
Unveiling the Mechanisms Underlying the Immunotherapeutic Potential of Gene–miRNA and Drugs in Head and Neck Cancer

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
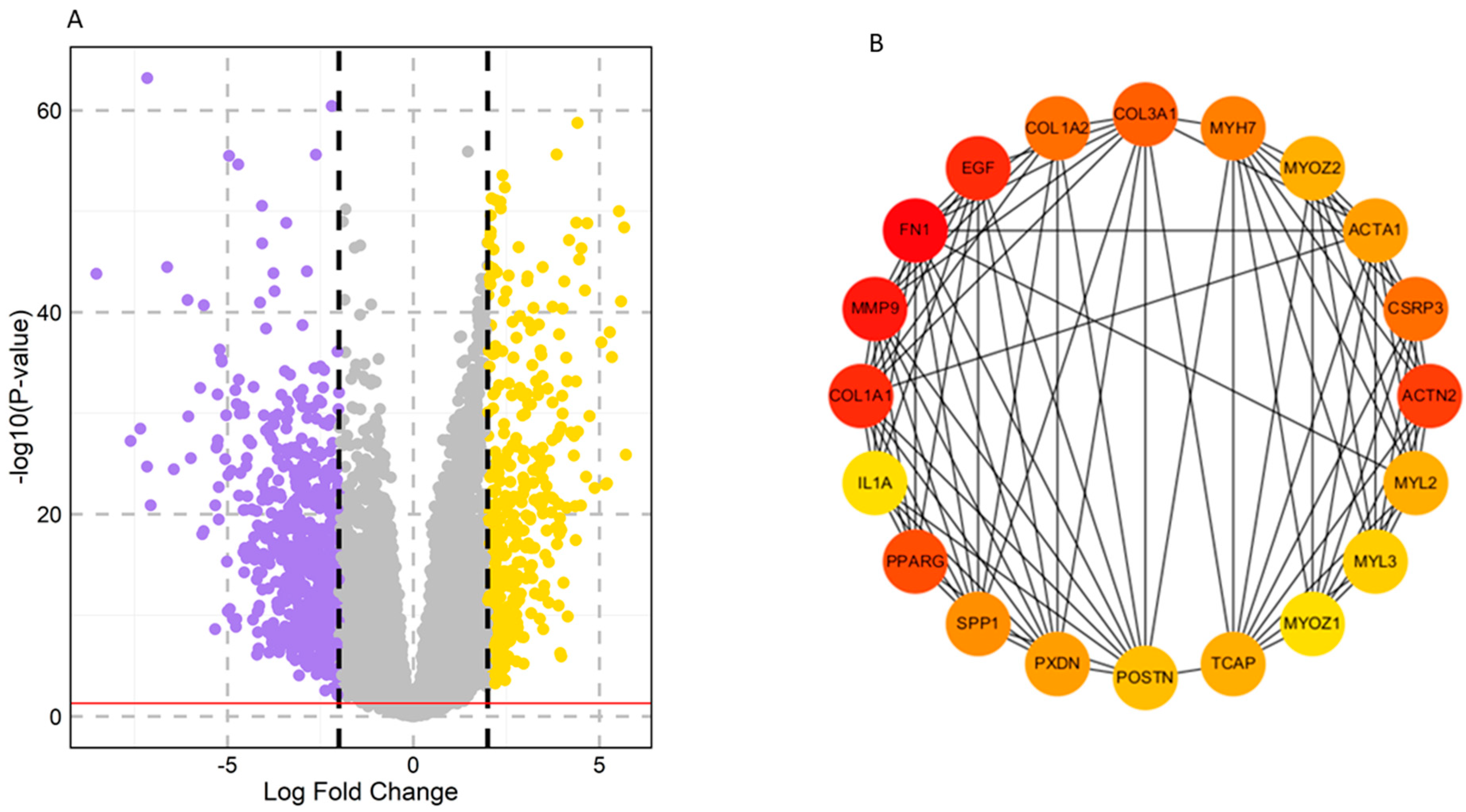
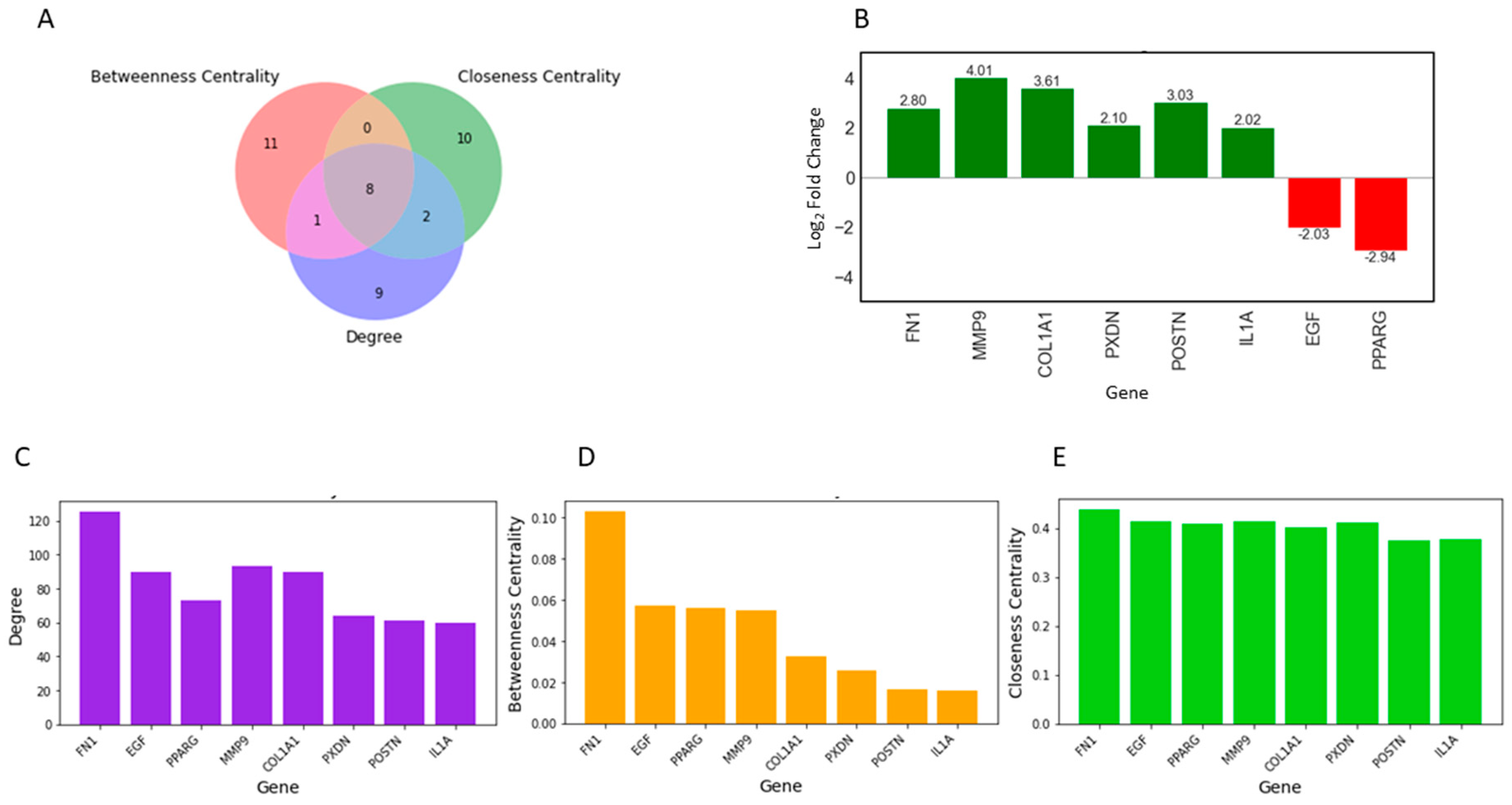
2.1. Identification of Key Regulators or Hub Genes
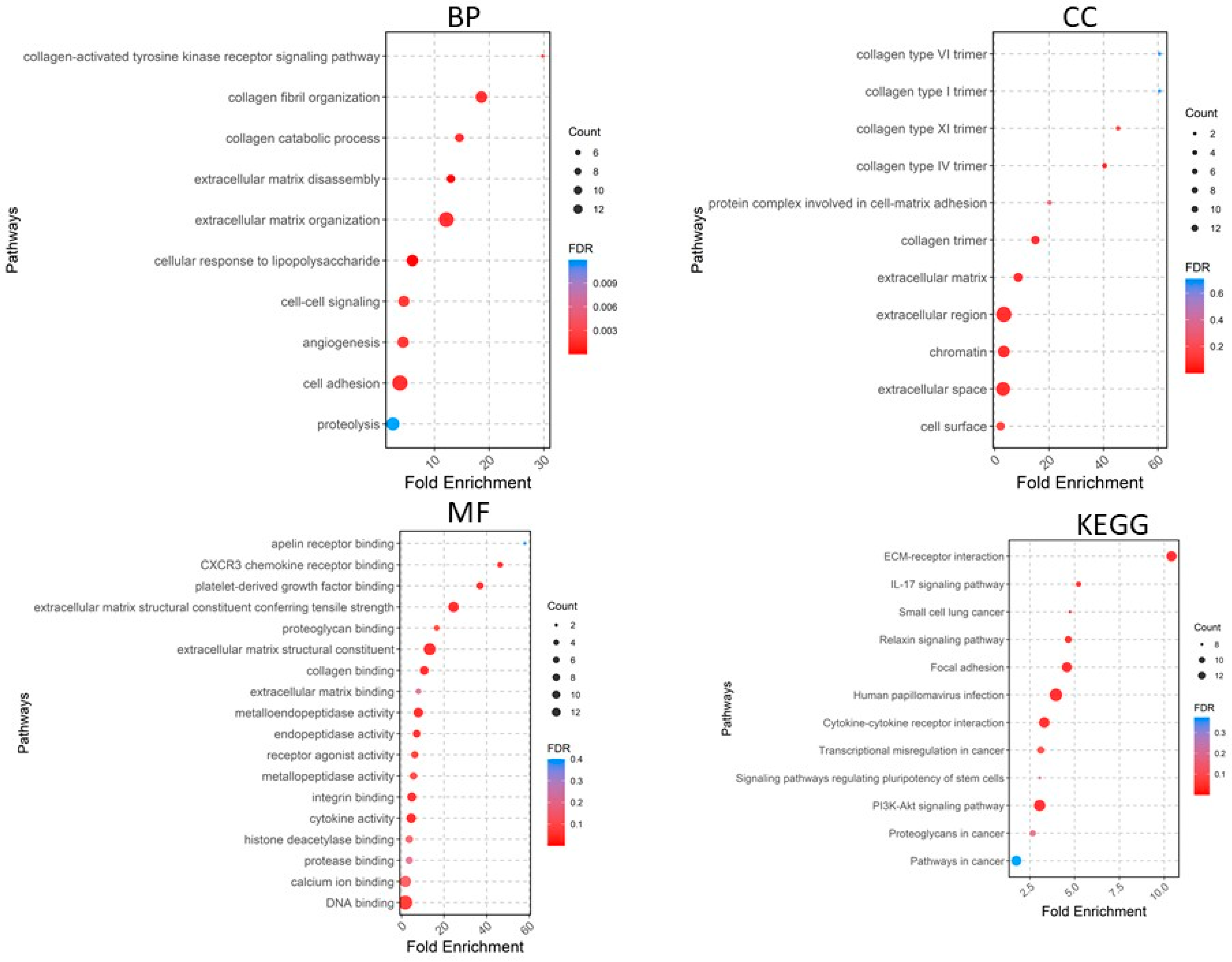
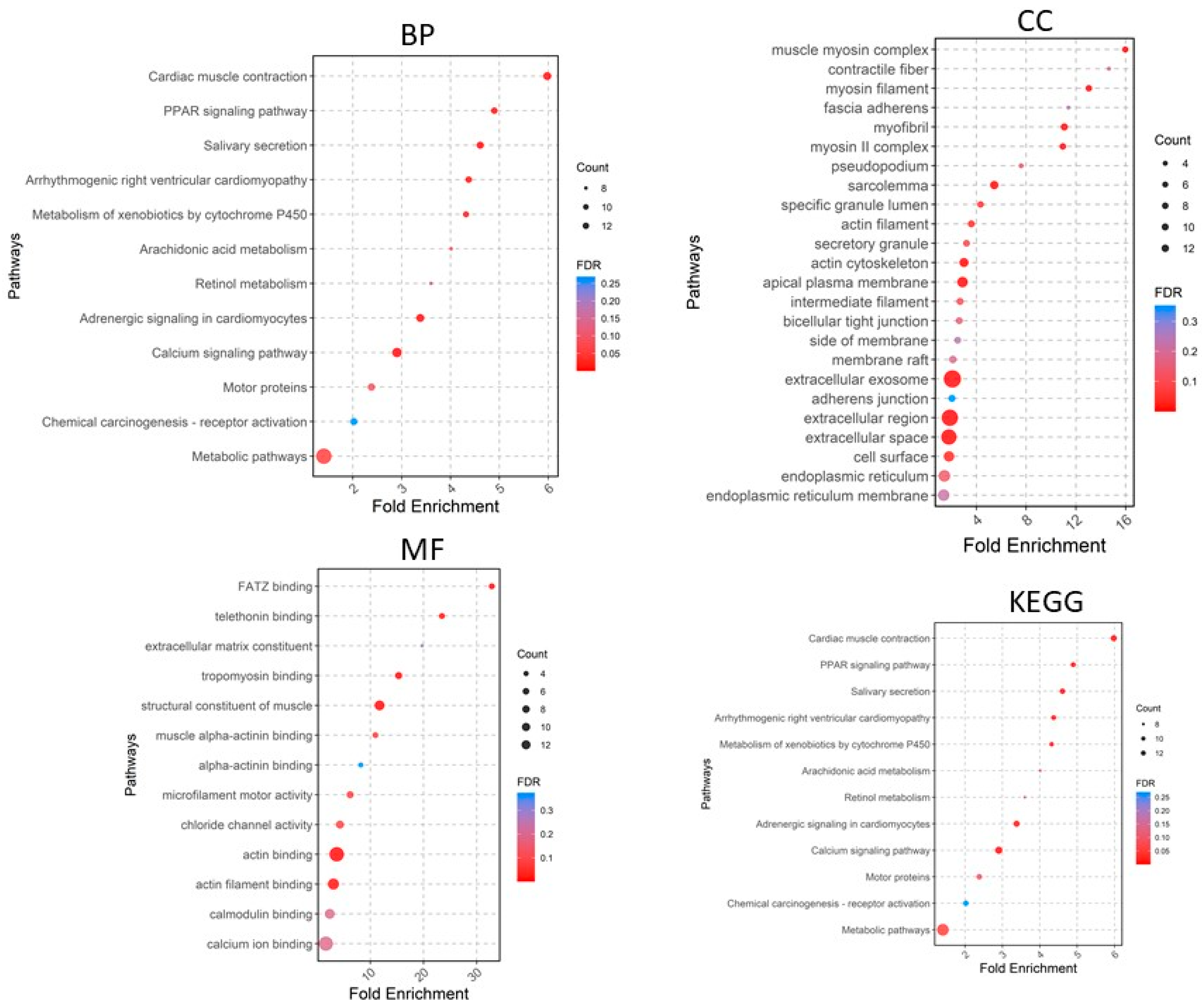
2.2. Functional Enrichment Analysis of DEGs
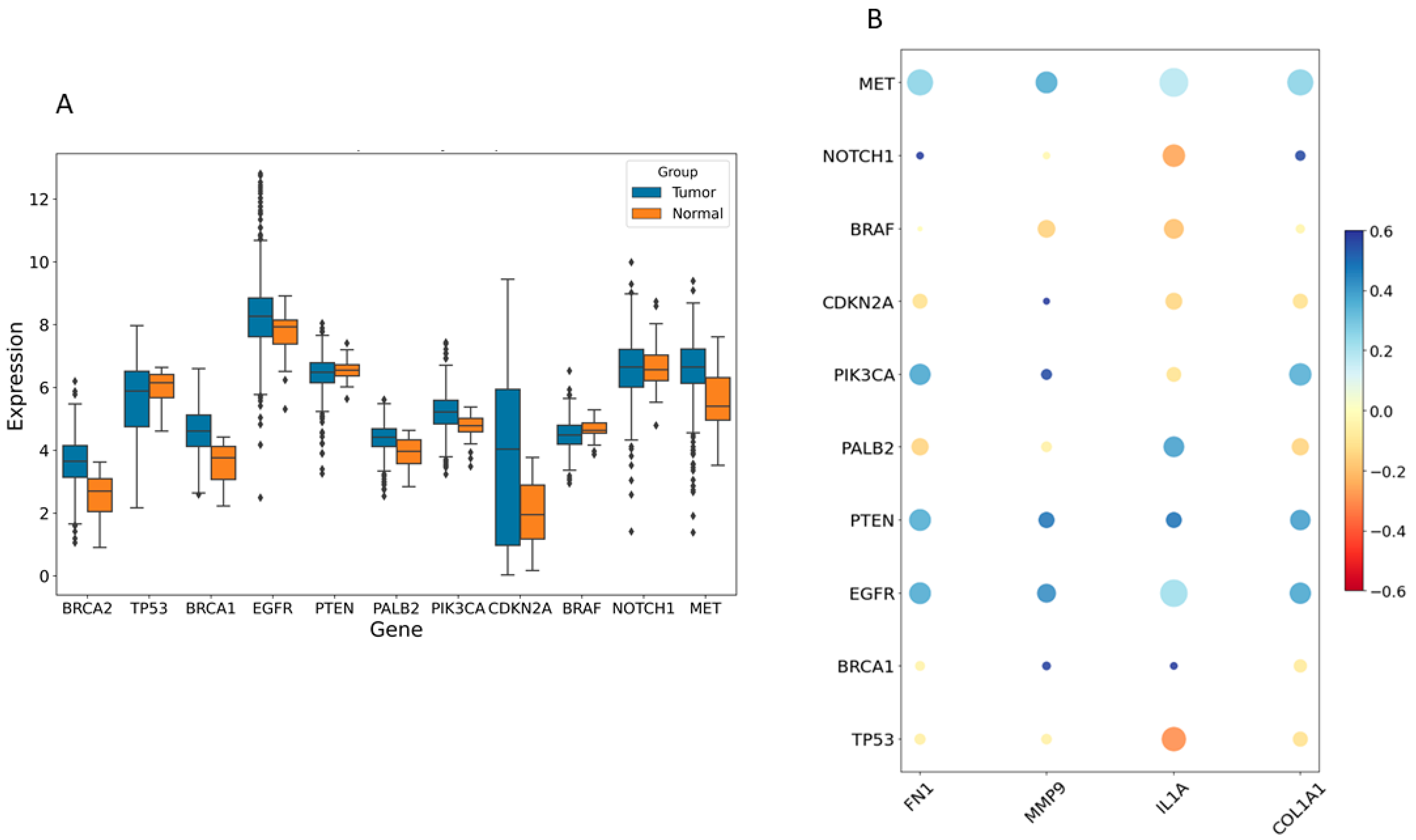
2.3. Relationship of Hub Genes and Disease-Related Genes
2.4. Gene Set Variation Analysis (GSVA) Analyses
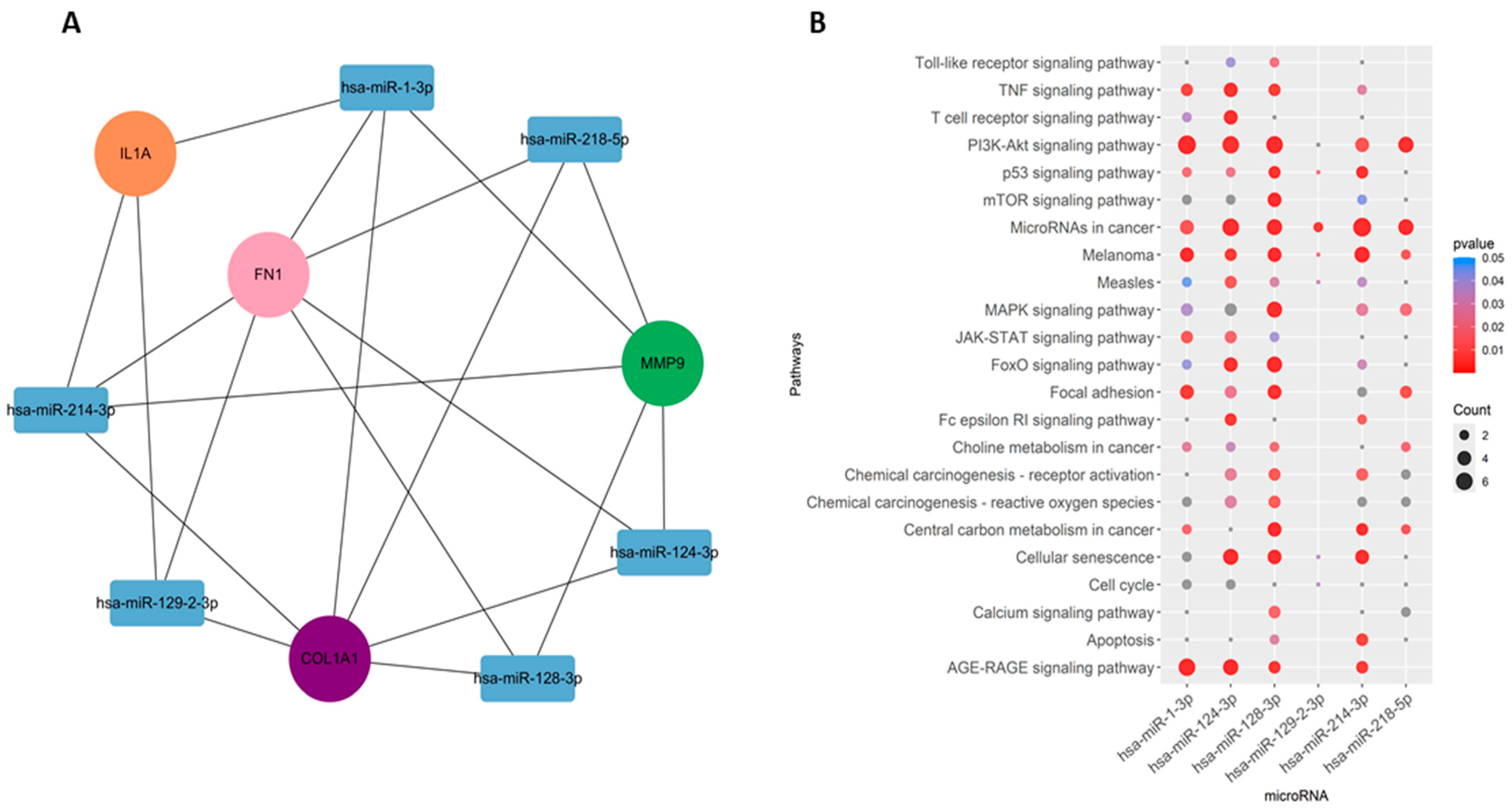
2.5. Prediction of miRNAs of Hub Genes and Enrichment Analyses
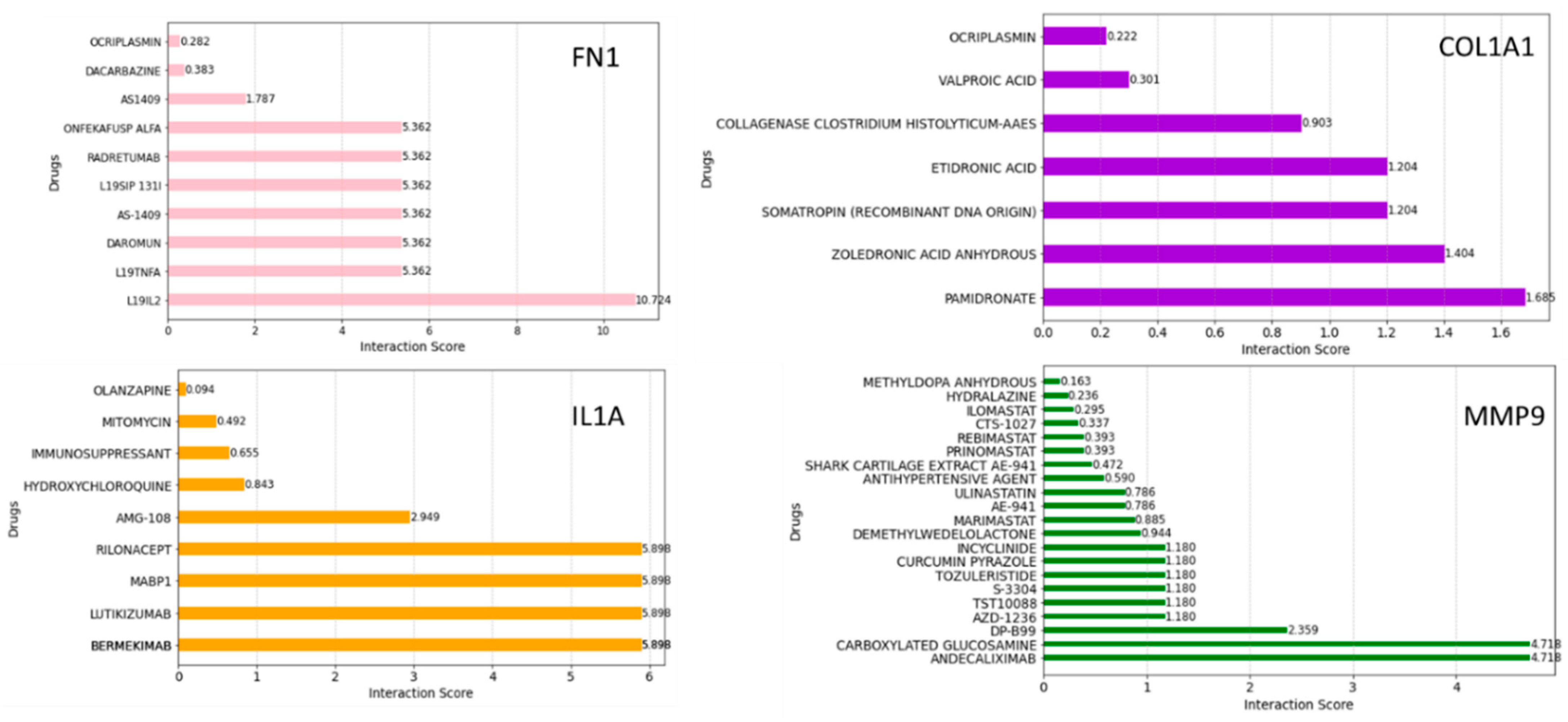
