
Hierarchical Virtual Screening of Potential New Antibiotics from Polyoxygenated Dibenzofurans against Staphylococcus aureus Strains


 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
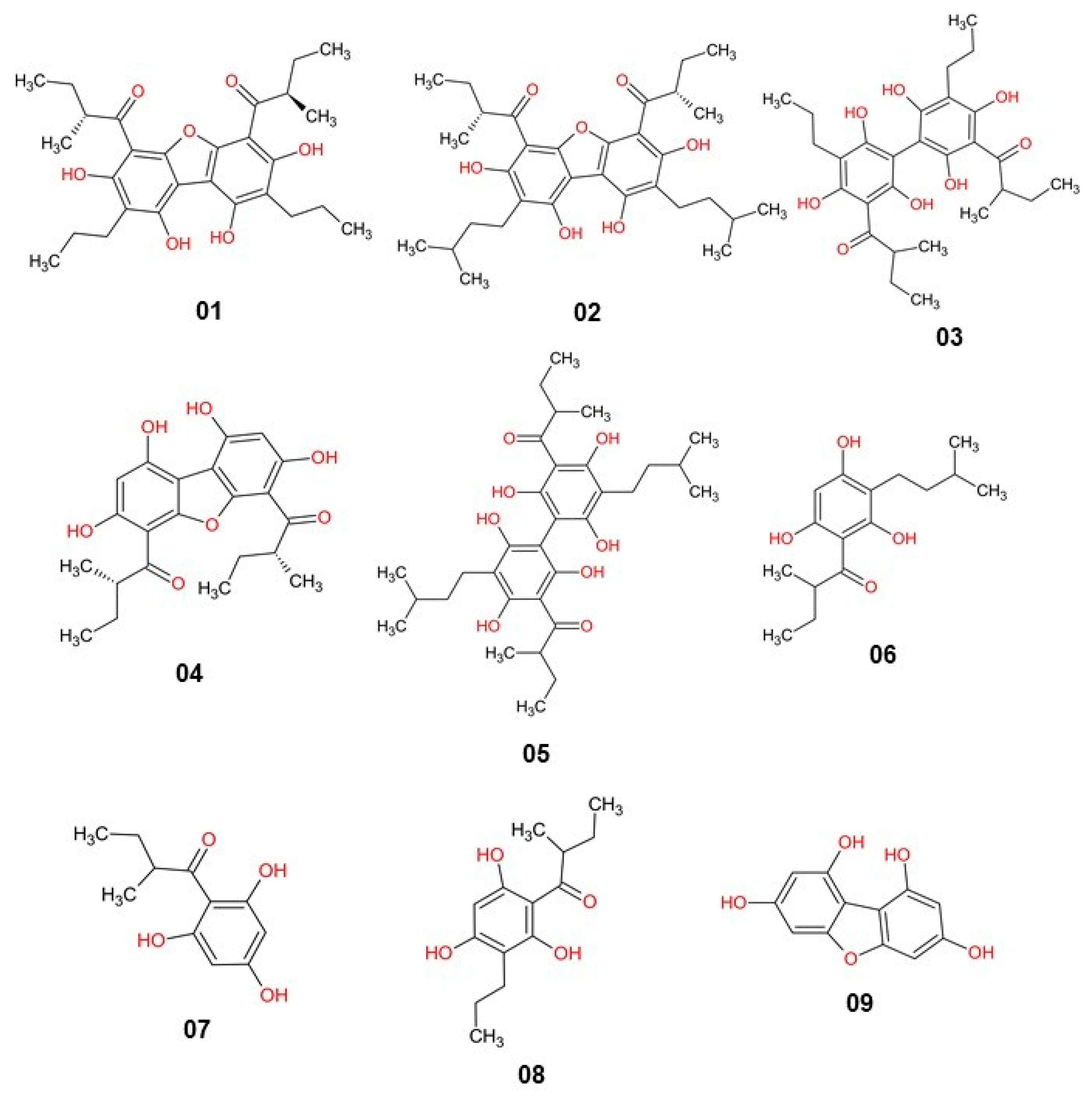
2. Results and Discussion
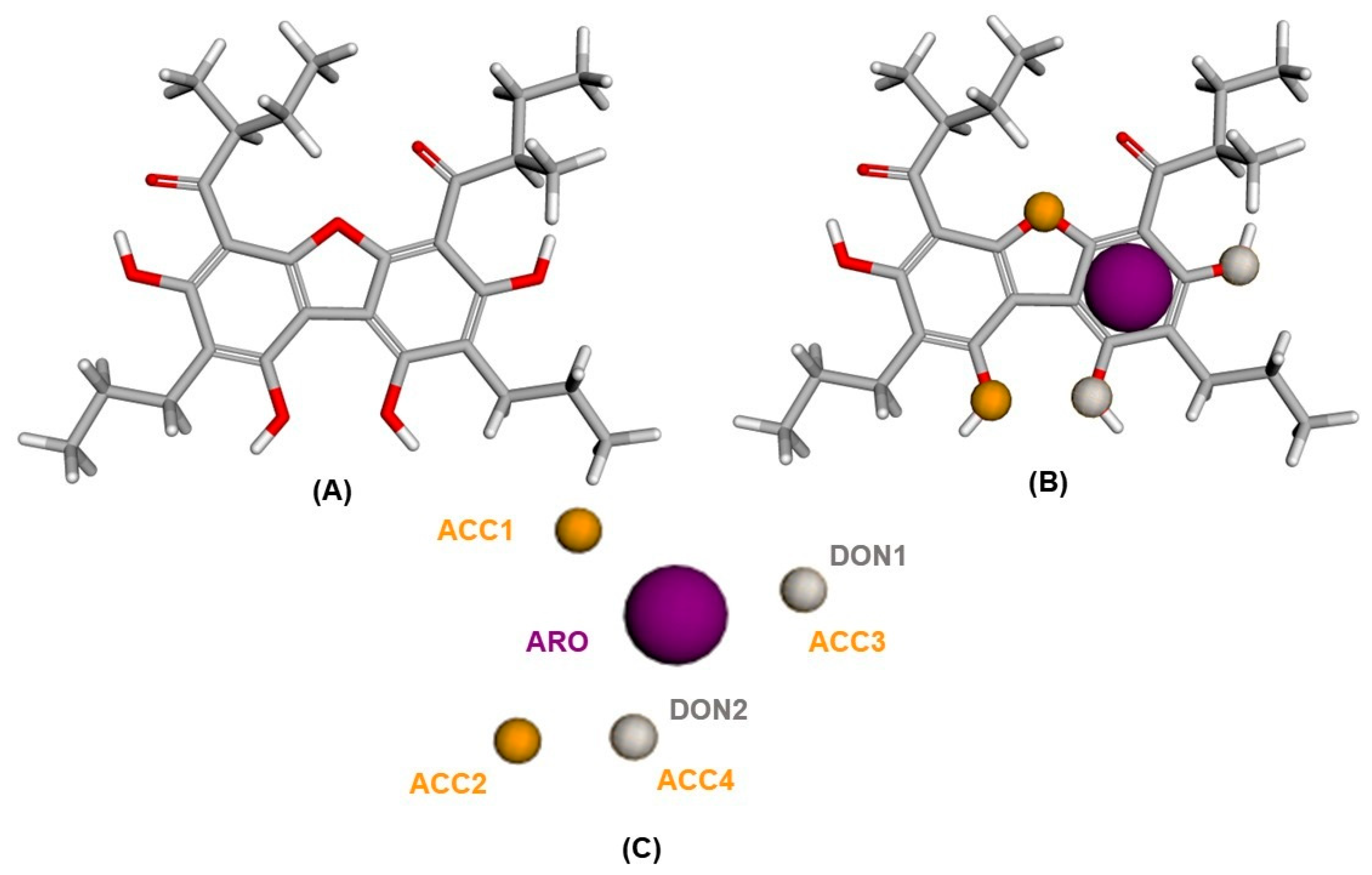
2.1. Pharmacophoric Model Generation
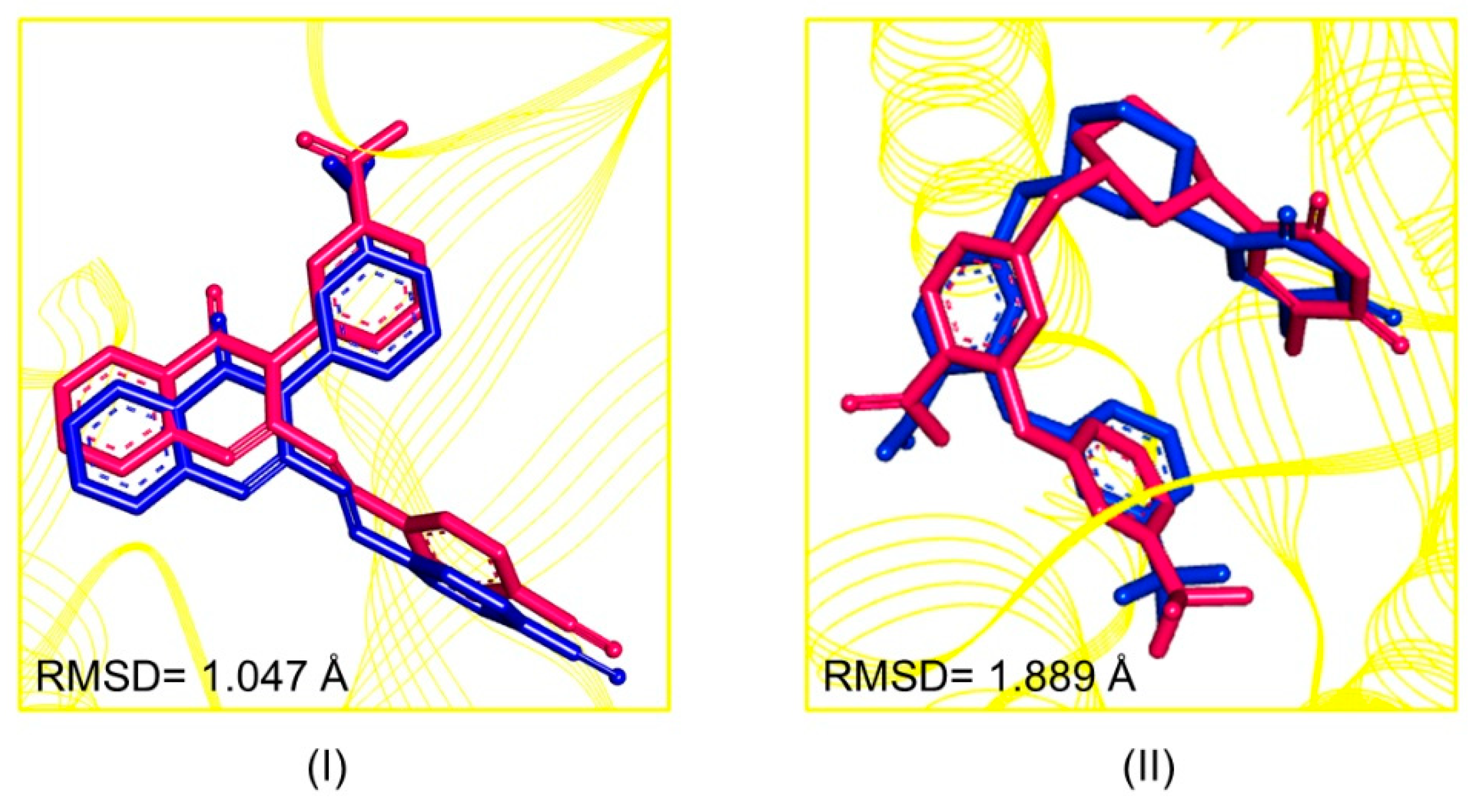
2.2. Pharmacophore-Based Virtual Screening
2.3. Similarity of Tanimoto
2.4. Predictions of Toxicological and Pharmacokinetic Properties of the New Hits
2.5. Biological Activity Prediction
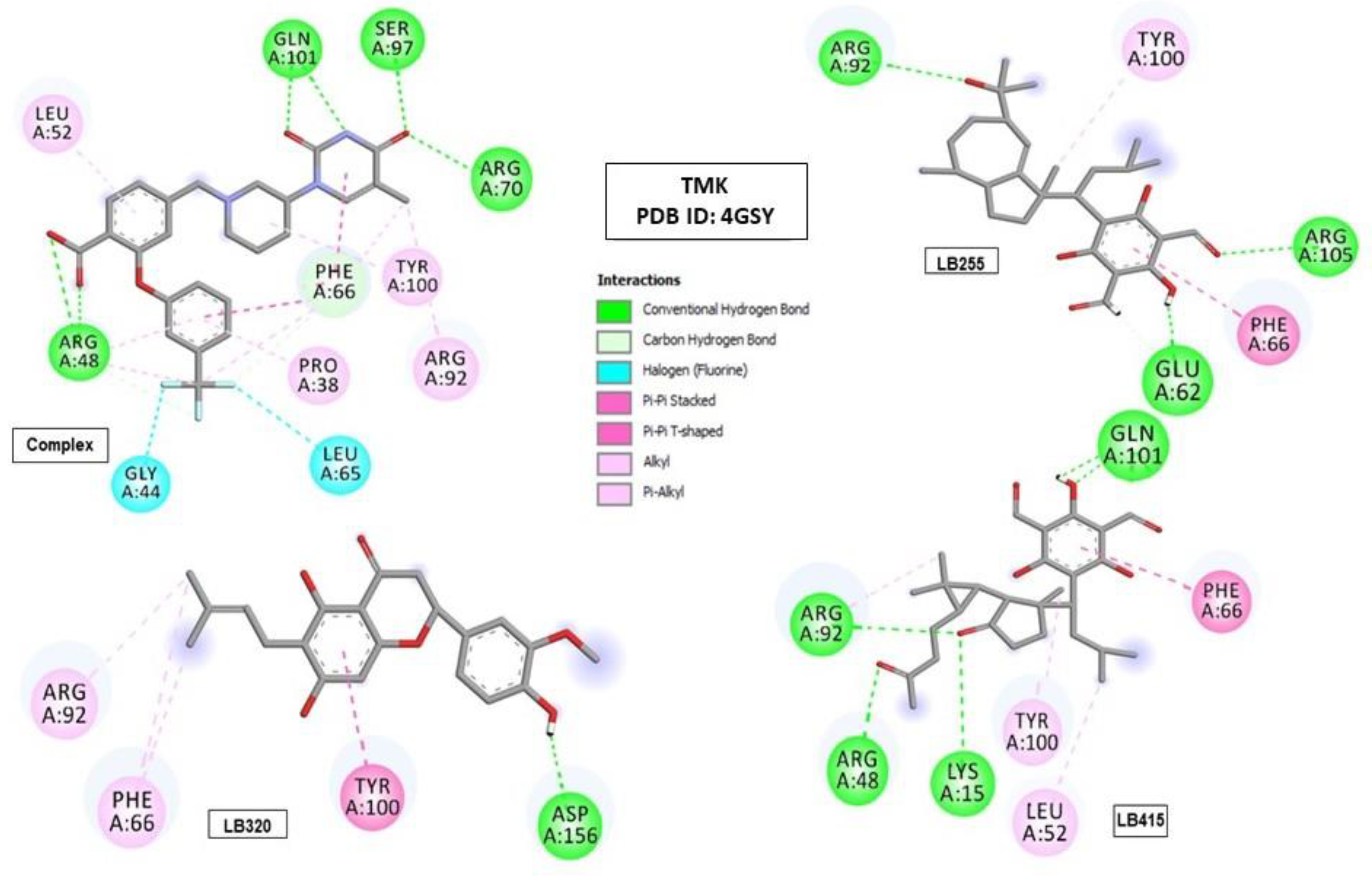
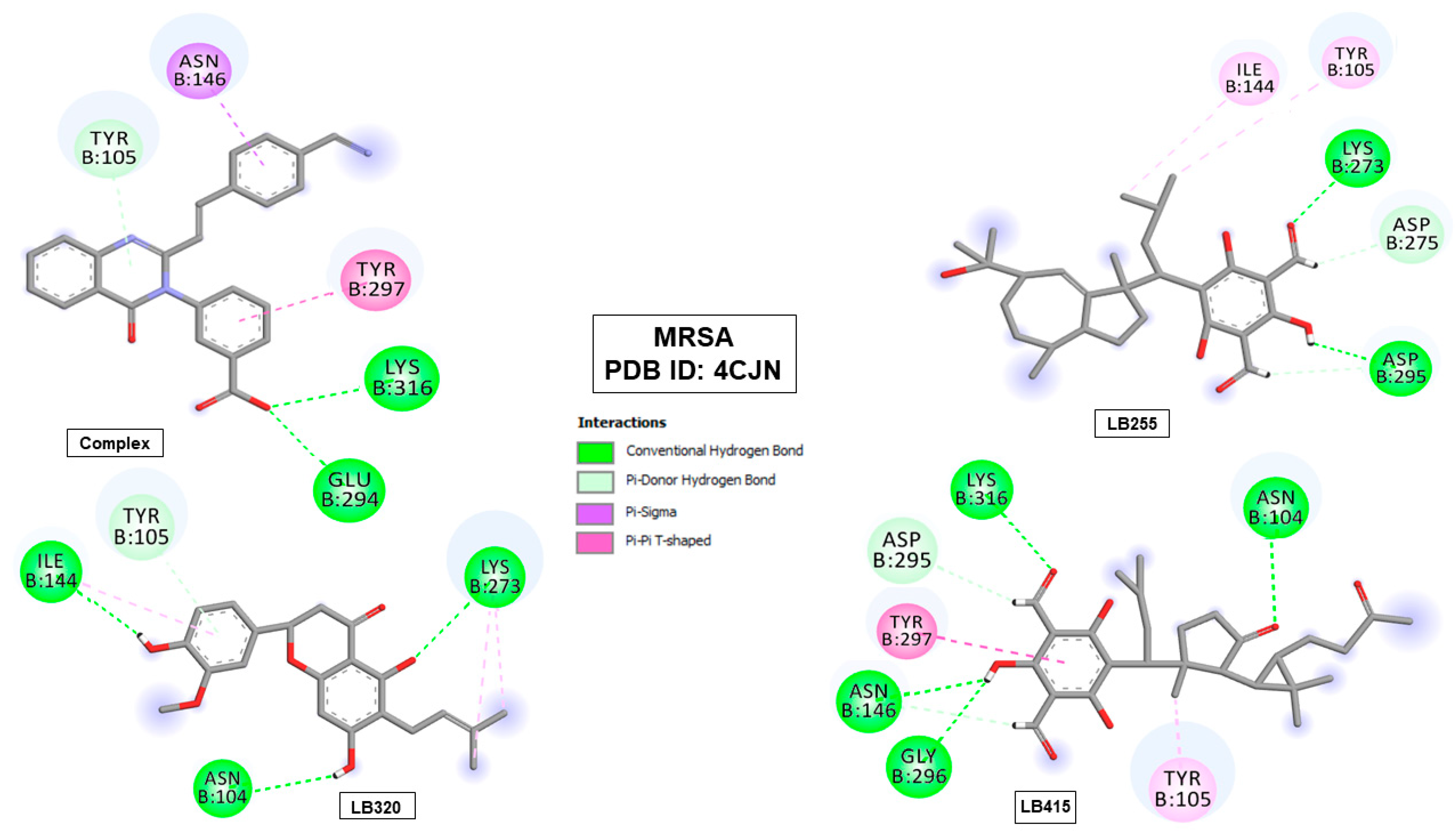
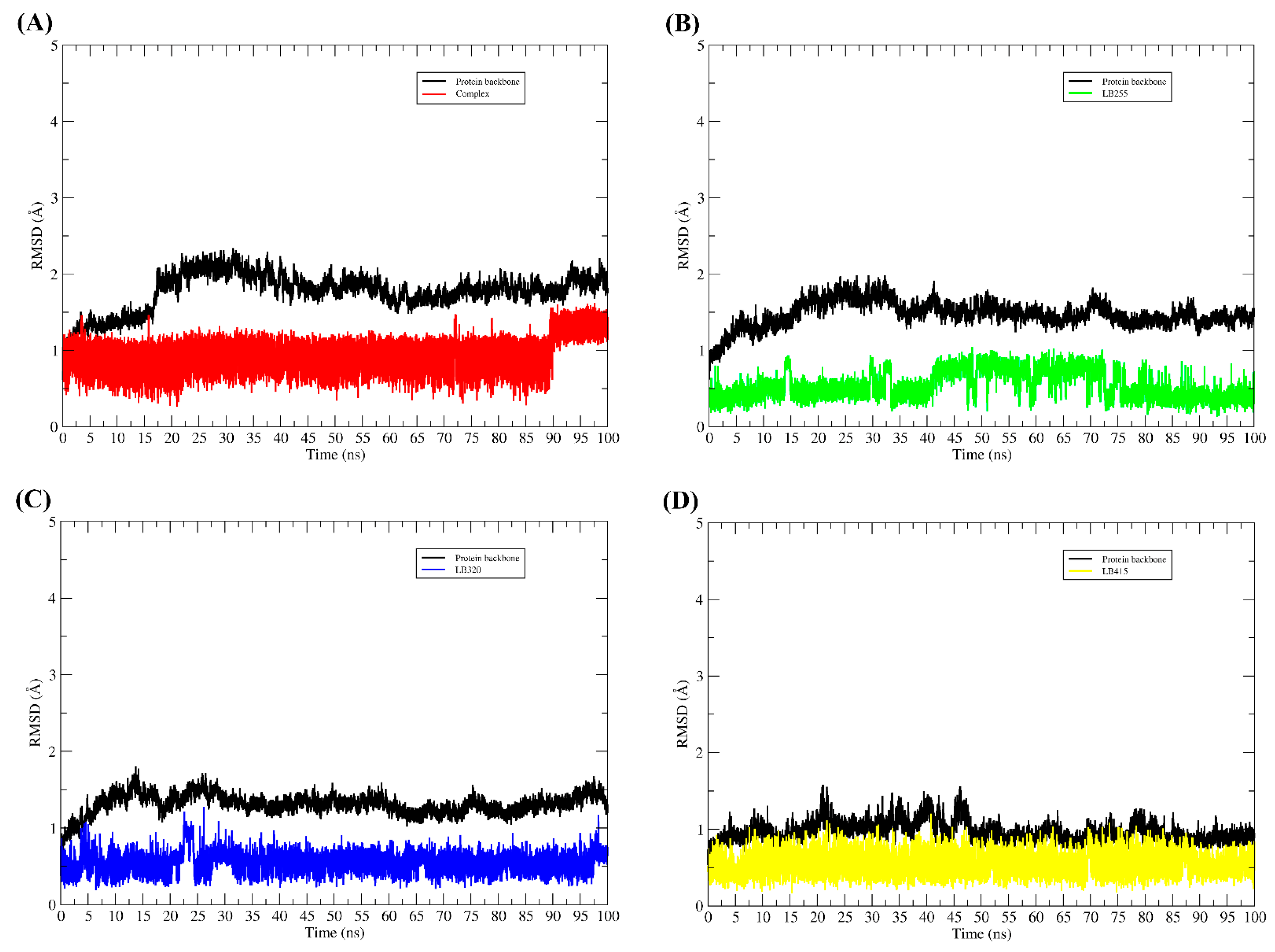
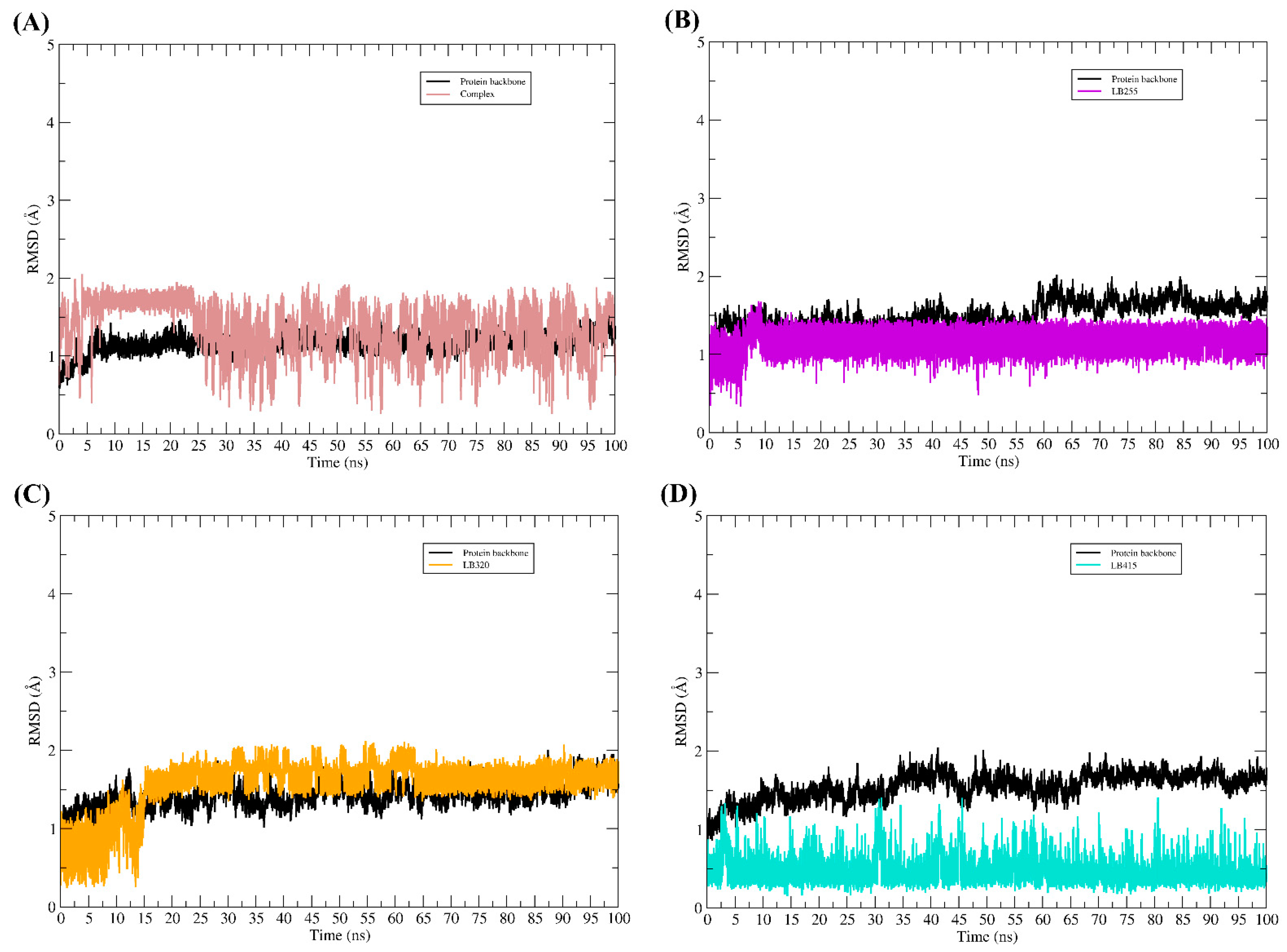
2.6. Molecular Binding Mode
2.7. Prediction of Synthetic Accessibility (SA)
2.8. Prediction of Lipophilicity and Water Solubility and Structure–Activity Relationship (SAR) of the Promising Molecule
3. Materials and Methods
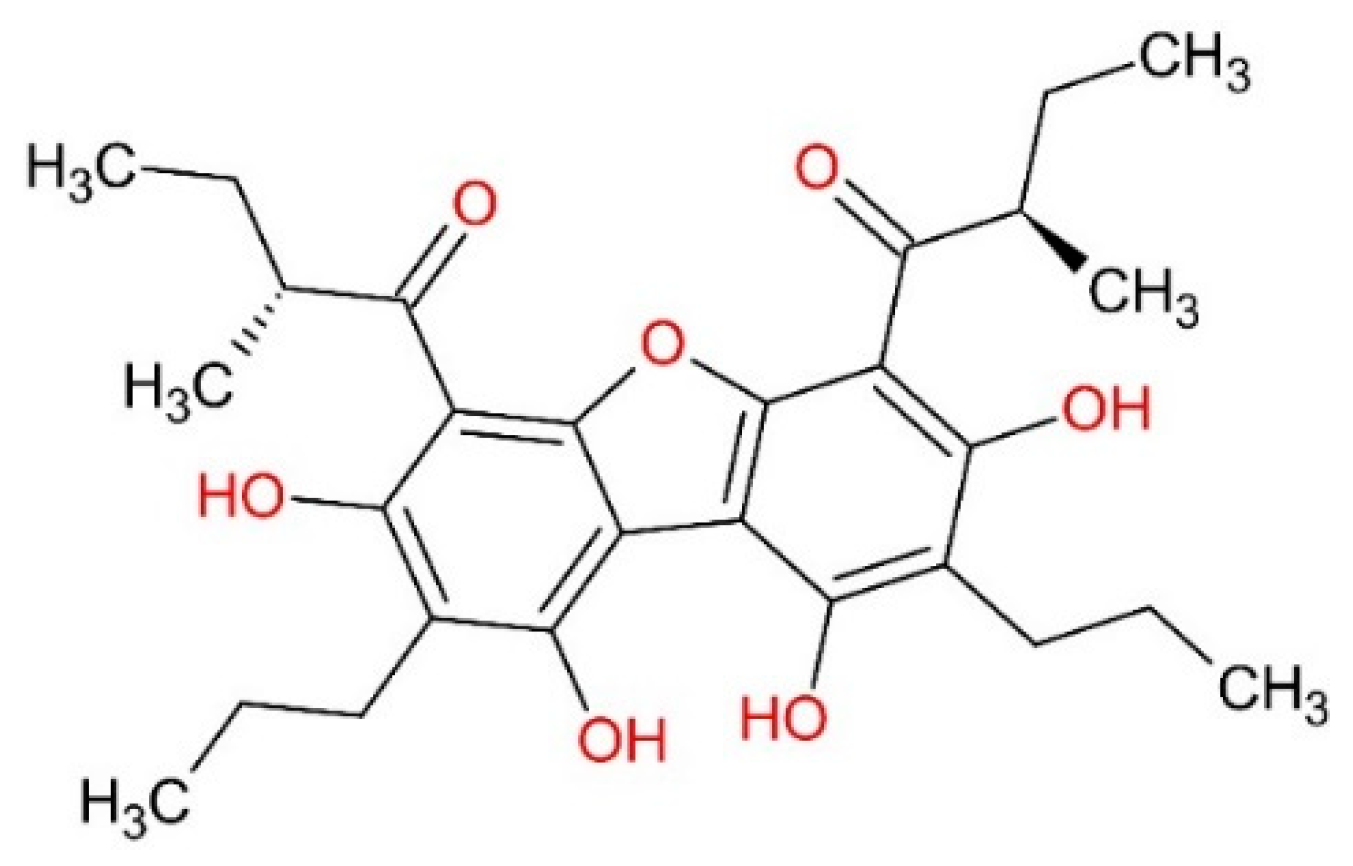
3.1. Selection of Molecules
3.2. Geometric Optimization of Selected Structures
3.3. Construction of the Pharmacophoric Model
3.4. Pharmacophore-Based Virtual Screening
3.5. Similarity of Tanimoto coefficient
3.6. Prediction of Pharmacokinetic and Toxicological Properties of the New Hits
3.7. Biological Activity Prediction of the New Hits
3.8. Molecular Docking
3.9. Molecular Dynamic Simulations
3.10. Free Energy Calculations
3.11. Synthetic Accessibility Prediction
3.12. Prediction of Lipophilicity and Water Solubility and SAR of the Promising Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Costa, M.D.S.; Rocha, J.E.; Campina, F.F.; Silva, A.R.P.; Da Cruz, R.P.; Pereira, R.L.S.; Quintans-Júnior, L.J.; De Menezes, I.R.; Adriano, A.D.S.; De Freitas, T.S.; et al. Comparative analysis of the antibacterial and drug-modulatory effect of D-limonene alone and complexed with β-cyclodextrin. Eur. J. Pharm. Sci. 2019, 128, 158–161. [Google Scholar] [CrossRef]
- Van Hoek, A.H.A.M.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J.M. Acquired Antibiotic Resistance Genes: An Overview. Front. Microbiol. 2011, 2, 203. [Google Scholar] [CrossRef]
- Casero, C.; Estévez-Braun, A.; Ravelo, Á.G.; Demo, M.; Méndez-Álvarez, S.; Machín, F. Achyrofuran is an antibacterial agent capable of killing methicillin-resistant vancomycin-intermediate Staphylococcus aureus in the nanomolar range. Phytomedicine 2013, 20, 133–138. [Google Scholar] [CrossRef]
- Casero, C.; Machín, F.; Méndez-Álvarez, S.; Demo, M.; Ravelo, Á.G.; Pérez-Hernández, N.; Joseph-Nathan, P.; Estévez-Braun, A. Structure and Antimicrobial Activity of Phloroglucinol Derivatives from Achyrocline satureioides. J. Nat. Prod. 2014, 78, 93–102. [Google Scholar] [CrossRef]
- de Almeida, R.B.M.; Barbosa, D.B.; do Bomfim, M.R.; Amparo, J.A.O.; Andrade, B.S.; Costa, S.L.; Campos, J.M.; Cruz, J.N.; Santos, C.B.; Leite, F.H.; et al. Identification of a Novel Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase: In Vitro and In Silico Studies. Pharmaceuticals 2023, 16, 95. [Google Scholar] [CrossRef]
- Almeida, V.M.; Dias, Ê.R.; Souza, B.C.; Cruz, J.N.; Santos, C.B.R.R.; Leite, F.H.A.A.; Queiroz, R.F.; Branco, A. Methoxylated flavonols from Vellozia dasypus Seub ethyl acetate active myeloperoxidase extract: In vitro and in silico assays. J. Biomol. Struct. Dyn. 2023, 40, 7574–7583. [Google Scholar] [CrossRef]
