, B.-G.; Ma, C.-G. The Neuroprotective Effects and Transdifferentiation of Astrocytes into Dopaminergic Neurons of Ginkgolide K on Parkinson’ Disease Mice. J. Neuroimmunol. 2022, 364, 577806. [Google Scholar] [CrossRef]
- Dey, A.; Nath De, J. Possible Anti-Parkinson’s Disease Therapeutics from Nature: A Review. Stud. Nat. Prod. Chem. 2015, 44, 447–520. [Google Scholar]
- Hinderliter, P.; Saghir, S.A. Pharmacokinetics. In Encyclopedia of Toxicology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 849–855. [Google Scholar]
- Maddison, J.E.; Page, S.W.; Dyke, T.M. Clinical Pharmacokinetics. In Small Animal Clinical Pharmacology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 27–40. [Google Scholar]
- Bolger, G.T. Routes of Drug Administration. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Rech, M.A.; Gottlieb, M. Intravenous Push Antibiotics Should Be Administered in the Emergency Department. Ann. Emerg. Med. 2021, 78, 384–385. [Google Scholar] [CrossRef]
- Garakani, A.; Murrough, J.W.; Freire, R.C.; Thom, R.P.; Larkin, K.; Buono, F.D.; Iosifescu, D.V. Pharmacotherapy of Anxiety Disorders: Current and Emerging Treatment Options. Front. Psychiatry 2020, 11, 1412. [Google Scholar] [CrossRef]
- Loane, C.; Politis, M. Buspirone: What Is It All About? Brain Res. 2012, 1461, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Strawn, J.R.; Mills, J.A.; Cornwall, G.J.; Mossman, S.A.; Varney, S.T.; Keeshin, B.R.; Croarkin, P.E. Buspirone in Children and Adolescents with Anxiety: A Review and Bayesian Analysis of Abandoned Randomized Controlled Trials. J. Child. Adolesc. Psychopharmacol. 2018, 28, 2–9. [Google Scholar] [CrossRef]
