
Arylpiperazine Derivatives and Cancer: A New Challenge in Medicinal Chemistry


 ,
,  , ,
, ,  ,
, 

Abstract

1. Introduction
2. The Evidence of N-Arylpiperazine Derivatives in Carcinogenic Pathways
2.1. N-Arylpiperazine Derivatives in Prostate Cancer
2.2. N-Arylpiperazine Derivatives in Colorectal Cancer
2.3. N-Arylpiperazine Derivatives in Pancreatic Cancer
2.4. N-Arylpiperazine Derivatives in Breast Cancer
2.5. N-Arylpiperazine Derivatives in Cervical Carcinoma
2.6. N-Arylpiperazine Derivatives in Leukemia
2.7. N-Arylpiperazine Derivatives in Melanoma
2.8. Other Examples of Arylpiperazines in Cancer
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Faizan, M.; Kumar, R.; Mazumder, A.; Salahuddin; Kukreti, N.; Kumar, A.; Chaitanya, M.V.N.L. The medicinal chemistry of piperazines: A review. Chem. Biol. Drug Des. 2024, 103, e14537. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, M.N.; Manetti, D.; Braconi, L.; Dei, S.; Gabellini, A.; Teodori, E. The piperazine scaffold for novel drug discovery efforts: The evidence to date. Expert Opin. Drug Discov. 2022, 17, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Popli, P.; Tiwary, N.; Swami, R. Small Molecules as Cancer Targeting Ligands: Shifting the Paradigm. J. Control. Release 2023, 355, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wu, J.; Liu, B. Therapeutic Strategies of Dual-Target Small Molecules to Overcome Drug Resistance in Cancer Therapy. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188866. [Google Scholar] [CrossRef] [PubMed]
- Karolak-Wojciechowska, J.; Fruzinski, A.; Mokrosz, M.J. Structure and Conformational Analysis of New Arylpiperazines Containing N-Butyl Chain. Elsevier 2001, 542, 47–56. [Google Scholar] [CrossRef]
- Weber, K.C.; Salum, L.B.; Honorio, K.M.; Andricopulo, A.D.; da Silva, A.B.F. Pharmacophore-Based 3D QSAR Studies on a Series of High Affinity 5-HT1A Receptor Ligands. Eur. J. Med. Chem. 2010, 45, 1508–1514. [Google Scholar] [CrossRef]
- Quaglia, W.; Cifani, C.; Del Bello, F.; Giannella, M.; Giorgioni, G.; Di Bonaventura, M.V.M.; Piergentili, A. 4WD to Travel Inside the 5-HT1A Receptor World. In Serotonin-A Chemical Messenger Between All Types of Living Cells; InTech: Tokyo, Japan, 2017; pp. 67–108. [Google Scholar]
- Caliendo, G.; Santagada, V.; Perissutti, E.; Fiorino, F. Derivatives as 5HT 1A Receptor Ligands-Past and Present. Curr. Med. Chem. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Fiorino, F.; Severino, B.; Magli, E.; Ciano, A.; Caliendo, G.; Santagada, V.; Frecentese, F.; Perissutti, E. 5-HT1A Receptor: An Old Target as a New Attractive Tool in Drug Discovery from Central Nervous System to Cancer. J. Med. Chem. 2014, 57, 4407–4426. [Google Scholar] [CrossRef]