- Rego, C.M.A.; Francisco, A.F.; Boeno, C.N.; Paloschi, M.V.; Lopes, J.A.; Silva, M.D.S.; Santana, H.M.; Serrath, S.N.; Rodrigues, J.E.; Lemos, C.T.; et al. Inflammasome NLRP3 activation induced by Convulxin, a C-type lectin-like isolated from Crotalus durissus terrificus snake venom. Sci. Rep. 2022, 12, 4706. [Google Scholar] [CrossRef]
- Oramas-Royo, S.; Pantoja, K.D.; Amesty, Á.; Romero, C.; Lorenzo-Castrillejo, I.; Machín, F.; Estévez-Braun, A. Synthesis and antibacterial activity of new symmetric polyoxygenated dibenzofurans. Eur. J. Med. Chem. 2017, 141, 178–187. [Google Scholar] [CrossRef]
- Olmedo, D.A.; Durant-Archibold, A.A.; López-Pérez, J.L.; Medina-Franco, J.L. Design and Diversity analysis of chemical libraries in drug discovery. Comb. Chem. High Throughput Screen 2023, 26. [Google Scholar] [CrossRef]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging ligand-based and structure-based methods in drug discovery: An overview of combined virtual screening approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef]
- Willet, P. The calculation of molecular structural similarity: Principles and practice. Mol. Inform. 2014, 33, 403–413. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Padilha, E.C.; Serafim, R.B.; Sarmiento, D.Y.R.; Santos, C.F.; Santos, C.B.R.; Silva, C.H.T.P. New PPARα/γ/δ optimal activator rationally designed by computational methods. J. Braz. Chem. Soc. 2016, 27, 1636–1647. [Google Scholar] [CrossRef]
- Pereira, A.L.E.E.; dos Santos, G.B.; Franco, M.S.F.F.; Federico, L.B.; da Silva, C.H.T.P.T.P.; Santos, C.B.R.R. Molecular modeling and statistical analysis in the design of derivatives of human dipeptidyl peptidase IV. J. Biomol. Struct. Dyn. 2017, 36, 318–334. [Google Scholar] [CrossRef]
- Joshi, M.; Purohit, M.; Shah, D.P.; Patel, D.; Depani, P.; Moryani, P.; Krishnakumar, A. Pathogenomic in silico approach identifies NSP-A and Fe-IIISBP as possible drug targets in Neisseria meningitidis MC58 and development of pharmacophores as novel therapeutic candidates. Mol. Divers. 2023, 27, 1163–1184. [Google Scholar] [CrossRef]
- Kwon, S. Structural insight into the working mechanism of the FAD synthetase from the human pathogen Streptococcus pneumoniae: A molecular docking simulation study. Int. J. Mol. Sci. 2023, 24, 3121. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, 442–448. [Google Scholar] [CrossRef]
- Cruz, J.V.; Serafim, R.B.; da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Araújo Neto, M.F.; Leite, F.H.A.; Taft, C.A.; da Silva, C.H.T.P. Santos Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef]
- Verhoeckx, K.; Cotter, P.; López-Expósito, I.; Kleiveland, C.; Lea, T.; Mackie, A.; Requena, T.; Swiatecka, D.; Wichers, H. The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–327. [Google Scholar] [CrossRef]
- Ferreira, E.F.B.; Silva, L.B.; Costa, G.V.; Costa, J.S.; Fujishima, M.A.T.; Leão, R.P.; Ferreira, A.L.S.; Federico, L.B.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of new inhibitors with potential antitumor activity from polypeptide structures via hierarchical virtual screening. Molecules 2019, 24, 2943. [Google Scholar] [CrossRef]