2.6. Drug–Gene Interaction Analysis
3. Discussion
3.1. Key Regulators Gene Analyses
3.2. GSVA Analyses
3.3. Gene–Disease-Related Gene Interaction Analyses
3.4. Gene–miRNAs Interaction Analyses
3.5. Gene–Drugs Interaction Analyses
3.6. Advantages, Medical Applications, and Limitations of this Study
4. Material and Methods
4.1. Dataset
4.2. Functional Enrichment Analysis of the DEGs
4.3. Key Regulators (KR)
4.4. Topological Properties of the Network
4.5. Degree
4.6. Betweenness Centrality
4.7. Closeness Centrality
4.8. ROC Curve Analyses
4.9. Gene Set Variation Analysis (GSVA)
4.10. Identification of miRNAs and Functional Enrichment Analysis
4.11. Drug–Genes Interaction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Parker, J.S.; Karaca, G.; Wu, J.; Funkhouser, W.K.; Moore, D.; Butterfoss, D.; Xiang, D.; Zanation, A.; Yin, X.; et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell 2004, 5, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Li, T.; Beeraka, N.M.; Zheng, Y.; Zhang, X.; Song, R.; Zhou, R.; Wang, X.; Sukocheva, O.; Fan, R.; et al. Molecular classification of human papilloma virus-negative head and neck squamous cell carcinomas: Cell cycle-based classifier and prognostic signature. PLoS ONE 2023, 18, e0286414. [Google Scholar] [CrossRef] [PubMed]
- Badwelan, M.; Muaddi, H.; Ahmed, A.; Lee, K.T.; Tran, S.D. Oral Squamous Cell Carcinoma and Concomitant Primary Tumors, What Do We Know? A Review of the Literature. Curr. Oncol. 