- Agrawal, M.; Ajazuddin; Tripathi, D.K.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Mourtas, S.; Hammarlund-Udenaes, M.; Alexander, A. Recent Advancements in Liposomes Targeting Strategies to Cross Blood-Brain Barrier (BBB) for the Treatment of Alzheimer’s Disease. J. Control. Release 2017, 260, 61–77. [Google Scholar] [CrossRef]
- Spuch, C.; Navarro, C. Liposomes for Targeted Delivery of Active Agents against Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). J. Drug Deliv. 2011, 2011, 469679. [Google Scholar] [CrossRef]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-Targeted Liposome Design: Challenges and Fundamental Considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanosystem Type | Main Composition | Encapsulated Molecule | Disease Intended to Treat | Intended Administration Route | Particle Size (nm) | PDI | ZP (mV) | EE (%) | pH | Viscosity (cP) | Main PK and/or PD Results | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethosomes | Ethanol, egg phosphatidylcholine, and carbomer gel matrix | Ligustrazine | Alzheimer’s disease | Transdermal | 146.3 ± 24.6 | 0.034 ± 0.009 | NR | 70.23 ± 1.20 | 5.9 | NR | Completely reversed memory deficits and decreased escape latency (rats). | [106] |
| Transfersomes | L-α phosphatidylcholine and sodium deoxycholate | Curcumin and berberine | Alzheimer’s disease | Intranasal | 130 to 170 | 0.054 to 0.120 | −7 to −32 | 65 to 68 | NR | NR | Higher brain Cmax, AUC, and MRT than non-encapsulated drugs, with synergy in dual loading and improved spatial memory and locomotor activity (mice). | [107] |
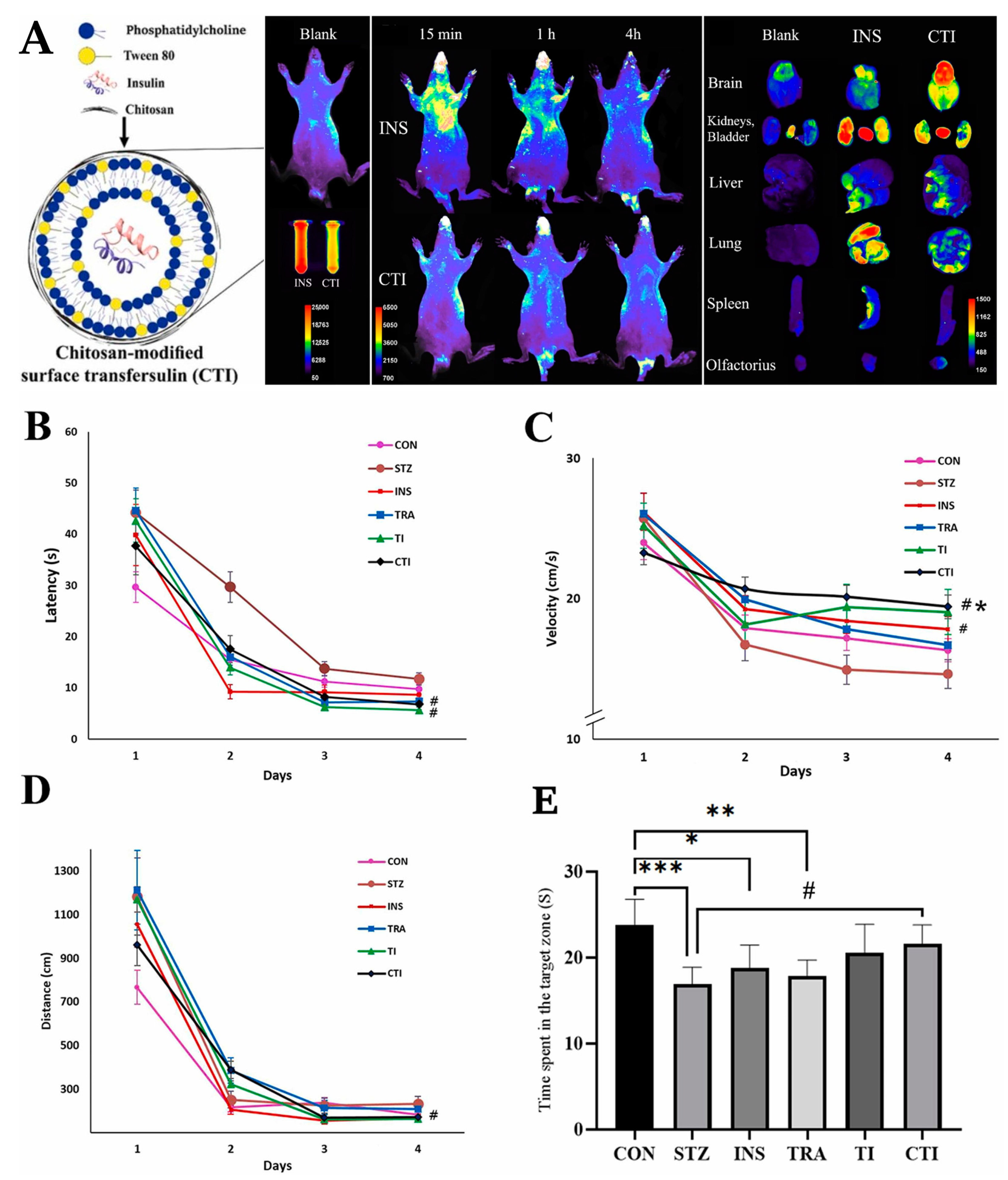
| Transfersomes | Soy lecithin, Tween 80, and chitosan | Insulin | Alzheimer’s disease | Intranasal | 137.9 ± 28.2 | 0.20 | +23.4 | 65.1 ± 0.9 | NR | NR | Higher brain drug targeting and retention compared to controls, and substantial improvement in movement, learning, and memory performance (rats). | [108] |