- Lambrinidis, G.; Vallianatou, T.; Tsantili-Kakoulidou, A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. A review. Adv. Drug Deliv. Rev. 2015, 86, 27–45. [Google Scholar] [CrossRef]
- da Costa, G.V.; Ferreira, E.F.B.; da S. Ramos, R.; da Silva, L.B.; de Sá, E.M.F.; da Silva, A.K.P.; Lobato, C.M.; Souto, R.N.P.; de P. da Silva, C.H.T.; Federico, L.B.; et al. Hierarchical virtual screening of potential insectides inhibitors of acetylcholinesterase and juvenile hormone from temephos. Pharmaceuticals 2019, 12, 61. [Google Scholar] [CrossRef]
- Ramos, R.S.; Macêdo, W.J.C.; Costa, J.S.; da Silva, C.H.T.d.P.; Rosa, J.M.C.; da Cruz, J.N.; Mozaniel, S.D.O.; de Aguiar Andrade, E.H.; Silva, R.B.L.; Souto, R.N.P.; et al. Potential inhibitors of the enzyme acetylcholinesterase and juvenile hormone with insecticidal activity: Study of the binding mode via docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 38, 4687–4709. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A.; Ihlenfeldt, W.-D.D.; Gloriozova, T.A.; Lagunin, A.A.; Borodina, Y.V.; Stepanchikova, A.V.; Nicklaus, M.C. PASS Biological Activity Spectrum Predictions in the Enhanced Open NCI Database Browser. J. Chem. Inf. Comput. Sci. 2003, 43, 228–236. [Google Scholar] [CrossRef]
- Sharaf, M.H.; El-Sherbiny, G.M.; Moghannem, S.A.; Abdelmonem, M.; Elsehemy, I.A.; Metwaly, A.M.; Kalaba, M.H. New combination approaches to combat methicillin-resistant Staphylococcus aureus (MRSA). Sci. Rep. 2021, 11, 4240. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Pogodin, P.V.; Lagunin, A.A.; Rudik, A.V.; Druzhilovskiy, D.S.; Filimonov, D.A.; Poroikov, V.V. AntiBac-Pred: A web aplication for predicting antibacterial activity of chemical compounds. J. Chem. Inf. Model. 2019, 59, 4513–4518. [Google Scholar] [CrossRef]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef]
- Bouley, R.; Kumarasiri, M.; Peng, Z.; Otero, L.H.; Song, W.; Suckow, M.A.; Schroeder, V.A.; Wolter, W.R.; Lastochkin, E.; Antunes, N.T.; et al. Discovery of antibiotic (E)-3-(3-Carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one. J. Am. Chem. Soc. 2015, 137, 1738–1741. [Google Scholar] [CrossRef]
- Mahasenan, K.V.; Molina, R.; Bouley, R.; Batuecas, M.T.; Fisher, J.F.; Hermoso, J.A.; Chang, M.; Mobashery, S. Conformational dynamics in penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus, allosteric communication network and enablement of catalysis. J. Am. Chem. Soc. 2017, 139, 2102–2110. [Google Scholar] [CrossRef]
- Martinez-Botella, G.; Breen, J.; Duffy, J.; Dumas, J.; Geng, B.; Gowers, I.; Green, O.; Guler, S.; Hentemann, M.; Hernandez-Juan, F.; et al. Discovery of selective and potent inhibitors of Gram-positive bacterial thymidylate kinase (TMK): Compund 16. J. Med. Chem. 2012, 55, 10010–10021. [Google Scholar] [CrossRef]
- Vanheusden, V.; Van Rompaey, P.; Munier-Lehmann, H.; Pochet, S.; Herdewijn, P.; Van Calenbergh, S. Thymidine and thymidine-5′-O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg. Med. Chem. Lett. 2003, 13, 3045–3048. [Google Scholar] [CrossRef]
- Lim, D.; Strynadka, N.C.J. Structural basis for the β lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 2002, 9, 870–876. [Google Scholar] [CrossRef]
- Metersky, M.L.; Kalil, A.C. New guidelines for nosocomial pneumonia. Curr. Opin. Pulm. Med. 2017, 23, 211–217. [Google Scholar] [CrossRef]