- Ikwu, F.A.; Shallangwa, G.A.; Mamza, P.A. Ligand Based Design, ADMET and Molecular Docking Studies of Arylpiperazine Derivatives as Potent Anti-Proliferate Agents Against LNCAP Prostate Cancer Cell Lines. Chem. Afr. 2021, 4, 71–84. [Google Scholar] [CrossRef]
- Corvino, A.; Fiorino, F.; Severino, B.; Saccone, I.; Frecentese, F.; Perissutti, E.; Di Vaio, P.; Santagada, V.; Caliendo, G.; Magli, E. The Role of 5-HT1A Receptor in Cancer as a New Opportunity in Medicinal Chemistry. Curr. Med. Chem. 2018, 25, 3214–3227. [Google Scholar] [CrossRef]
- Cowen, D.S.; Sowers, R.S.; Manning, D.R. Activation of a Mitogen-Activated Protein Kinase (ERK2) by the 5-Hydroxytryptamine 1A Receptor Is Sensitive Not Only to Inhibitors of Phosphatidylinositol 3-Kinase, but to an Inhibitor of Phosphatidylcholine Hydrolysis. J. Biol. Chem. 1996, 271, 22297–22300. [Google Scholar] [CrossRef] [PubMed]
- Blesen, T.; Hawes, B.E.; Luttrell, D.K.; Krueger, K.M.; Touhara, K.; Porflrlt, E.; Sakauet, M.; Luttrell, L.M.; Lefkowitz, R.J. Receptor-tyrosine-kinase- and Gpy-mediated MAP kinase activation by a common signalling pathway. Lett. Nat. 1995, 376, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Della Rocca, G.J.; Mukhin, Y.V.; Garnovskaya, M.N.; Daaka, Y.; Clark, G.J.; Luttrell, L.M.; Lefkowitz, R.J.; Raymond, J.R. Serotonin 5-HT1A Receptor-mediated Erk Activation Requires Calcium/Calmodulin-dependent Receptor Endocytosis. J. Biol. Chem. 1999, 274, 4749–4753. [Google Scholar] [CrossRef] [PubMed]
- Shinka, T.; Onodera, D.; Tanaka, T.; Shoji, N.; Miyazaki, T.; Moriuchi, T.; Fukumoto, T. Serotonin Synthesis and Metabolism-Related Molecules in a Human Prostate Cancer Cell Line. Oncol. Lett. 2011, 2, 211–215. [Google Scholar] [CrossRef]
- Tutton, P.J.M.; Steel, G.G. Influence of biogenic amines on the growth of xenografted human colorectal carcinomas. Br. J. Cancer 1979, 40, 743–749. [Google Scholar] [CrossRef]
- Matsuda, T.; Seong, Y.H.; Aono, H.; Kanda, T.; Baba, A.; Saito, K.; Tobe, A.; Iwata, H. Agonist Activity of a Novel Compound, 1-[3-(3,4-Methylenedioxyphenoxy) Propyl]-4-Phenyl Piperazine (BP-554), at Central 5-HT1A Receptors. Eur. J. Pharmacol. 1989, 170, 75–82. [Google Scholar] [CrossRef]
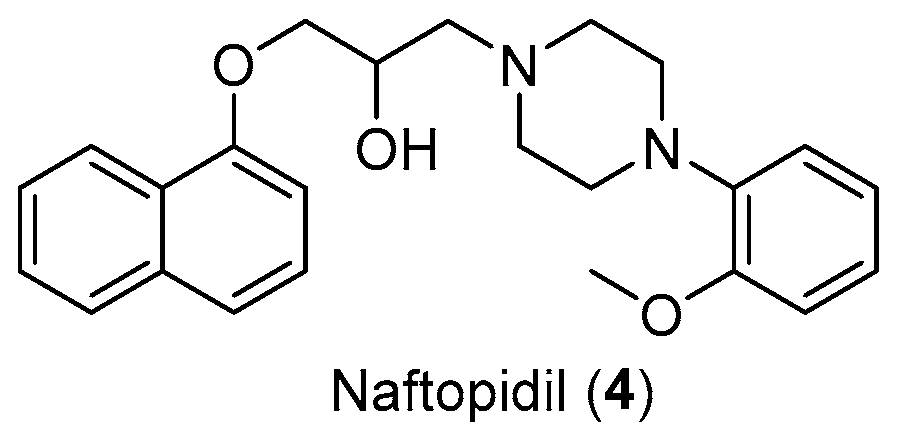
- Florent, R.; Poulain, L.; N’Diaye, M. Drug Repositioning of the A1-Adrenergic Receptor Antagonist Naftopidil: A Potential New Anti-Cancer Drug? Int. J. Mol. Sci. 2020, 21, 5339. [Google Scholar] [CrossRef]