| Transfersomes | Phosphatidylcholine, sodium deoxycholate, pectin, Pluronic® F-127, and Pluronic® F-68 | Rasagiline | Parkinson’s disease | Intranasal | 198.635 ± 34.98 | 0.45 ± 0.079 | −33.45 ± 4.73 | 95.735 ± 0.091 | NR | NR | Brain Cmax and AUC values significantly higher than controls (rats). | [109] |
| Transfersomes | Soybean lecithin, sodium deoxychola, and gellan gum | Aripiprazole | Schizophrenia and bipolar disorders (main therapy); major depressive disorders (adjuvant therapy) | Intranasal | 72.12 ± 0.72 | 0.19 ± 0.07 | −55.6 ± 1.9 | 97.06 ± 0.10 | NR | NR | Reduction in locomotor activity and immobility, swimming, and climbing times, higher than controls (mice). | [98] |
| Transfersomes | Soy phosphatidylcholine, sodium deoxycholate, ethanol, and Carbopol 934P | Asenapine | Schizophrenia and bipolar disorder | Transdermal | 126.0 | 0.232 | −43.7 | 54.96 | NR | NR | Higher Cmax, AUC, Tmax, t1/2, and MRT compared to controls (rats). | [110] |
| Niosomes | Cholesterol and Span 60 | Asenapine | Schizophrenia and bipolar disorder | Oral | 84 ± 5 | 0.27 | −17.53 | 70 | NR | NR | Higher Cmax, AUC, and t1/2 values than controls, and significantly improved locomotor activity (rats). | [111] |
| Niosomes | Cholesterol, Span 80 and chitosan | Olanzapine | Schizophrenia and related psychotic disorders | Intranasal | 250.1 ± 5.0 | NR | NR | 71.9 | NR | 8.4 ± 1.2 | Higher brain Cmax and AUC, t1/2, and MRT (rats). | [112] |
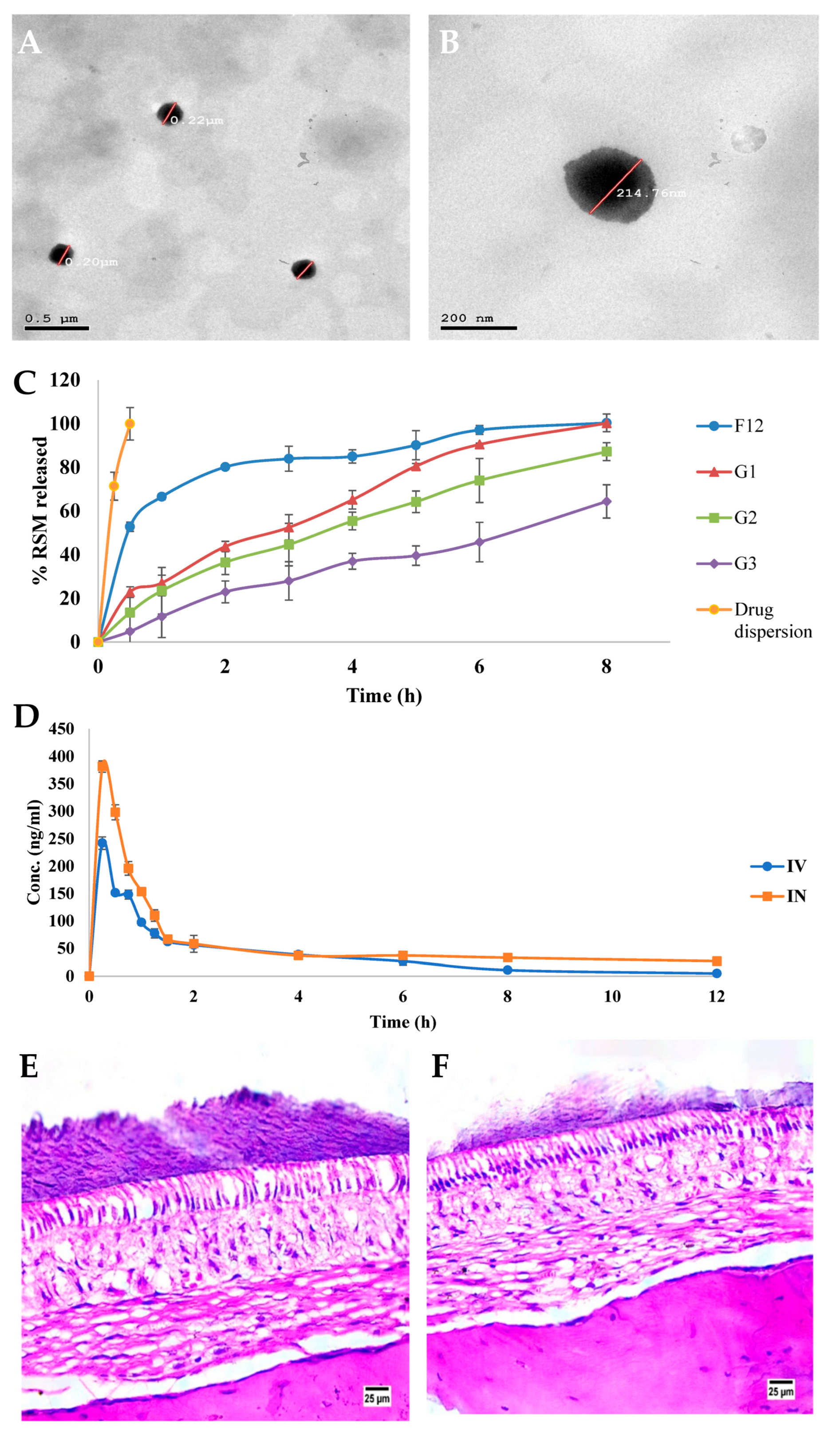
| Niosomes | Cholesterol and Span 20 | Rivastigmine and N-acetyl cysteine | Alzheimer’s disease | Intranasal or intravenous | 162.7 | <0.1 | −24.8 | 85.9 to 97.7 | NR | NR | Higher AUC, Cmax, t1/2, and MRT values (rats). | [113] |
| Niosomes | Cholesterol and Span 60 | Carnosine | Alzheimer’s disease | NR | 560 ± 203 | NR | NR | NR | NR | 32.4 ± 5 | NR | [114] |