- Knezevic, P.; Aleksic, V.; Simin, N.; Svircev, E.; Petrovic, A.; Mimica-Dukic, N. Antimicrobial activity of Eucalyptus camaldulensis essential oils and their interactions with conventional antimicrobial agents against multi-drug resistant Acinetobacter baumannii. J. Ethnopharmacol. 2016, 178, 125–136. [Google Scholar] [CrossRef]
- Šmejkal, K.; Chudík, S.; Klouček, P.; Marek, R.; Cvačka, J.; Urbanová, M.; Julínek, O.; Kokoška, L.; Šlapetová, T.; Holubová, P.; et al. Antibacterial C-geranylflavonoids from Paulownia tomentosa fruits. J. Nat. Prod. 2008, 71, 706–709. [Google Scholar] [CrossRef]
- Navrátilová, A.; Nešuta, O.; Vančatová, I.; Čížek, A.; Varela-M, R.E.; López-Abán, J.; Villa-Pulgarin, J.A.; Mollinedo, F.; Muro, A.; Žemličková, H.; et al. C-Geranylated flavonoids from Paulownia tomentosa fruits with antimicrobial potential and synergistic activity with antibiotics. Pharm. Biol. 2016, 54, 1398–1407. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, F.; Jiang, X.; Zhang, W.; Liu, J.; Liu, W.; Fu, L. Synthesis and antimicrobial evaluation of 3-methanone-6-substituted-benzofuran derivatives. Eur. J. Med. Chem. 2012, 54, 879–886. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef]
- Wingert, B.M.; Camacho, C.J. Improving small molecule virtual screening strategies for the next generation of therapeutics. Curr Opin Chem Biol. 2018, 44, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, I.C.F.; Borges, R.S.; Silva, G.M.; Lima, L.R.; Bastos, R.S.; Ramos, R.S.; Silva, L.B.; da Silva, C.H.T.P.; dos Santos, C.B.R. Hierarchical virtual screening based on rocaglamide derivatives to discover new potential anti-skin cancer agents. Front. Mol. Biosci. 2022, 9, 836572. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, 198–201. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment. Dassault Syst. 2015, 4. Available online: https://www.scirp.org/(S(351jmbntv-nsjt1aadkposzje))/reference/referencespapers.aspx?referenceid=2450411 (accessed on 30 September 2023).
- Guedes, I.A.; Barreto, A.M.S.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miiteya, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198. [Google Scholar] [CrossRef]
- Dos Santos, K.L.B.; Cruz, J.N.; Silva, L.B.; Ramos, R.S.; Neto, M.F.A.; Lobato, C.C.; Ota, S.S.B.; Leite, F.H.A.; Borges, R.S.; Silva, C.H.T.P.d.; et al. Identification of novel chemical entities for adenosine receptor type 2a using molecular modeling approaches. Molecules 2020, 25, 1245. [Google Scholar] [CrossRef]
- de Melo Lima, A.; Siqueira, A.S.; Möller, M.L.S.; de Souza, R.C.; Cruz, J.N.; Lima, A.R.J.; da Silva, R.C.; Aguiar, D.C.F.; da Silva Gonçalves Vianez Junior, J.L.; Gonçalves, E.C. In silico improvement of the cyanobacterial lectin microvirin and mannose interaction. J. Biomol. Struct. Dyn. 2022, 40, 1064–1073. [Google Scholar] [CrossRef]
- da Silva, D.F.; de Souza, J.L.; da Costa, D.M.; Costa, D.B.; Moreira, P.O.L.; da Fonseca, A.L.; de Pilla Varotti, F.; Cruz, J.N.; Santos, C.B.R.; Alves, C.Q.; et al. Antiplasmodial activity of coumarins isolated from Polygala boliviensis: In vitro and in silico studies. J. Biomol. Struct. Dyn. 2023, 1–21. [Google Scholar] [CrossRef]
