

Simultaneous Analysis of Thirteen Compounds in Yeokwisan Using High-Performance Liquid Chromatography–Photodiode Array Detection and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry and Their Antioxidant Effects


Abstract
:1. Introduction
2. Results and Discussion
2.1. Development of Simultaneous Analysis Method Using the HPLC–PDA System
2.1.1. Selection of Marker Compounds for Development of the Simultaneous Analysis Method by HPLC–PDA
2.1.2. Optimization of HPLC Conditions for Development of the Simultaneous Analysis Method
2.1.3. Method Validation of the Developed HPLC Analysis Assay
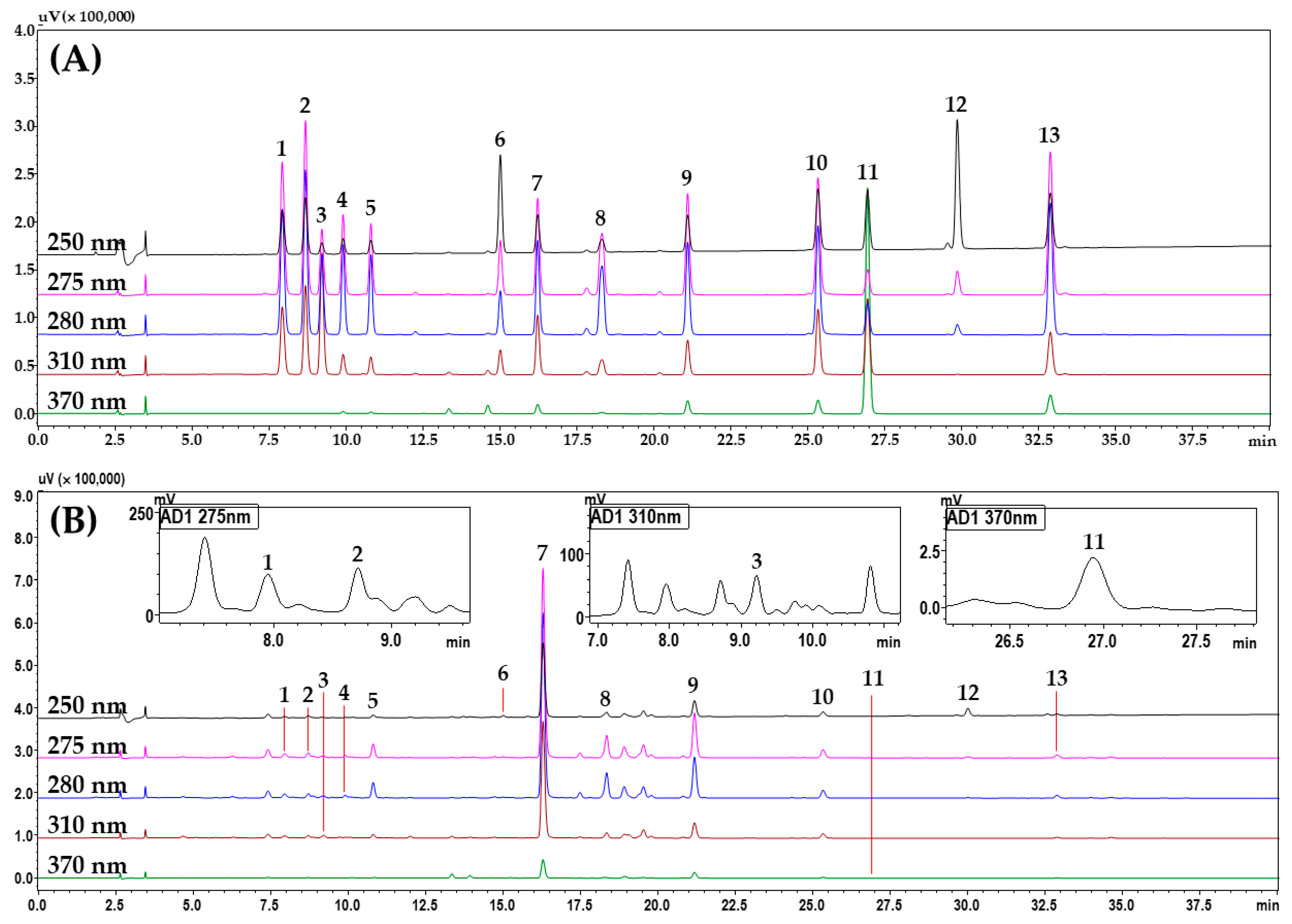
2.1.4. Simultaneous Determination of 13 Marker Compounds in a YWS Sample by the HPLC–PDA Assay
2.2. Development of Simultaneous Analysis Method Using the UPLC–MS/MS System
2.2.1. Conditions for UPLC–MS/MS Multiple Reaction Monitoring (MRM) Method for Simultaneous Analysis of the 13 Marker Compounds in a YWS Sample
2.2.2. Method Validation of the Developed UPLC–MS/MS Analysis Assay
2.2.3. Quantification of the 13 Marker Components in a YWS Sample by the Developed UPLC–MS/MS MRM Analysis Method
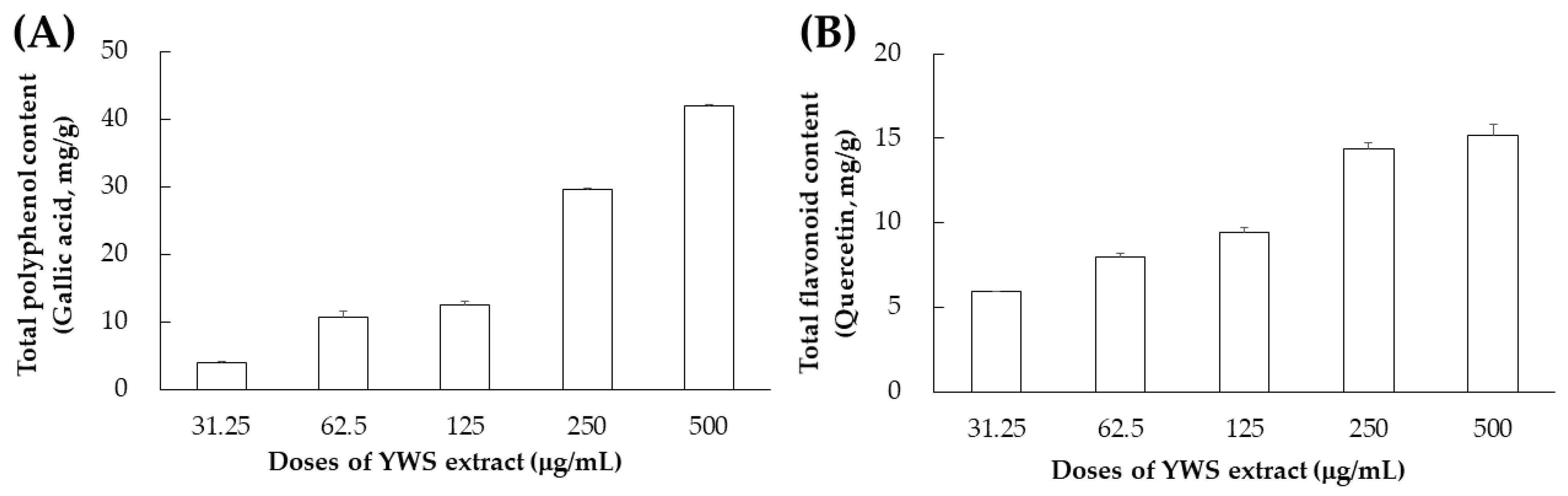
2.3. Total Polyphenol and Total Flavonoid Contents
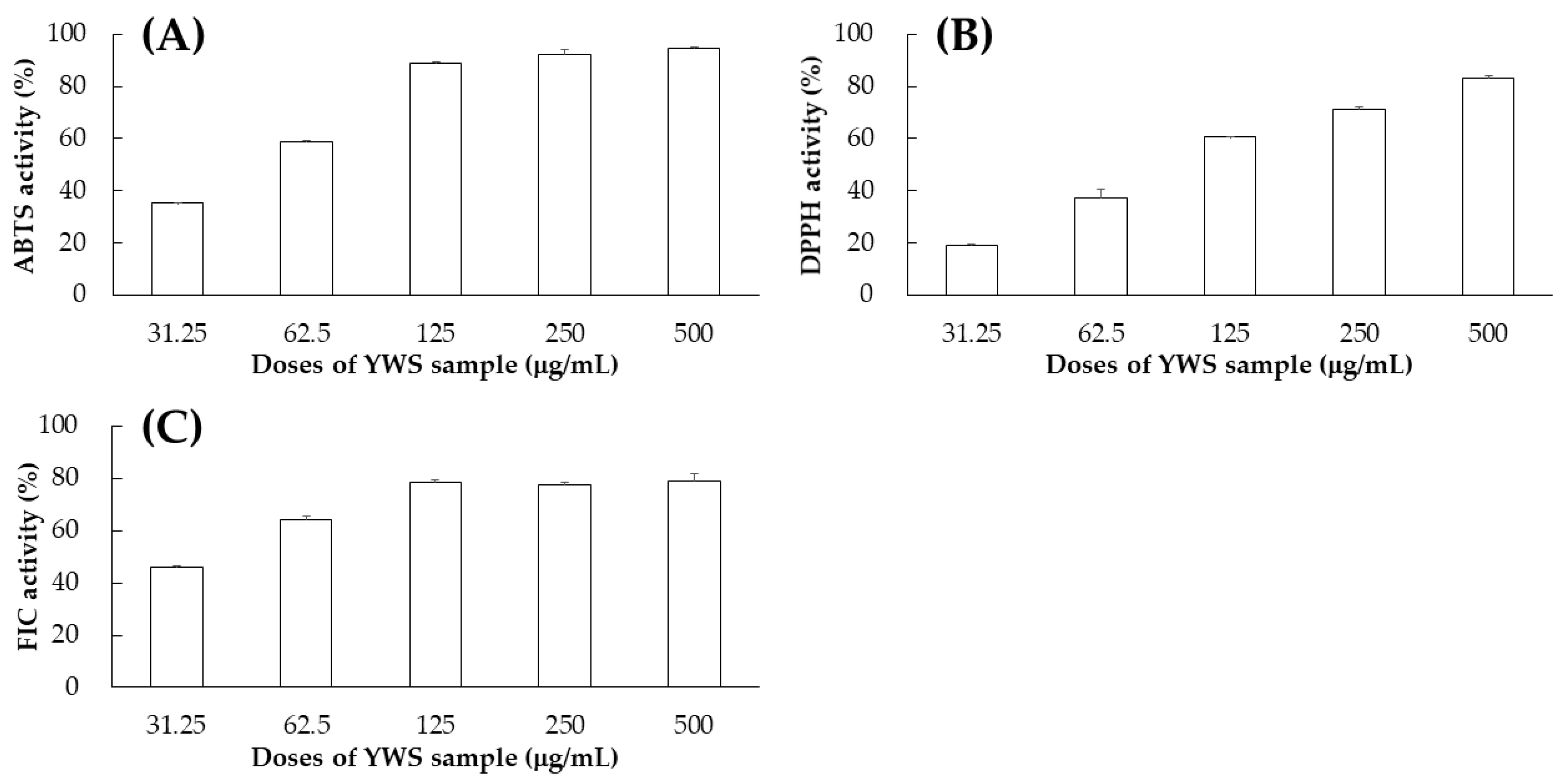
2.4. Antioxidant Effects of YWS Sample and Its Marker Compounds
3. Materials and Methods
3.1. Plant Materials
3.2. Chemicals and Reagents
3.3. Preparation of YWS Sample
3.4. Instrumentation and Analytical Conditions for Development of the HPLC–PDA Assay
3.5. Instrumentation and Analytical Conditions for Development of the UPLC–MS/MS Assay
3.6. Method Validation of Optimized Assays in HPLC–PDA and UPLC–MS/MS Systems
3.7. Total Polyphenol and Total Flavonoid Contents
3.8. Antioxidant Effects of YWS Sample and Its Marker Compounds
3.8.1. DPPH Radical Scavenging Assay
3.8.2. ABTS Activity
3.8.3. FIC Activity
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, D.; Zhang, Y.; Gao, Y.; Chai, X.; Pi, R.; Chan, G.; Hu, Y. Translating traditional herbal formulas into modern drugs: A network-based analysis of Xiaoyao decoction. Chin. Med. 2020, 15, 25. [Google Scholar] [CrossRef]
- Yang, L.; Liu, W.; Hu, Z.; Yang, M.; Li, J.; Fan, X.; Pan, H. A systems pharmacology approach for identifying the multiple mechanisms of action of the Wei Pi Xiao decoction for the treatment of gastric precancerous lesions. Evid. Based Complement. Altern. Med. 2019, 2019, 1562707. [Google Scholar] [CrossRef]
- Xue, Z.; Li, Y.; Zhou, M.; Liu, Z.; Fan, G.; Wang, X.; Zhu, Y.; Yang, J. Traditional herbal medicine discovery for the treatment and prevention of pulmonary arterial hypertension. Front. Pharmacol. 2021, 12, 720873. [Google Scholar] [CrossRef]
- Liu, S.; Yi, L.Z.; Liang, Y.Z. Traditional Chinese medicine and separation science. J. Sep. Sci. 2008, 31, 2113–2137. [Google Scholar] [CrossRef]