2023, 30, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Revesz, M.; Oberna, F.; Slezak, A.; Ferenczi, O.; Kenessey, I.; Takacsi-Nagy, Z. The characteristics of head and neck squamous cell cancer in young adults: A retrospective single-center study. Pathol. Oncol. Res. POR 2023, 29, 1611123. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Bhayani, M.; Kuchta, K.; Galloway, T.; Fundakowski, C. Patterns of distant metastasis in head and neck cancer at presentation: Implications for initial evaluation. Oral Oncol. 2019, 88, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Vander Broek, R.; Mohan, S.; Eytan, D.F.; Chen, Z.; Van Waes, C. The PI3K/Akt/mTOR axis in head and neck cancer: Functions, aberrations, cross-talk, and therapies. Oral Dis. 2015, 21, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Gallo, O.; Masini, E.; Bianchi, B.; Bruschini, L.; Paglierani, M.; Franchi, A. Prognostic significance of cyclooxygenase-2 pathway and angiogenesis in head and neck squamous cell carcinoma. Hum. Pathol. 2002, 33, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Langevin, S.M.; Eliot, M.; Nelson, H.H.; Pawlita, M.; McClean, M.D.; Kelsey, K.T. High-risk HPV types and head and neck cancer. Int. J. Cancer 2014, 135, 1653–1661. [Google Scholar] [CrossRef]
- Stein, A.P.; Saha, S.; Kraninger, J.L.; Swick, A.D.; Yu, M.; Lambert, P.F.; Kimple, R.J. Prevalence of Human Papillomavirus in Oropharyngeal Cancer: A Systematic Review. Cancer J. 2015, 21, 138–146. [Google Scholar] [CrossRef]