- Guedes, I.A.; Costa, L.S.C.C.; dos Santos, K.B.; Karl, A.L.M.M.; Rocha, G.K.; Teixeira, I.M.; Galheigo, M.M.; Medeiros, V.; Krempser, E.; Custódio, F.L.; et al. Drug design and repurposing with DockThor-VS Web Server: Virtual screening focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci. Rep. 2020, 11, 5543. [Google Scholar] [CrossRef]
- Santos, K.B.; Guedes, I.A.; Karl, A.L.M.; Dardenne, L.E. Highly flexible ligand docking: Benchmarking of the DockThor program on the LEADS-PEP protein–peptide data Set. J. Chem. Inf. Model. 2020, 60, 667–683. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Computat. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Sgobba, M.; Caporuscio, F.; Anighoro, A.; Portioli, C.; Rastelli, G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 2012, 58, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Kochev, N.; Jeliazkova, N.; Tsakovska, I. CHAPTER 3. Chemoinformatics representation of chemical structures—A milestone for successful Big Data modelling in predictive toxicology. In Big Data in Predictive Toxicology; Royal Society of Chemistry: London, UK, 2019; pp. 69–107. [Google Scholar] [CrossRef]
- Silva, L.B.; Ferreira, E.F.B.; Maryam; Espejo-Román, J.M.; Costa, G.V.; Cruz, J.V.; Kimani, N.M.; Costa, J.S.; Bittencourt, J.A.H.M.; Cruz, J.N.; et al. Galantamine based novel acetylcholinesterase enzyme inhibitors: A molecular modeling design approach. Molecules 2023, 28, 1035. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Sepay, N.N.N.; Sepay, N.N.N.; Al Hoque, A.; Mondal, R.; Halder, U.C.; Muddassir, M. In silico fight against novel coronavirus by finding chromone derivatives as inhibitor of coronavirus main proteases enzyme. Struct. Chem. 2020, 31, 1831–1840. [Google Scholar] [CrossRef]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of LogP Methods on more than 96,000 Compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Structure | MW 1 | RotB 2 | PSA 3 | LogP (a) | Donor (HBD) | Acceptor (HBA) | Aromatic (b) |
|---|---|---|---|---|---|---|---|
| 01 | 484.58 | 10 | 128.20 | 7.12 | 4 | 3 | |
| 7 | |||||||
| 02 | 540.69 | 12 | 128.20 | 8.51 | 4 | 3 | |
| 7 | |||||||
| 03 | 502.60 | 11 | 155.50 | 6.75 | 6 | 2 | |
| 8 | |||||||
| 04 | 400.42 | 6 | 128.20 | 5.07 | 3 | ||
| 4 | 7 | ||||||
| 05 | 558.71 | 13 | 155.50 | 8.26 | 6 | 2 | |
| 8 | |||||||
| 06 | 280.36 | 6 | 77.70 | 4.71 | 3 | 1 | |
| 4 | |||||||
| 07 | 210.23 | 3 | 77.70 | 2.44 | 3 | 1 | |
| 4 | |||||||
| 08 | 252.31 | 5 | 77.70 | 3.93 | 3 | 1 | |
| 4 | |||||||
| 09 | 232.19 | 0 | 94.00 | 1.98 | 4 | 3 | |
| 5 | |||||||
| Minimum | 210.23 | 0 | 77.70 | 1.98 | 3 | 4 | 1 |
| Maximum | 558.71 | 13 | 155.50 | 8.51 | 6 | 8 | 3 |
| Chemical Structure | Tanimoto Index | ||
|---|---|---|---|
| Tetrahydroxybenzofuran (Pivot) | Methicillin | Oxacillin | |
| MolPort-039-052-415 | 0.426 | 0.228 | 0.199 |
| MolPort-001-741-320 | 0.421 | 0.262 | 0.250 |
| MolPort-039-339-001 | 0.416 | 0.254 | 0.251 |
| MolPort-001-742-504 | 0.416 | 0.253 | 0.249 |
| MolPort-039-338-719 | 0.409 | 0.252 | 0.251 |
| MolPort-039-338-651 | 0.409 | 0.251 | 0.250 |
| MolPort-039-052-414 | 0.403 | 0.226 | 0.200 |
| MolPort-039-338-750 | 0.401 | 0.275 | 0.269 |
| MolPort-039-339-000 | 0.399 | 0.254 | 0.256 |
| MolPort-005-945-435 | 0.397 | 0.251 | 0.238 |
| MolPort-039-052-600 | 0.397 | 0.227 | 0.204 |
| MolPort-035-706-258 | 0.394 | 0.225 | 0.205 |