- Hwang, S.J.; Wang, J.H.; Lee, J.S.; Lee, H.D.; Choi, T.J.; Choi, S.H.; Son, C.G. Yeokwisan, a standardized herbal formula, enhances gastric emptying via modulation of the ghrelin pathway in a loperamide-induced functional dyspepsia mouse model. Front. Pharmacol. 2021, 22, 753153. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kim, J.Y.; Kwon, O.J.; Jung, S.Y.; Joung, J.Y.; Yang, C.S.; Lee, J.H.; Cho, J.H.; Son, C.G. Efficacy of a traditional herbal formula, Banha-sasim-tang in functional dyspepsia classified as excess pattern. Front. Pharmacol. 2021, 12, 698887. [Google Scholar] [CrossRef]
- Setia, G.; Bhavanani, A.B.; Ramanathan, M.; Ananthakrishnan, N.; Vinoth, V.; Prabu, B.S.M.; Subramanian, B. Yoga therapy in functional dyspepsia. A narrative review. J. Gastrointest. Liver Dis. 2023, 32, 513–525. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Wang, Y.; Yang, K.; Yeertai, Y.; Jia, Q.; Li, L.; Jiang, K.; Du, G.; Ling, J. Chaihu Shugan powder inhibits interstitial cells of cajal mitophagy through USP30 in the treatment of functional dyspepsia. J. Ethnopharmacol. 2024, 323, 117695. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yuan, H.; Li, Q.; Li, Y.; Geng, S.; Zhu, Y.; Liao, M.; Jiang, H. Global trends in research related to functional dyspepsia and anxiety or depression over the past two decades: A bibliometric analysis. Front. Neurosci. 2023, 17, 1218001. [Google Scholar] [CrossRef]
- Shen, X.; Xie, A.; Li, Z.; Jiang, C.; Wu, J.; Li, M.; Yue, X. Research progress for probiotics regulating intestinal flora to improve functional dyspepsia: A review. Foods 2024, 13, 151. [Google Scholar] [CrossRef]
- Sun, J.; Gou, J.; Qin, L.; Liu, T.; Huang, Y.; Lu, Y.; Wang, Y.; Liu, C.; Li, Y. Screening of anti-functional dyspepsia compounds in Cynanchum auriculatum: A spectrum-effect relationship analysis, and ATP-binding cassette transporters inhibitor evaluation. J. Ethnopharmacol. 2024, 318, 116867. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.R.; Jung, D.; Kim, S.C.; Park, J.W.; Choi, W.G.; Kim, B.J. Analysis of network pharmacological efficacy and therapeutic effectiveness in animal models for functional dyspepsia of Foeniculi fructus. Nutrients 2023, 15, 2644. [Google Scholar] [CrossRef] [PubMed]
- Aregawi, L.G.; Shokrolahi, M.; Gebremeskel, T.G.; Zoltan, C. The effect of ginger supplementation on the improvement of dyspeptic symptoms in patients with functional dyspepsia. Cureus 2023, 15, e46061. [Google Scholar] [CrossRef]
- Tu, Y.; Luo, X.; Liu, D.; Li, H.; Xia, H.; Ma, C.; Zhang, D.; Yang, Y.; Pan, X.; Wang, T.; et al. Extracts of Poria cocos improve functional dyspepsia via regulating brain–gut peptides, immunity and repairing of gastrointestinal mucosa. Phytomedicine 2022, 95, 153875. [Google Scholar] [CrossRef] [PubMed]