- Chen, H.; Liang, X.; Xu, F.; Xu, B.; He, X.; Huang, B.; Yuan, M. Synthesis and Cytotoxic Activity Evaluation of Novel Arylpiperazine Derivatives on Human Prostate Cancer Cell Lines. Molecules 2014, 19, 12048–12064. [Google Scholar] [CrossRef]
- Xu, F.; Chen, H.; Xu, J.; Liang, X.; He, X.; Shao, B.; Sun, X.; Li, B.; Deng, X.; Yuan, M. Synthesis, Structure-Activity Relationship and Biological Evaluation of Novel Arylpiperzines as A1A/1D-AR Subselective Antagonists for BPH. Bioorganic Med. Chem. 2015, 23, 7735–7742. [Google Scholar] [CrossRef]
- Hanušová, V.; Skálová, L.; Kralova, V.; Matouskova, P. Potential Anti-cancer Drugs Commonly Used for Other Indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef]
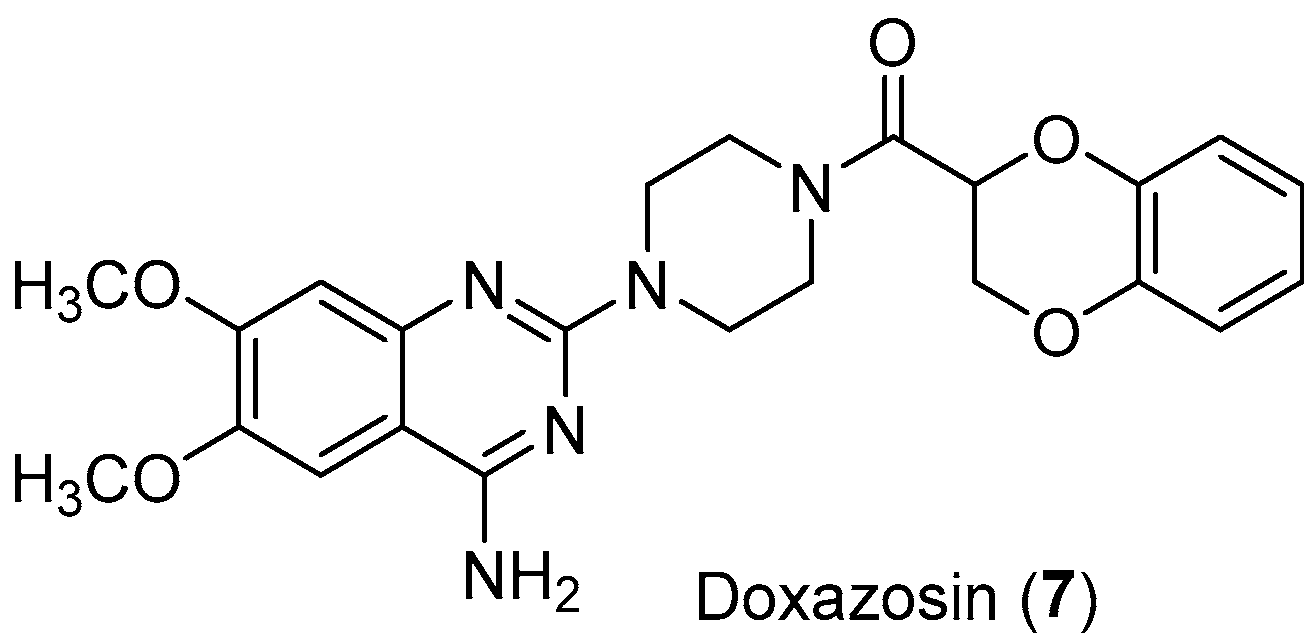
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Okada, M.; Yoshioka, T.; Kitanaka, C. Doxazosin, a Classic Alpha 1-Adrenoceptor Antagonist, Overcomes Osimertinib Resistance in Cancer Cells via the Upregulation of Autophagy as Drug Repurposing. Biomedicines 2020, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Chen, H.; Chen, S.; Shen, J.; Li, J. Synthesis, Bioactivity, and Molecular Docking of Novel Arylpiperazine Derivatives as Potential AR Antagonists. Front. Chem. 2022, 10, 947065. [Google Scholar] [CrossRef] [PubMed]
- Kinoyama, I.; Taniguchi, N.; Yoden, T.; Koutoku, H.; Furutani, T.; Kudoh, M.; Okada, M. Synthesis and Pharmacological Evaluation of Novel Arylpiperazine Derivatives as Nonsteroidal Androgen Receptor Antagonists. Chem. Pharm. Bull. 2004, 52, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.; Moore, C.M.; Chong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate Cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schroder, F.; Tomlins, S.A.; Tannok, I.; Drake, C.G.; de Bono, J.S. Prostate Cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.L.; Sun, T.; Zhou, Z.; Niu, J.; Tian, X.; Yuan, M. Synthesis, Biological Evaluation and SAR of Naftopidil-Based Arylpiperazine Derivatives. Bioorganic Med. Chem. Lett. 2018, 28, 1534–1539. [Google Scholar] [CrossRef]
- Kinoyama, I.; Taniguchi, N.; Kawaminami, E.; Nozawa, E.; Koutoku, H.; Furutani, T.; Kudoh, M.; Okada, M. N-Arylpiperazine-1-Carboxamide Derivatives: A Novel Series of Orally Active Nonsteroidal Androgen Receptor Antagonists. Chem. Pharm. Bull. 2005, 53, 402–409. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.M.; Mogilski, S.; Wigluszc, R.J.; Janczak, J.; Maniewska, J.; Malinka, W.; Filipek, B. Synthesis and Pharmacological Evaluation of Novel Arylpiperazine Oxicams Derivatives as Potent Analgesics without Ulcerogenicity. Bioorganic Med. Chem. 2019, 27, 1619–1628. [Google Scholar] [CrossRef]