| MolPort-035-706-259 | 0.394 | 0.225 | 0.205 |
| MolPort-028-610-187 | 0.393 | 0.250 | 0.240 |
| MolPort-028-610-188 | 0.392 | 0.252 | 0.242 |
| MolPort-005-945-312 | 0.388 | 0.266 | 0.276 |
| MolPort-035-706-255 | 0.376 | 0.255 | 0.240 |
| MolPort-035-706-257 | 0.371 | 0.234 | 0.225 |
| Chemical Structure | Carcinogenicity (a) | Ames Test (a) | LD50 (b) (mg/kg) | Class (b) | |
|---|---|---|---|---|---|
| Rat | Mouse | Mutagenicity | |||
| Tetrahydroxybutanefuran (pivot) | Negative | Positive | Mutagenic | 1000 | 4 |
| Oxacillin | Positive | Negative | Not mutagenic | 5000 | 5 |
| Methicillin | Negative | Negative | Not mutagenic | 2880 | 5 |
| MolPort-001-741-320 | Negative | Positive | Mutagenic | 2000 | 4 |
| MolPort-001-742-504 | Negative | Positive | Mutagenic | 2000 | 4 |
| MolPort-005-945-312 | Negative | Negative | Not mutagenic | 2000 | 4 |
| MolPort-005-945-435 | Negative | Negative | Not mutagenic | 10 | 2 |
| MolPort-028-610-187 | Negative | Negative | Mutagenic | 10 | 2 |
| MolPort-028-610-188 | Negative | Negative | Not mutagenic | 10 | 2 |
| MolPort-035-706-255 | Negative | Positive | Not mutagenic | 690 | 4 |
| MolPort-035-706-257 | Positive | Positive | Not mutagenic | 690 | 4 |
| MolPort-035-706-258 | Negative | Positive | Mutagenic | 400 | 4 |
| MolPort-035-706-259 | Negative | Positive | Mutagenic | 400 | 4 |
| MolPort-039-052-414 | Negative | Positive | Mutagenic | 1060 | 4 |
| MolPort-039-052-415 | Positive | Negative | Not mutagenic | 1060 | 4 |
| MolPort-039-052-600 | Negative | Positive | Not mutagenic | 400 | 4 |
| MolPort-039-338-651 | Negative | Positive | Mutagenic | 2000 | 4 |
| MolPort-039-338-719 | Negative | Positive | Mutagenic | 2000 | 4 |
| MolPort-039-338-750 | Positive | Positive | Not mutagenic | 2000 | 4 |
| MolPort-039-339-000 | Positive | Negative | Mutagenic | 2000 | 4 |
| MolPort-039-339-001 | Negative | Positive | Not mutagenic | 2000 | 4 |
| Chemical Structure | HIA% (a) | PCaco-2 (nm/s) (b) |
|---|---|---|
| Tetrahydroxybutanefuran (pivot) | 90.04 | 20.23 |
| Oxacillin | 0.42 | 20.15 |
| Methicillin | 87.32 | 14.99 |
| MolPort-001-741-320 | 90.45 | 7.82 |
| MolPort-001-742-504 | 90.29 | 10.95 |
| MolPort-005-945-312 | 88.58 | 12.02 |
| MolPort-005-945-435 | 88.40 | 13.14 |
| MolPort-028-610-187 | 92.38 | 17.18 |
| MolPort-028-610-188 | 92.39 | 14.70 |
| MolPort-035-706-255 | 86.88 | 18.19 |
| MolPort-035-706-257 | 86.88 | 17.80 |
| MolPort-035-706-258 | 86.88 | 17.80 |
| MolPort-035-706-259 | 86.88 | 17.80 |
| MolPort-039-052-414 | 91.59 | 20.06 |
| MolPort-039-052-415 | 89.24 | 18.72 |
| MolPort-039-052-600 | 86.05 | 20.02 |
| MolPort-039-338-651 | 84.27 | 18.20 |
| MolPort-039-338-719 | 88.40 | 18.58 |
| MolPort-039-338-750 | 91.20 | 20.48 |
| MolPort-039-339-000 | 92.40 | 15.95 |
| MolPort-039-339-001 | 92.40 | 16.91 |
| Chemical Structure | Distribution | |
|---|---|---|
| PPB(%) (a) | CBrain/CBlood (b) | |
| Tetrahydroxybutanefuran (pivot) | 97.73 | 5.09 |
| Oxacillin | 61.92 | 0.03 |
| Methicillin | 56.05 | 0.11 |
| MolPort-001-741-320 | 100.00 | 0.76 |
| MolPort-001-742-504 | 100.00 | 2.38 |
| MolPort-005-945-312 | 100.00 | 2.35 |
| MolPort-005-945-435 | 100.00 | 3.20 |
| MolPort-028-610-187 | 100.00 | 2.78 |
| MolPort-028-610-188 | 98.76 | 3.43 |
| MolPort-035-706-255 | 100.00 | 0.83 |