- Ha, N.Y.; Ko, S.J.; Park, J.W.; Kim, J. Efficacy and safety of the herbal formula Naesohwajung-tang for functional dyspepsia: A randomized, double-blind, placebo-controlled, multi-center trial. Front. Pharmacol. 2023, 14, 1157535. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Bao, Y.; Wang, S.; Li, T.; Meng, X. Rapid analysis of components in Qizhiweitong tablets and plasma after oral administration in rats by UPLC–Q–TOF–MS/MS based on a self-developed database. Biomed. Chromagtogr. 2022, 36, e5460. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Ahn, E.K.; Yang, C. Herbal medicine prescriptions for functional dyspepsia: A nationwide population-based study in Korea. Evid. Based Complement. Altern. Med. 2022, 2022, 3306420. [Google Scholar] [CrossRef]
- Bai, Y.; Zheng, M.; Fu, R.; Du, J.; Wang, J.; Zhang, M.; Fan, Y.; Huang, X.; Li, Z. Effect of Massa Medicata Fermentata on the intestinal flora of rats with functional dyspepsia. Microb. Pathog. 2023, 174, 105927. [Google Scholar] [CrossRef]
- Kim, H.; Choi, Y.; An, Y.; Jung, Y.R.; Lee, J.Y.; Lee, H.J.; Jeong, J.; Kim, Z.; Kim, K. Development of p-coumaric acid analysis in human plasma and its clinical application to PK/PD study. J. Clin. Med. 2020, 10, 108. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, E.K.; Shim, W.S. Phytotherapeutic effects of the fruits of Poncirus trifoliata (L.) Raf. on cancer, inflammation, and digestive dysfunction. Phytother. Res. 2018, 32, 616–624. [Google Scholar] [CrossRef]
- Li, H.; Wu, C.; Liu, Y.; Zhang, S.; Gao, X. Baihe Zhimu formula attenuates the efficacy of tamoxifen against breast cancer in mice through modulation of CYP450 enzymes. BMC Complement. Altern. Med. 2019, 19, 240. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.S. Simultaneous quantification of nine target compounds in traditional Korean medicine, Bopyeo-tang, using high-performance liquid chromatography–photodiode array detector and ultra-performance liquid chromatography–tandem mass spectrometry. Molecules 2024, 29, 1171. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.S.; Shin, H.K. Quantitative analysis of eight compounds in traditional Korean medicine, Gongjindan using HPLC, UPLC–MS/MS, and GC–MS/MS systems. Separations 2023, 10, 231. [Google Scholar] [CrossRef]
- Shu, Y.; Chen, Y.; Qin, K.; Liu, X.; Cai, B. A study on the chemical compositions of the Yinqiaosan (Lonicerae and Forsythiae powder) at different time of later-decoction by gas chromatography mass spectrometry. Pharmacogn. Mag. 2016, 12, 134–138. [Google Scholar] [PubMed]
- Bose, A. HPLC calibration process parameters in terms of system suitability test. Austin Chromatogr. 2014, 1, 1–4. [Google Scholar]
- Epshtein, N.A. System suitability requirements for liquid chromatography methods: Controlled parameters and their recommended values (Review). Pharm. Chem. J. 2020, 54, 518–525. [Google Scholar] [CrossRef]