| Niosomes | Cholesterol, polyoxyethylene monostearate, and borneol | Ginkgolide B and puerarin | Alzheimer’s disease and Parkinson’s disease | Intravenous | 142.65 | 0.261 | NR | 49.90 | NR | NR | Higher Cmax, AUC, t1/2, and MRT values (rats). | [115] |
| Niosomes | Buspirone | Cholesterol, Span 60, Carbopol 934P, hydroxypropyl methylcellulose K4M, and benzalkonium chloride | Anxiety disorders | Intranasal | 181.9 ± 0.36 | NR | −15.4 | 87.7 ± 0.66 | NR | 2600 ± 0.48 to 7800 ± 0.56 | NR | [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires, P.C.; Paiva-Santos, A.C.; Veiga, F. Liposome-Derived Nanosystems for the Treatment of Behavioral and Neurodegenerative Diseases: The Promise of Niosomes, Transfersomes, and Ethosomes for Increased Brain Drug Bioavailability. Pharmaceuticals 2023, 16, 1424. https://doi.org/10.3390/ph16101424
Pires PC, Paiva-Santos AC, Veiga F. Liposome-Derived Nanosystems for the Treatment of Behavioral and Neurodegenerative Diseases: The Promise of Niosomes, Transfersomes, and Ethosomes for Increased Brain Drug Bioavailability. Pharmaceuticals. 2023; 16(10):1424. https://doi.org/10.3390/ph16101424
Chicago/Turabian StylePires, Patrícia C., Ana Cláudia Paiva-Santos, and Francisco Veiga. 2023. "Liposome-Derived Nanosystems for the Treatment of Behavioral and Neurodegenerative Diseases: The Promise of Niosomes, Transfersomes, and Ethosomes for Increased Brain Drug Bioavailability" Pharmaceuticals 16, no. 10: 1424. https://doi.org/10.3390/ph16101424
APA StylePires, P. C., Paiva-Santos, A. C., & Veiga, F. (2023). Liposome-Derived Nanosystems for the Treatment of Behavioral and Neurodegenerative Diseases: The Promise of Niosomes, Transfersomes, and Ethosomes for Increased Brain Drug Bioavailability. Pharmaceuticals, 16(10), 1424. https://doi.org/10.3390/ph16101424