| MolPort-035-706-257 | 100.00 | 1.28 |
| MolPort-035-706-258 | 100.00 | 1.78 |
| MolPort-035-706-259 | 100.00 | 1.78 |
| MolPort-039-052-414 | 100.00 | 3.33 |
| MolPort-039-052-415 | 100.00 | 0.19 |
| MolPort-039-052-600 | 100.00 | 1.41 |
| MolPort-039-338-651 | 100.00 | 1.49 |
| MolPort-039-338-719 | 100.00 | 3.83 |
| MolPort-039-338-750 | 100.00 | 6.62 |
| MolPort-039-339-000 | 100.00 | 4.66 |
| MolPort-039-339-001 | 100.00 | 5.63 |
| Antibacterial Activity | ||||||
|---|---|---|---|---|---|---|
| Pass Online | Antibac-Pred | |||||
| Structure | Pa (a) | Pi (b) | Code | Name | Conf. (c) | ChEMBL ID |
| Tetrahydroxybenzofuran | 0.465 | 0.020 | - | S. aureus | 0.116 | CHEMBL352 |
| RESISTANT S. aureus | 0.062 | CHEMBL352 | ||||
| RESISTANT S. aureus subsp. aureus RN4220 | 0.948 | CHEMBL2366906 | ||||
| Oxacillin | RESISTANT S. aureus subsp. aureus RN4220 | 0.948 | CHEMBL2366906 | |||
| 0.684 | 0.005 | - | S. aureus | 0.398 | CHEMBL352 | |
| S. aureus subsp. aureus RN4220 | 0.213 | CHEMBL2366906 | ||||
| Methicillin | 0.671 | 0.005 | - | RESISTANT S. aureus subsp. aureus RN4220 | 0.878 | CHEMBL2366906 |
| S. aureus | 0.344 | CHEMBL352 | ||||
| S. aureus subsp. aureus RN4220 | 0.190 | CHEMBL2366906 | ||||
| RESISTANT S. aureus | 0.032 | CHEMBL352 | ||||
| MolPort-001-741-320 | 0.487 | 0.018 | LB320 | RESISTANT S. aureus subsp. aureus RN4220 | 0.059 | CHEMBL2366906 |
| Staphylococcus simulans | 0.362 | CHEMBL612425 | ||||
| Staphylococcus sciuri | 0.353 | CHEMBL613150 | ||||
| MolPort-035-706-255 | 0.344 | 0.045 | LB255 | RESISTANT S. simulans | 0.310 | CHEMBL612425 |
| S. sciuri | 0.210 | CHEMBL613150 | ||||
| S. simulans | 0.155 | CHEMBL612425 | ||||
| MolPort-039-052-415 | 0.400 | 0.30 | LB415 | - | - | - |
| Structures | ΔG (a) | Hydrogen Bond (Å Distance) | Hydrophobic Interactions |
|---|---|---|---|
| Complex (0Y5) | −9.185 | ARG48 (2.93) (3.02), PHE66 (5.36), ARG70 (2.89), SER97 (2.67) and GLN101 (2.79) (2.83) | PRO38, ARG48, LEU52, PHE66, ARG92 and TYR100 |
| LB255 | −7.870 | GLU62 (2.15), ARG92 (3.14) and ARG105 (4.08) | PHE66 and TYR100 |
| LB320 | −8.184 | ASP156 (1.61) | PHE66, ARG92 and TYR100 |
| LB415 | −8.048 | LYS15 (3.37), ARG48 (5.08), ARG92 (5.13) and GLN101 (2.68) (2.71) | LEU52, PHE66, ARG92 and TYR100 |
| Structures | ΔG (a) | Hydrogen Bond (Å Distance) | Hydrophobic Interactions |
|---|---|---|---|
| Complex (QZN) | −8.046 | TYR105 (4.18), GLU294 (2.64) and LYS316 (2.94) | ASN146 and TYR297 |
| LB255 | −7.199 | LYS273 (2.84), ASP275 (2.69) and ASP295 (1.66) (2.93) | TYR105 and TYR144 |
| LB320 | −8.019 | ASN104 (2.82), TYR105 (4.14), ILE144 (2.77) and LYS273 (2.87) | ILE144 and LYS273 |
| LB415 | −7.691 | ASN104 (3.37), ASN146 (2.49) (2.74), ASP295 (2.53), GLY296 (2.88) and LYS316 (2.81) | TYR105 and TYR297 |
| Compounds | System TMK | System PBP2a |
|---|---|---|
| Complex | −30.54 | −32.87 |
| LB255 | −35.84 | −29.15 |
| LB320 | −38.54 | −36.52 |
| LB415 | −33.42 | −35.76 |
| Compound | SA (%) (a) | SA SCORE (%) (b) |
|---|---|---|
| Pivot | 51.31 | 40.98 |
| LB255 | 36.58 | 50.17 |
| LB320 | 65.08 | 30.88 |
| LB415 | 38.88 | 50.06 |
| Chemical Structure | iLOGP | XLOGP3 | WLOGP | MLOGP | SILICOS-IT | Mean |
|---|---|---|---|---|---|---|
| Methicillin | 2.24 | 1.22 | 0.57 | 1.01 | 0.78 | 1.16 |