- Li, Z.; Wen, R.; Du, Y.; Zhao, S.; Zhao, P.; Jiang, H.; Rong, R.; Lv, Q. Simultaneous quantification of fifteen compounds in rat plasma by LC–MS/MS and its application to a pharmacokinetic study of Chaihu-Guizhi decoction. J. Chromatogr. B 2019, 1105, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, S.; Lu, M.; Zhang, Z.; Tian, X.; Lv, D. Simultaneous determination and pharmacokinetic study of three isoflavones from Trifolium pratense extract in rat plasma by LC–MS/MS. Biomed. Chromatogr. 2015, 29, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Wan, M.; Zhang, L.; Zhu, Y.; Sun, H.; Bi, K. Simultaneous determination of baicalin, wogonoside, baicalein, wogonin, oroxylin A and chrysin of Radix Scutellariae extract in rat plasma by liquid chromatography tandem mass spectrometry. J. Pharm. Biomed. Anal. 2012, 70, 6–12. [Google Scholar] [CrossRef]
- Zhou, S.; Cao, J.; Qiu, F.; Kong, W.; Yang, S.; Yang, M. Simultaneous determination of five bioactive components in Radix Glycyrrhizae by pressurized liquid extraction combined with UPLC–PDA and UPLC/ESI–QTOF–MS confirmation. Phytochem. Anal. 2013, 24, 527–533. [Google Scholar] [CrossRef]
- Tan, G.; Zhu, Z.; Jing, J.; Lv, L.; Lou, Z.; Zhang, G.; Chai, Y. Characterization of constituents in Sini decoction and rat plasma by high-performance liquid chromatography with diode array detection coupled to time-of-flight mass spectrometry. Biomed. Chromatogr. 2011, 25, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pan, Y.; Fan, G.; Chai, Y.; Wu, Y. Application of an efficient strategy based on MAE, HPLC–DAD–MS/MS and HSCCC for the rapid extraction, identification, separation and purification of flavonoids from Fructus Aurantii Immaturus. Biomed. Chromatogr. 2010, 24, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, Z.; Yang, J.; Li, Y.; Wang, Y.; Chai, X. Chemical material basis study of Xuefu Zhuyu decoction by ultra-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. J. Food Drug Anal. 2015, 23, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yue, Y.D.; Tang, F.; Sun, J. Screening and analysis of the potential bioactive components in rabbit plasma after oral administration of hot-water extracts from leaves of Bambusa textilis McClure. Molecules 2012, 17, 8872–8885. [Google Scholar] [CrossRef] [PubMed]
- Perron, N.R.; Brumaghim, J.L. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem. Biophys. 2009, 53, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Dragsted, L.O. Antioxidant actions of polyphenols in humans. Int. J. Vitam. Nutr. Res. 2003, 73, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Vijaya, K.; Ananthan, S.; Nalini, R. Antibacterial effect of theaflavin, polyphenon 60 (Camellia sinensis) and Euphorbia hirta on Shigella spp.—A cell culture study. J. Ethnopharmacol. 1995, 49, 115–118. [Google Scholar] [CrossRef] [PubMed]
- WFO Plant List. Available online: http://www.wfoplantlist.org (accessed on 1 May 2024).