- Szczuka, I.; Wierzbicki, J.; Serek, P.; Szczesniak-Siega, B.M.; Krzystek-Korpacka, M. Heat Shock Proteins Hspa1 and Hsp90aa1 Are Upregulated in Colorectal Polyps and Can Be Targeted in Cancer Cells by Anti-Inflammatory Oxicams with Arylpiperazine Pharmacophore and Benzoyl Moiety Substitutions at Thiazine Ring. Biomolecules 2021, 11, 1588. [Google Scholar] [CrossRef]
- Xie, W.; Chu, M.; Song, G.; Zuo, Z.; Han, Z.; Chen, C.; Li, Y.; Wang, Z. Emerging Roles of Long Noncoding RNAs in Chemoresistance of Pancreatic Cancer. Semin. Cancer Biol. 2022, 83, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic Cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Xue, Z.; Feng, Y.; Xie, Y.; Deng, B.; Yao, Y.; Tian, X.; An, Q.; Yang, L.; Yao, Q.; et al. N-Arylpiperazine-Containing Compound (C2): An Enhancer of Sunitinib in the Treatment of Pancreatic Cancer, Involving D1DR Activation. Toxicol. Appl. Pharmacol. 2019, 384, 114789. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Niu, M.; Zhou, Z.; Zheng, X.; Zhang, L.; Tian, Y.; Yu, X.; Bu, G.; Xu, H.; Ma, Q.; et al. VPS35 Regulates Cell Surface Recycling and Signaling of Dopamine Receptor D1. Neurobiol. Aging 2016, 46, 22–31. [Google Scholar] [CrossRef]
- Hao, F.; Wang, S.; Zhu1, X.; Xue, J.; Li, J.; Wang, L.; Li, J.; Lu, W.; Zhou, T. Pharmacokinetic-Pharmacodynamic Modeling of the Anti-Tumor Effect of Sunitinib Combined with Dopamine in the Human Non-Small Cell Lung Cancer Xenograft. Pharm. Res. 2017, 34, 408–418. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, S.; Ren, Y.; Li, J.; Guo, T.; Lu, W.; Zhou, T. Antitumor Effect of Axitinib Combined with Dopamine and PK-PD Modeling in the Treatment of Human Breast Cancer Xenograft. Acta Pharmacol. Sin. 2019, 40, 243–256. [Google Scholar] [CrossRef]
- Zubair, M.; Wang, S.; Ali, N. Advanced Approaches to Breast Cancer Classification and Diagnosis. Front. Pharmacol. 2021, 11, 632079. [Google Scholar] [CrossRef]
- Ambrosio, M.R.; Magli, E.; Caliendo, G.; Sparaco, R.; Massarelli, P.; D’Esposito, V.; Migliaccio, T.; Mosca, G.; Fiorino, F.; Formisano, P. Serotoninergic Receptor Ligands Improve Tamoxifen Effectiveness on Breast Cancer Cells. BMC Cancer 2022, 22, 171. [Google Scholar] [CrossRef]
- Fiorino, F.; Perissutti, E.; Severino, B.; Santagada, V.; Cirillo, D.; Terracciano, S.; Massarelli, P.; Bruni, G.; Collavoli, E.; Renner, C.; et al. New 5-Hydroxytryptamine1A Receptor Ligands Containing a Norbornene Nucleus: Synthesis and in Vitro Pharmacological Evaluation. J. Med. Chem. 2005, 48, 5495–5503. [Google Scholar] [CrossRef]