| Oxacillin | 2.23 | 2.38 | 1.51 | 1.56 | 1.59 | 1.85 |
| LB255 | 3.31 | 1.48 | 1.68 | 0.78 | 1.69 | 1.79 |
| LB320 | 2.98 | 4.61 | 3.23 | 2.13 | 3.38 | 3.27 |
| LB415 | 1.14 | 2.56 | 2.62 | 1.14 | 4.26 | 2.34 |
| Chemical Structure | ESOL | Ali | SILICOS-IT | Mean |
|---|---|---|---|---|
| Methicillin | −2.74 | −3.56 | −2.75 | −3.01 |
| Oxacillin | −3.79 | −4.92 | −4.23 | −4.31 |
| LB255 | −2.83 | −3.11 | −1.57 | −2.50 |
| LB320 | −5.26 | −6.75 | −2.32 | −4.77 |
| LB415 | −3.85 | −4.92 | −3.14 | −3.97 |
| Nº | Code SMILES | MIC (a) |
|---|---|---|
| 01 | C[C@H](CC)C(=O)c1c2oc3c(c2c(O)c(CCC)c1O)c(O)c(CCC)c(O)c3C(=O)[C@H](C)CC | 0.24 |
| 02 | Oc1c(c(O)c(c(O)c1CCC)c1c(O)c(CCC)c(O)c(C(=O)C(C)CC)c1O)C(=O)C(C)CC | 2.00 |
| 03 | C[C@@H](CC)C(=O)c1c2oc3c(c2c(O)c(CCC(C)C)c1O)c(O)c(CCC(C)C)c(O)c3C(=O)[C@H](C)CC | 2.16 |
| 04 | C[C@H](CC)C(=O)c1c2oc3c(c2c(O)cc1O)c(O)cc(O)c3C(=O)[C@@H](C)CC | 3.20 |
| 05 | Oc1c(c(O)c(c(O)c1CCC(C)C)c1c(O)c(CCC(C)C)c(O)c(C(=O)C(C)CC)c1O)C(=O)C(C)CC | 4.64 |
| 06 | Oc1c(c(O)cc(O)c1CCC(C)C)C(=O)C(C)CC | 8.96 |
| 07 | O=C(c1c(O)cc(O)cc1O)C(C)CC | 13.44 |
| 08 | Oc1c(c(O)cc(O)c1CCC)C(=O)C(C)CC | 16.13 |
| 09 | Oc1cc2oc3cc(O)cc(O)c3c2c(O)c1 | >30 |
| Receptor | Ligand/ID | Grid Center Coordinates | Grid Box Dimensions |
|---|---|---|---|
| Penicillin-binding protein (PBP2a-MRSA) PDB ID: 4CJN | (E)-3-(2-(4-Cyanostyryl)-4-oxoquinazolin-3(4H)-yl)benzoic acid/QZN | X= 8.992933 Y= −1.203300 Z= −69.561400 | 20x 20y 20z |
| Thymidylate kinase enzyme (TMK) PDB ID: 4GSY | 4-{[(3S)-3-(5-Methyl-2,4-dio-xo-3,4-dihydropyr-midi-1(2H)yl)piperidin-1-yl]methyl}-2-[3(triflu-oromethyl)phenoxy]benzoic acid/0Y5 | X= 8.577139 Y= 0.252556 Z= 27.171667 | 20x 20y 20z |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, L.P.S.; Lima, L.R.; Silva, L.B.; Cruz, J.N.; Ramos, R.S.; Lima, L.S.; Cardoso, F.M.N.; Silva, A.V.; Rodrigues, D.P.; Rodrigues, G.S.; et al. Hierarchical Virtual Screening of Potential New Antibiotics from Polyoxygenated Dibenzofurans against Staphylococcus aureus Strains. Pharmaceuticals 2023, 16, 1430. https://doi.org/10.3390/ph16101430
Oliveira LPS, Lima LR, Silva LB, Cruz JN, Ramos RS, Lima LS, Cardoso FMN, Silva AV, Rodrigues DP, Rodrigues GS, et al. Hierarchical Virtual Screening of Potential New Antibiotics from Polyoxygenated Dibenzofurans against Staphylococcus aureus Strains. Pharmaceuticals. 2023; 16(10):1430. https://doi.org/10.3390/ph16101430
Chicago/Turabian StyleOliveira, Lana P. S., Lúcio R. Lima, Luciane B. Silva, Jorddy N. Cruz, Ryan S. Ramos, Luciana S. Lima, Francy M. N. Cardoso, Aderaldo V. Silva, Dália P. Rodrigues, Gabriela S. Rodrigues, and et al. 2023. "Hierarchical Virtual Screening of Potential New Antibiotics from Polyoxygenated Dibenzofurans against Staphylococcus aureus Strains" Pharmaceuticals 16, no. 10: 1430. https://doi.org/10.3390/ph16101430
APA StyleOliveira, L. P. S., Lima, L. R., Silva, L. B., Cruz, J. N., Ramos, R. S., Lima, L. S., Cardoso, F. M. N., Silva, A. V., Rodrigues, D. P., Rodrigues, G. S., Proietti-Junior, A. A., dos Santos, G. B., Campos, J. M., & Santos, C. B. R. (2023). Hierarchical Virtual Screening of Potential New Antibiotics from Polyoxygenated Dibenzofurans against Staphylococcus aureus Strains. Pharmaceuticals, 16(10), 1430. https://doi.org/10.3390/ph16101430