- Folin, O.; Dennis, W. On phosphotungstic-phosphomolybdic compounds as color reagents. J. Biol. Chem. 1912, 12, 239–243. [Google Scholar] [CrossRef]
- Moreno, M.I.; Isla, M.I.; Sampietro, A.R.; Vattuone, M.A. Comparison of the free radical-scavenging activity of propolis from several regions of Argentina. J. Ethnopharmacol. 2000, 71, 109–114. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant determinations by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte 1 | Detected Wavelength (nm) | Linear Range (μg/mL) | r2 | LOD (μg/mL) | LOQ (μg/mL) | |
|---|---|---|---|---|---|---|
| 1 | 275 | 0.31–20.00 | y = 18,211.69x + 1008.30 | 1.0000 | 0.04 | 0.11 |
| 2 | 275 | 0.31–20.00 | y = 28,044.42x + 1604.28 | 1.0000 | 0.01 | 0.04 |
| 3 | 310 | 0.31–20.00 | y = 100,148.87x + 5544.60 | 1.0000 | 0.03 | 0.08 |
| 4 | 280 | 0.78–50.00 | y = 28,290.54x + 4655.94 | 0.9999 | 0.06 | 0.17 |
| 5 | 280 | 0.47–30.00 | y = 18,281.87x + 1530.74 | 1.0000 | 0.03 | 0.09 |
| 6 | 250 | 0.47–30.00 | y = 36,397.22x + 2960.46 | 1.0000 | 0.04 | 0.12 |
| 7 | 275 | 2.34–150.00 | y = 36,882.40x + 12,832.74 | 1.0000 | 0.28 | 0.85 |
| 8 | 280 | 0.78–50.00 | y = 18,071.54x + 2503.17 | 1.0000 | 0.07 | 0.21 |
| 9 | 275 | 0.47–30.00 | y = 58,815.90x + 4399.17 | 1.0000 | 0.04 | 0.12 |
| 10 | 275 | 0.78–50.00 | y = 65,548.02x + 83,066.84 | 0.9999 | 0.09 | 0.28 |
| 11 | 370 | 0.31–20.00 | y = 73,467.19x + 3382.49 | 1.0000 | 0.02 | 0.08 |
| 12 | 250 | 0.78–50.00 | y = 8533.07x + 731.10 | 1.0000 | 0.04 | 0.13 |
| 13 | 275 | 0.31–20.00 | y = 84,077.35x + 4664.54 | 1.0000 | 0.02 | 0.08 |
| Analyte 1 | Original Amount (μg/mL) | Spiked Amount (μg/mL) | Found Amount (μg/mL) | Recovery (%) | SD 2 | RSD 3 (%) |
|---|---|---|---|---|---|---|
| 1 | 6.93 | 1.00 | 7.96 | 103.62 | 2.02 | 1.95 |
| 3.00 | 9.87 | 98.24 | 0.79 | 0.81 | ||
| 6.00 | 12.73 | 96.68 | 0.60 | 0.62 | ||
| 2 | 4.13 | 1.00 | 5.13 | 99.49 | 2.51 | 2.53 |
| 2.00 | 6.14 | 100.56 | 2.21 | 2.19 | ||
| 4.00 | 8.10 | 99.21 | 1.33 | 1.35 | ||
| 3 | 6.44 | 1.00 | 7.45 | 101.14 | 2.66 | 2.63 |
| 3.00 | 9.44 | 99.81 | 1.06 | 1.06 | ||
| 6.00 | 12.20 | 96.01 | 0.33 | 0.34 | ||
| 4 | 2.31 | 1.00 | 3.33 | 101.95 | 2.00 | 1.96 |
| 2.00 | 4.42 | 105.31 | 0.71 | 0.68 | ||
| 4.00 | 6.43 | 102.94 | 0.39 | 0.38 | ||
| 5 | 18.44 | 3.00 | 21.47 | 101.06 | 2.16 | 2.14 |
| 7.50 | 25.59 | 95.26 | 0.91 | 0.95 | ||
| 15.00 | 32.89 | 96.29 | 0.19 | 0.19 | ||
| 6 | 0.94 | 1.00 | 1.91 | 97.50 | 0.86 | 0.88 |
| 2.00 | 3.05 | 105.52 | 0.97 | 0.92 | ||
| 4.00 | 5.09 | 103.83 | 0.54 | 0.52 | ||
| 7 | 8.94 | 2.00 | 11.02 | 103.86 | 2.37 | 2.29 |
| 5.00 | 13.96 | 100.22 | 0.47 | 0.47 | ||
| 10.00 | 18.67 | 97.22 | 0.49 | 0.50 | ||
| 8 | 31.81 | 6.00 | 37.83 | 100.39 | 0.76 | 0.75 |
| 15.00 | 46.54 | 98.20 | 0.34 | 0.34 | ||
| 30.00 | 61.08 | 97.58 | 0.18 | 0.19 | ||
| 9 | 15.29 | 3.00 | 18.25 | 98.71 | 0.43 | 0.44 |
| 7.50 | 22.43 | 95.20 | 0.06 | 0.06 | ||
| 15.00 | 29.76 | 96.47 | 0.13 | 0.14 | ||
| 10 | 26.22 | 4.00 | 30.19 | 99.20 | 2.62 | 2.64 |
| 10.00 | 36.10 | 98.81 | 2.07 | 2.09 | ||
| 20.00 | 47.31 | 105.46 | 1.07 | 1.02 | ||
| 11 | 0.28 | 1.00 | 1.30 | 102.26 | 0.24 | 0.23 |
| 2.00 | 2.39 | 105.38 | 0.23 | 0.22 | ||
| 4.00 | 4.45 | 104.35 | 0.07 | 0.07 | ||
| 12 | 19.48 | 4.00 | 23.53 | 101.15 | 0.89 | 0.88 |
| 10.00 | 29.13 | 96.50 | 0.20 | 0.20 | ||
| 20.00 | 39.33 | 99.25 | 0.33 | 0.33 | ||
| 13 | 9.24 | 2.00 | 11.27 | 101.34 | 1.81 | 1.78 |
| 5.00 | 14.33 | 101.69 | 1.12 | 1.10 | ||
| 10.00 | 19.24 | 100.00 | 0.17 | 0.17 |
| Analyte 1 | Conc. (μg/mL) | Intraday | Interday | ||||
|---|---|---|---|---|---|---|---|
| Observed Conc. (μg/mL) | Precision (RSD, %) | Accuracy (%) | Observed Conc. (μg/mL) | Precision (RSD, %) | Accuracy (%) | ||
| 1 | 5.0 | 5.11 | 0.50 | 102.27 | 5.21 | 1.79 | 104.23 |
| 10.0 | 10.10 | 0.21 | 101.01 | 10.23 | 1.17 | 102.26 | |