- Fiorino, F.; Magli, E.; Kędzierska, E.; Ciano, A.; Corvino, A.; Severino, B.; Perissutti, E.; Frecentese, F.; Di Vaio, P.; Saccone, I.; et al. New 5-HT1A, 5HT2A and 5HT2C Receptor Ligands Containing a Picolinic Nucleus: Synthesis, in Vitro and in Vivo Pharmacological Evaluation. Bioorganic Med. Chem. 2017, 25, 5820–5837. [Google Scholar] [CrossRef]
- Andreozzi, G.; Ambrosio, M.R.; Magli, E.; Maneli, G.; Severino, B.; Corvino, A.; Sparaco, R.; Perissutti, E.; Le’sniak, A.; Frecentese, F.; et al. Design, Synthesis and Biological Evaluation of Novel N-Arylpiperazines Containing a 4,5-Dihydrothiazole Ring. Pharmaceuticals 2023, 16, 1483. [Google Scholar] [CrossRef] [PubMed]
- Gimmelli, R.; Persico, M.; Imperatore, C.; Saccoccia, F.; Guidi, A.; Casertano, M.; Luciano, P.; Pietrantoni, A.; Bertuccini, L.; Paladino, A.; et al. Thiazinoquinones as New Promising Multistage Schistosomicidal Compounds Impacting Schistosoma mansoni and Egg Viability. ACS Infect Dis. 2020, 6, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Ferrall, L.; Lin, K.Y.; Roden, R.B.S.; Hung, C.-F.; Wu, T.-C. Cervical Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2021, 27, 4953–4973. [Google Scholar] [CrossRef] [PubMed]
- Small, W.; Bacon, M.A.; Bajaj, A.; Chuang, L.T.; Fisher, B.J.; Harkenrider, M.M.; Jhingran, A.; Kitchener, H.C.; Mileshkin, L.R.; Viswanathan, A.N.; et al. Cervical Cancer: A Global Health Crisis. Cancer 2017, 123, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.W.; Zheng, X.; Lin, Y.-P.; Hu, C.-Y.; Wang, X.-L.; Wan, C.-P.; Rao, G.-X. Design, Synthesis and Anticancer Activity of Novel Hybrid Compounds between Benzofuran and N-Aryl Piperazine. Bioorganic Med. Chem. Lett. 2016, 26, 3421–3424. [Google Scholar] [CrossRef]
- Snyder, R. Leukemia and Benzene. Int. J. Environ. Res. Public Health 2012, 9, 2875–2893. [Google Scholar] [CrossRef]
- Choi, M.J.; No, E.S.; Thorat, D.A.; Jang, J.W.; Yang, H.; Lee, J.; Choo, H.; Kim, S.J.; Lee, C.S.; Ko, S.Y.; et al. Synthesis and Biological Evaluation of Aryloxazole Derivatives as Antimitotic and Vascular-Disrupting Agents for Cancer Therapy. J. Med. Chem. 2013, 56, 9008–9018. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef]
- Hayward, N.K. Genetics of Melanoma Predisposition. Oncogene 2003, 22, 3053–3062. [Google Scholar] [CrossRef]
- Romagnoli, R.; Oliva, P.; Prencipe, F.; Manfredini, S.; Germano, M.P.; De Luca, L.; Ricci, F.; Corallo, D.; Aveic, S.; Mariotto, E.; et al. Cinnamic Acid Derivatives Linked to Arylpiperazines as Novel Potent Inhibitors of Tyrosinase Activity and Melanin Synthesis. Eur. J. Med. Chem. 2022, 231, 114147. [Google Scholar] [CrossRef]