| 20.0 | 20.13 | 0.52 | 100.66 | 20.51 | 1.89 | 102.56 | |
| 2 | 5.0 | 5.11 | 0.42 | 102.28 | 5.21 | 1.80 | 104.26 |
| 10.0 | 10.09 | 0.22 | 100.94 | 10.22 | 1.16 | 102.20 | |
| 20.0 | 20.11 | 0.57 | 100.57 | 20.49 | 1.87 | 102.44 | |
| 3 | 5.0 | 5.11 | 0.43 | 102.17 | 5.20 | 1.77 | 104.09 |
| 10.0 | 10.09 | 0.23 | 100.94 | 10.22 | 1.16 | 102.17 | |
| 20.0 | 20.09 | 0.58 | 100.47 | 20.47 | 1.84 | 102.34 | |
| 4 | 12.5 | 12.81 | 0.52 | 102.46 | 13.09 | 2.01 | 104.72 |
| 25.0 | 25.27 | 0.23 | 101.06 | 25.79 | 0.86 | 103.17 | |
| 50.0 | 50.28 | 0.66 | 100.56 | 51.27 | 1.97 | 102.54 | |
| 5 | 7.5 | 7.68 | 0.44 | 102.34 | 7.82 | 1.76 | 104.32 |
| 15.0 | 15.15 | 0.26 | 101.01 | 15.34 | 1.15 | 102.29 | |
| 30.0 | 30.17 | 0.59 | 100.58 | 30.74 | 1.86 | 102.48 | |
| 6 | 7.5 | 7.66 | 0.43 | 102.20 | 7.81 | 1.75 | 104.11 |
| 15.0 | 15.14 | 0.26 | 100.95 | 15.33 | 1.17 | 102.20 | |
| 30.0 | 30.15 | 0.64 | 100.51 | 30.71 | 1.84 | 102.36 | |
| 7 | 37.5 | 38.34 | 0.41 | 102.24 | 39.05 | 1.74 | 104.12 |
| 75.0 | 75.64 | 0.24 | 100.86 | 76.57 | 1.16 | 102.09 | |
| 150.0 | 150.76 | 0.63 | 100.51 | 153.51 | 1.82 | 102.34 | |
| 8 | 5.0 | 5.11 | 0.50 | 102.27 | 5.21 | 1.92 | 104.27 |
| 12.5 | 12.77 | 0.46 | 102.16 | 13.02 | 1.34 | 102.24 | |
| 25.0 | 25.23 | 0.26 | 100.92 | 25.57 | 1.95 | 102.37 | |
| 9 | 50.0 | 50.34 | 0.65 | 100.68 | 51.29 | 1.79 | 104.12 |
| 7.5 | 7.66 | 0.46 | 102.14 | 7.80 | 1.29 | 102.27 | |
| 15.0 | 15.12 | 0.25 | 100.81 | 15.31 | 1.92 | 102.58 | |
| 10 | 30.0 | 30.18 | 0.64 | 100.59 | 30.73 | 1.75 | 104.03 |
| 12.5 | 12.86 | 0.86 | 102.91 | 13.09 | 1.19 | 102.06 | |
| 25.0 | 25.34 | 0.24 | 101.36 | 25.64 | 1.83 | 102.44 | |
| 11 | 50.0 | 50.41 | 0.56 | 100.82 | 51.28 | 1.74 | 104.74 |
| 5.0 | 5.10 | 0.48 | 102.06 | 5.20 | 1.12 | 102.55 | |
| 10.0 | 10.08 | 0.24 | 100.79 | 10.21 | 1.74 | 102.56 | |
| 12 | 20.0 | 20.10 | 0.64 | 100.52 | 20.48 | 1.78 | 103.98 |
| 12.5 | 12.78 | 0.88 | 102.23 | 12.99 | 1.20 | 102.09 | |
| 25.0 | 25.11 | 0.32 | 100.43 | 25.43 | 1.85 | 102.39 | |
| 13 | 50.0 | 49.97 | 0.69 | 99.93 | 50.91 | 1.71 | 103.93 |
| 5.0 | 5.11 | 0.42 | 102.13 | 5.20 | 1.20 | 101.72 | |
| 10.0 | 10.10 | 0.27 | 101.00 | 10.22 | 1.89 | 101.83 | |
| Analyte 1 | HPLC–PDA Assay | UPLC–MS/MS MRM Assay | Source 2 | ||||
|---|---|---|---|---|---|---|---|
| Mean (mg/g) | SD | RSD (%) | Mean (mg/g) | SD | RSD (%) | ||
| 1 | 5.94 | 0.06 | 1.06 | 5.93 | 0.01 | 0.13 | GRR |
| 2 | 3.89 | 0.03 | 0.74 | 0.65 | 0.01 | 1.02 | GRR |
| 3 | 0.62 | 0.01 × 10−2 | 0.01 | 0.57 | 0.03 | 4.98 | PCT |
| 4 | 2.23 | 0.01 | 0.29 | 2.52 | 0.11 | 4.33 | PFI |
| 5 | 15.02 | 0.12 | 0.80 | 14.67 | 1.09 | 7.42 | PFI |
| 6 | 1.29 | 0.01 × 10−1 | 0.08 | 0.74 | 0.02 | 2.93 | GRR |
| 7 | 99.03 | 0.21 | 0.21 | 100.36 | 1.96 | 1.96 | SR |
| 8 | 27.32 | 0.05 | 0.19 | 6.94 | 0.17 | 2.41 | PFI |
| 9 | 15.96 | 0.04 | 0.23 | 19.90 | 0.19 | 0.93 | SR |
| 10 | 1.98 | 0.03 × 10−1 | 0.15 | 2.74 | 0.04 × 10−2 | 1.53 | SR |
| 11 | 0.02 | 0.02 × 10−2 | 0.67 | 0.02 | 0.04 | 1.82 | GRR |
| 12 | 18.31 | 0.10 | 0.55 | 19.35 | 0.33 | 1.72 | GRR |
| 13 | 0.95 | 0.03 × 10−1 | 0.27 | 1.20 | 0.01 | 1.11 | SR |
| Analyte 1 | Ion Mode | Exact Mass | MRM Transition | Cone Voltage (V) | Collision Energy (eV) | |
|---|---|---|---|---|---|---|
| Precursor Ion | Production Ion | |||||
| 1 | − | 550.17 | 549.01 | 255.02 | 78 | 30 |
| 2 | − | 418.13 | 416.97 | 254.99 | 52 | 18 |
| 3 | − | 164.05 | 162.89 | 119.39 | 36 | 15 |
| 4 | + | 580.18 | 581.02 | 273.04 | 40 | 24 |
| 5 | + | 580.18 | 581.02 | 273.04 | 40 | 20 |
| 6 | + | 430.13 | 430.97 | 269.04 | 42 | 20 |
| 7 | + | 446.08 | 447.00 | 271.00 | 26 | 18 |
| 8 | + | 592.18 | 595.03 | 287.03 | 32 | 18 |
| 9 | + | 460.10 | 461.00 | 285.03 | 42 | 18 |
| 10 | + | 284.07 | 270.95 | 123.00 | 80 | 32 |
| 11 | + | 270.05 | 256.91 | 137.01 | 36 | 18 |
| 12 | + | 256.07 | 823.24 | 453.18 | 56 | 26 |
| 13 | + | 822.40 | 284.97 | 269.97 | 54 | 22 |
| Analyte 1 | Retention Time (min) | Linear Range (μg/L) | r2 | LOD (μg/L) | LOQ (μg/L) | |
|---|---|---|---|---|---|---|
| 1 | 3.80 | 145.00–2320.00 | y = 117.84x − 98.42 | 0.9998 | 0.39 | 1.18 |
| 2 | 4.00 | 17.50–280.00 | y = 188.23x − 336.34 | 0.9991 | 0.03×10−1 | 0.01 |
| 3 | 4.20 | 18.75–300.00 | y = 31.85x − 11.87 | 0.9982 | 4.60 | 13.81 |
| 4 | 4.26 | 65.00–1040.00 | y = 167.05x − 798.50 | 0.9977 | 0.29 | 0.87 |
| 5 | 4.47 | 300.00–4800.00 | y = 73.74x − 2306.46 | 0.998 | 2.47 | 7.40 |
| 6 | 5.40 | 17.50–280.00 | y = 5342.60x + 2610.58 | 0.9993 | 0.08 | 0.23 |
| 7 | 5.83 | 1750.00–28,000.00 | y = 1920.04x + 1.27 × 106 | 0.9965 | 0.80 | 2.39 |
| 8 | 5.89 | 67.50–1080.00 | y = 222.63x + 7550.99 | 0.9971 | 0.19 | 0.57 |
| 9 | 6.92 | 475.00–7600.00 | y = 3788.22x + 242,543.00 | 0.9991 | 0.01 | 0.02 |
| 10 | 7.92 | 87.50–1400.00 | y = 1201.74x − 15,570.10 | 0.9989 | 0.27 | 0.81 |
| 11 | 8.43 | 0.75–12.00 | y = 3636.80x − 160.47 | 0.9994 | 0.08 | 0.23 |
| 12 | 8.73 | 500.00–8000.00 | y = 220.35x + 22,317.90 | 0.9972 | 3.50 | 10.51 |
| 13 | 9.39 | 32.50–520.00 | y = 4444.41x + 17,728.90 | 0.9985 | 0.07 | 0.20 |
| Analyte 1 | Spiked Amount (μg/L) | Found Amount (μg/L) | Recovery (%) | SD | RSD (%) |
|---|---|---|---|---|---|
| 1 | 120.00 | 117.68 | 98.07 | 8.07 | 1.14 |
| 300.00 | 287.17 | 95.72 | 10.57 | 1.20 | |
| 600.00 | 638.11 | 106.35 | 114.80 | 9.33 | |
| 2 | 14.00 | 13.15 | 93.93 | 1.48 | 1.90 |
| 35.00 | 33.46 | 95.60 | 1.26 | 1.28 | |
| 70.00 | 67.61 | 96.59 | 1.41 | 1.06 | |
| 3 | 14.00 | 15.15 | 108.21 | 4.46 | 6.16 |
| 35.00 | 36.80 | 105.14 | 2.09 | 2.22 | |
| 70.00 | 70.65 | 100.93 | 2.45 | 1.92 | |
| 4 | 50.00 | 43.83 | 87.66 | 25.13 | 8.45 |
| 125.00 | 101.39 | 81.11 | 30.64 | 8.63 | |
| 250.00 | 253.36 | 101.34 | 44.33 | 8.74 | |
| 5 | 200.00 | 206.45 | 103.23 | 41.90 | 2.52 |
| 500.00 | 447.71 | 89.54 | 88.87 | 4.67 | |
| 1000.00 | 1080.73 | 108.07 | 54.58 | 2.15 | |
| 6 | 14.00 | 15.11 | 107.93 | 3.79 | 4.25 |
| 35.00 | 35.52 | 101.49 | 4.52 | 4.13 | |
| 70.00 | 72.06 | 102.94 | 4.12 | 2.82 | |
| 7 | 1400.00 | 1506.24 | 107.59 | 320.34 | 2.77 |
| 3500.00 | 3526.71 | 100.76 | 474.11 | 3.49 | |
| 7000.00 | 6826.44 | 97.52 | 718.95 | 4.26 | |
| 8 | 60.00 | 61.41 | 102.35 | 35.12 | 4.64 |
| 150.00 | 156.29 | 104.19 | 47.12 | 5.54 | |
| 300.00 | 251.27 | 83.76 | 88.21 | 9.32 | |
| 9 | 400.00 | 385.35 | 96.34 | 33.51 | 1.41 |
| 1000.00 | 936.03 | 93.60 | 52.04 | 1.78 | |
| 2000.00 | 1880.40 | 94.02 | 38.95 | 1.01 | |
| 10 | 80.00 | 86.27 | 107.84 | 9.30 | 2.58 |
| 200.00 | 188.89 | 94.45 | 12.71 | 2.75 | |
| 400.00 | 370.11 | 92.53 | 9.77 | 1.52 | |
| 11 | 0.60 | 0.62 | 103.33 | 0.03 | 0.83 |
| 1.50 | 1.49 | 99.33 | 0.09 | 2.39 | |
| 3.00 | 2.92 | 97.33 | 0.06 | 1.04 | |
| 12 | 400.00 | 402.46 | 100.62 | 137.30 | 5.87 |
| 1000.00 | 1058.19 | 105.82 | 203.91 | 6.81 | |
| 2000.00 | 2086.39 | 104.32 | 203.49 | 5.06 | |
| 13 | 26.00 | 26.40 | 101.54 | 2.05 | 1.40 |
| 65.00 | 64.93 | 99.89 | 4.24 | 2.29 | |
| 130.00 | 128.92 | 99.17 | 4.45 | 1.78 |
| Analyte 1 | Conc. (μg/L) | Intraday | Interday | ||||
|---|---|---|---|---|---|---|---|
| Observed Conc. (μg/L) | Precision (RSD, %) | Accuracy (%) | Observed Conc. (μg/L) | Precision (RSD, %) | Accuracy (%) | ||
| 1 | 145.00 | 144.49 | 2.06 | 99.65 | 146.51 | 1.37 | 101.04 |
| 580.00 | 583.88 | 1.94 | 100.67 | 582.12 | 1.34 | 100.37 | |
| 2320.00 | 2313.55 | 0.63 | 99.72 | 2330.49 | 0.63 | 100.45 | |
| 2 | 17.50 | 17.55 | 1.33 | 100.29 | 17.72 | 1.34 | 101.24 |
| 70.00 | 70.40 | 1.51 | 100.58 | 70.23 | 1.93 | 100.33 | |
| 280.00 | 278.51 | 0.92 | 99.47 | 279.65 | 0.39 | 99.88 | |
| 3 | 18.75 | 18.35 | 3.93 | 97.85 | 18.19 | 4.65 | 97.03 |
| 75.00 | 76.06 | 2.84 | 101.41 | 73.46 | 3.34 | 97.94 | |
| 300.00 | 297.32 | 1.68 | 99.11 | 300.84 | 1.33 | 100.28 | |