- Lee, Y.B.; Gong, Y.D.; Yoon, H.; Ahn, C.H.; Jeon, M.K.; Kong, J.Y. Synthesis and Anticancer Activity of New 1-[(5 or 6-Substituted 2-Alkoxyquinoxalin-3-Yl)Aminocarbonyl]-4-(Hetero)Arylpiperazine Derivatives. Bioorganic Med. Chem. 2010, 18, 7966–7974. [Google Scholar] [CrossRef] [PubMed]
- Miller-Moslin, K.; Peukert, S.; Jain, R.K.; McEwan, M.A.; Karki, R.; Llamas, L.; Yusuff, N.; He, F.; Li, Y.; Sun, Y.; et al. 1-amino-4-benzylphthalazines as orally bioavailable smoothened antagonists with antitumor activity. J. Med. Chem. 2009, 52, 3954–3968. [Google Scholar] [CrossRef] [PubMed]
- Peukert, S.; He, F.; Dai, M.; Zhang, R.; Sun, Y.; Miller-Moslin, K.; McEwan, M.; Lagu, B.; Wang, K.; Yusuff, N.; et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem 2013, 8, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.W.; Qiu, S.Q.; Zhang, G.J. Molecular and functional imaging in cancer-targeted therapy: Current applications and future directions. Sigal Transduct. Target. Ther. 2023, 8, 89. [Google Scholar] [CrossRef]
- Senapati, J.; Sasaki, K.; Issa, G.C.; Lipton, J.H.; Radich, J.P.; Jabbour, E.; Kantarjian, H.M. Management of chronic myeloid leukemia in 2023-common ground and common sense. Blood Cancer J. 2023, 13, 58. [Google Scholar] [CrossRef]
- Jindal, A.; Thadi, A.; Shailubhai, K. Hepatocellular Carcinoma: Etiology and Current and Future Drugs. J. Clin. Exp. Hepatol. 2019, 9, 221–232. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cancer | Cell Lines Expressing 5-HT1AR | Drugs | Effects |
|---|---|---|---|
| Prostate Cancer | PC3, DU145, LNCap | NAN-190 Pindobind | 5HT1A antagonists that inhibit cell growth in vitro, inducing apoptosis. |
| 6-nitroquipazine Zimelidine Fluoxetine | 5HT uptake inhibitors that cause dose-dependent inhibition of cells proliferation. | ||
| Bladder Carcinoma | SHT1376 | NAN 190 SB224289 | 5HT1A (NAN-190) and 5HT1B (SB224289) antagonists that show an inhibitory effect on the serotonin-induced growth cells. |
| Small Cell Lung Carcinoma | GLC8 | Spiperone SDZ 216–525 | 5-HT1A (spiperone) and 5-HT7 (SDZ 216–525) antagonists that inhibit 8-OH-DPAT-induced mitogenic effect. |
| Colorectal Carcinoma | HT29 | BW501C Citalopram Fluoxetine | Serotoninergic antagonists (BW501C) and SSRIs (Citalopram and Fluoxetine) that retard the tumor growth. |
| NAN 190 SB224289 | 5HT1A (NAN-190) and 5HT1B (SB224289) antagonists that reduce cell growth acting as antiproliferative agents. | ||