| 4 | 65.00 | 61.21 | 5.11 | 94.17 | 63.52 | 4.68 | 97.73 |
| 260.00 | 277.62 | 7.50 | 106.78 | 269.33 | 6.26 | 103.59 | |
| 1040.00 | 1017.95 | 0.81 | 97.88 | 1027.21 | 2.32 | 98.77 | |
| 5 | 300.00 | 297.86 | 5.16 | 99.29 | 302.65 | 2.02 | 100.88 |
| 1200.00 | 1237.03 | 6.42 | 103.09 | 1216.11 | 4.04 | 101.34 | |
| 4800.00 | 4818.62 | 1.66 | 100.39 | 4907.27 | 1.99 | 102.23 | |
| 6 | 17.50 | 16.77 | 1.07 | 95.81 | 16.78 | 0.59 | 95.87 |
| 70.00 | 72.02 | 1.17 | 102.89 | 72.15 | 0.15 | 103.06 | |
| 280.00 | 275.91 | 0.51 | 98.54 | 279.81 | 2.06 | 99.93 | |
| 7 | 1750.00 | 1601.09 | 5.55 | 91.49 | 1603.95 | 5.54 | 91.65 |
| 7000.00 | 7481.61 | 2.87 | 106.88 | 7542.56 | 0.76 | 107.75 | |
| 28000.00 | 26816.89 | 0.88 | 95.77 | 26707.39 | 0.71 | 95.38 | |
| 8 | 67.50 | 61.97 | 6.33 | 91.81 | 61.14 | 3.04 | 90.57 |
| 270.00 | 291.57 | 7.12 | 107.99 | 289.07 | 4.43 | 107.06 | |
| 1080.00 | 1041.33 | 2.77 | 96.42 | 1051.48 | 1.00 | 97.36 | |
| 9 | 475.00 | 453.76 | 1.24 | 95.53 | 458.77 | 1.43 | 96.58 |
| 1900.00 | 1955.38 | 1.25 | 102.91 | 1924.62 | 1.50 | 101.30 | |
| 7600.00 | 7500.00 | 0.32 | 98.68 | 7546.88 | 0.55 | 99.30 | |
| 10 | 87.50 | 91.19 | 1.71 | 104.21 | 92.36 | 1.41 | 105.55 |
| 350.00 | 338.49 | 2.97 | 96.71 | 332.45 | 1.93 | 94.99 | |
| 1400.00 | 1401.20 | 2.05 | 100.09 | 1416.33 | 2.60 | 101.17 | |
| 11 | 0.75 | 0.74 | 2.07 | 98.22 | 0.75 | 0.74 | 99.51 |
| 3.00 | 3.01 | 0.51 | 100.22 | 2.98 | 0.67 | 99.33 | |
| 12.00 | 11.94 | 0.63 | 99.47 | 12.01 | 0.49 | 100.05 | |
| 12 | 500.00 | 461.99 | 5.44 | 92.40 | 485.37 | 5.25 | 97.07 |
| 2000.00 | 1976.57 | 7.17 | 98.83 | 2000.69 | 1.20 | 100.03 | |
| 8000.00 | 7780.40 | 4.44 | 97.26 | 7828.61 | 2.40 | 97.86 | |
| 13 | 32.50 | 30.71 | 1.48 | 94.49 | 30.60 | 1.46 | 94.16 |
| 130.00 | 134.47 | 0.58 | 103.44 | 133.63 | 0.57 | 102.79 | |
| 520.00 | 511.09 | 0.45 | 98.29 | 511.99 | 0.24 | 98.46 | |
| Analyte 1 | IC50 2 | ||
|---|---|---|---|
| DPPH | ABTS | FIC | |
| 1 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 |
| 3 | 79.27 ± 3.86 | 49.51 ± 0.72 | >100 |
| 4 | >100 | >100 | >100 |
| 5 | >100 | >100 | >100 |
| 6 | >100 | >100 | >100 |
| 7 | 48.91 ± 2.37 | 33.03 ± 0.91 | 58.2 ± 0.3 |
| 8 | >100 | >100 | >100 |
| 9 | >100 | >100 | >100 |
| 10 | 14.17 ± 2.17 | 18.53 ± 0.6 | 31.3 ± 2.5 |
| 11 | >100 | >100 | >100 |
| 12 | >100 | >100 | >100 |
| 13 | >100 | >100 | >100 |
| Ascorbic acid 3 | 5.24 ± 1.89 | 3.95 ± 1.26 | - |
| EDTA 4 | - | - | 115.69 ± 1.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, C.-S.; Kim, S.-Y.; Kim, D.-S. Simultaneous Analysis of Thirteen Compounds in Yeokwisan Using High-Performance Liquid Chromatography–Photodiode Array Detection and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry and Their Antioxidant Effects. Pharmaceuticals 2024, 17, 727. https://doi.org/10.3390/ph17060727
Seo C-S, Kim S-Y, Kim D-S. Simultaneous Analysis of Thirteen Compounds in Yeokwisan Using High-Performance Liquid Chromatography–Photodiode Array Detection and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry and Their Antioxidant Effects. Pharmaceuticals. 2024; 17(6):727. https://doi.org/10.3390/ph17060727
Chicago/Turabian StyleSeo, Chang-Seob, So-Yeon Kim, and Dong-Seon Kim. 2024. "Simultaneous Analysis of Thirteen Compounds in Yeokwisan Using High-Performance Liquid Chromatography–Photodiode Array Detection and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry and Their Antioxidant Effects" Pharmaceuticals 17, no. 6: 727. https://doi.org/10.3390/ph17060727
APA StyleSeo, C.-S., Kim, S.-Y., & Kim, D.-S. (2024). Simultaneous Analysis of Thirteen Compounds in Yeokwisan Using High-Performance Liquid Chromatography–Photodiode Array Detection and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry and Their Antioxidant Effects. Pharmaceuticals, 17(6), 727. https://doi.org/10.3390/ph17060727