| Cholangiocarcinoma | Mz-chA1, HuH28, HUVV-T1, CCLP-1, SG231, TFK1. | - | - |
| Compd. | Structure | Selectivity Ratio | IC50 Values (DU145) |
|---|---|---|---|
| 9 |  | α1B/α1 Aratio = 16.7 | 0.93 ± 0.19 |
| 10 |  | α1B/α1D ratio = 10.9 | 0.90 ± 0.20 |
| Compd. | Structure | IC50 (μM) a | % inhibition b |
|---|---|---|---|
| YM-92088 (11) |  | 0.47 | - |
| 12 | 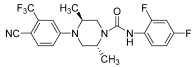 | 0.20 | 85% ** ED50 = 1.1 mg/kg |
| Compound | Structure | MCF-7 | MDA-MB231 |
|---|---|---|---|
| 21 |  | 14.70 ± 1.9 | 31.37 ± 5.1 |
| 22 |  | 15.93 ± 1.8 | 39.96 ± 9.8 |
| 23 |  | 19.47 ± 2.3 | 36.32 ± 7.7 |
| 24 |  | - | 23.27 ± 3.4 |
| 25 |  | - | 34.60 ± 5.4 |
| 26 |  | - | 47.15 ± 6.7 |
| Compound 27 |  | ||
|---|---|---|---|
| Cell Lines (IC50 μM) | |||
| A549 | Hela | MCF-7 | SGC7901 |
| 5.73 ± 1.22 | 0.03 ± 0.04 | 12.38 ± 3.62 | 6.17 ± 1.62 |
| Compound | Structure | IC50 (nM) |
|---|---|---|
| 28 |  | 60.2 |
| Compound | Structure | IC50 (μM) |
|---|---|---|
| 29 | 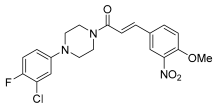 | 0.51 ± 0.10 |
| Human Cancer Cell Lines | IC50, μM |
|---|---|
| MDA-MB-231 | 0.012 |
| Caki-1 | 0.011 |
| UMRC2 | 0.013 |
| PANC-1 | 0.021 |
| A549 | 0.021 |
| MKN-45 | 0.020 |
| HepG2 | 0.019 |
| HCT116 | 0.020 |
| HT29 | 0.021 |
| PC-3 | 0.021 |
| U251 | 0.015 |
| HeLa | 0.021 |
| SK-MEL-28 | 0.020 |
| OVCAR-3 | 0.012 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreozzi, G.; Corvino, A.; Severino, B.; Magli, E.; Perissutti, E.; Frecentese, F.; Santagada, V.; Caliendo, G.; Fiorino, F. Arylpiperazine Derivatives and Cancer: A New Challenge in Medicinal Chemistry. Pharmaceuticals 2024, 17, 1320. https://doi.org/10.3390/ph17101320
Andreozzi G, Corvino A, Severino B, Magli E, Perissutti E, Frecentese F, Santagada V, Caliendo G, Fiorino F. Arylpiperazine Derivatives and Cancer: A New Challenge in Medicinal Chemistry. Pharmaceuticals. 2024; 17(10):1320. https://doi.org/10.3390/ph17101320
Chicago/Turabian StyleAndreozzi, Giorgia, Angela Corvino, Beatrice Severino, Elisa Magli, Elisa Perissutti, Francesco Frecentese, Vincenzo Santagada, Giuseppe Caliendo, and Ferdinando Fiorino. 2024. "Arylpiperazine Derivatives and Cancer: A New Challenge in Medicinal Chemistry" Pharmaceuticals 17, no. 10: 1320. https://doi.org/10.3390/ph17101320
APA StyleAndreozzi, G., Corvino, A., Severino, B., Magli, E., Perissutti, E., Frecentese, F., Santagada, V., Caliendo, G., & Fiorino, F. (2024). Arylpiperazine Derivatives and Cancer: A New Challenge in Medicinal Chemistry. Pharmaceuticals, 17(10), 1320. https://doi.org/10.3390/ph17101320