- Sabatini, M.E.; Chiocca, S. Human papillomavirus as a driver of head and neck cancers. Br. J. Cancer 2020, 122, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.A. Radiotherapy for head and neck cancer. Semin. Plast. Surg. 2010, 24, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Olmi, P.; Crispino, S.; Fallai, C.; Torri, V.; Rossi, F.; Bolner, A.; Amichetti, M.; Signor, M.; Taino, R.; Squadrelli, M.; et al. Locoregionally advanced carcinoma of the oropharynx: Conventional radiotherapy vs. accelerated hyperfractionated radiotherapy vs. concomitant radiotherapy and chemotherapy--a multicenter randomized trial. Int. J. Radiat. Oncol. Biol. Phys. 2003, 55, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, D.; Goncalves, J.P.; Ojeda, F.; de Moor, B.; Moreau, Y. Candidate gene prioritization by network analysis of differential expression using machine learning approaches. BMC Bioinform. 2010, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.K.; Priya, A.; Malik, M.Z.; Thanaraj, T.A.; Singh, A.K.; Mago, P.; Ghosh, C.; Shalimar; Tandon, R.; Chaturvedi, R. A bioinformatics approach to elucidate conserved genes and pathways in C. elegans as an animal model for cardiovascular research. Sci. Rep. 2024, 14, 7471. [Google Scholar] [CrossRef] [PubMed]
- Nazarieh, M.; Wiese, A.; Will, T.; Hamed, M.; Helms, V. Identification of key player genes in gene regulatory networks. BMC Syst. Biol. 2016, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Cai, B.; Ke, R.; Chen, L.; Ni, X.; Liu, H.; Lin, X.; Wang, B.; Shan, X. Integrative bioinformatics and experimental validation of hub genetic markers in acne vulgaris: Toward personalized diagnostic and therapeutic strategies. J. Cosmet. Dermatol. 2024, 23, 1777–1799. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Kan, C.; Chen, B.; Qu, N.; Hou, N.; Liu, Y.; Han, F. COL1A1: A novel oncogenic gene and therapeutic target in malignancies. Pathol. Res. Pract. 2022, 236, 154013. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Hu, Y.; Gan, L.; Lai, J.; Zheng, P.; Zhang, Y.; Shuai, L.; Jiang, Y.; Chen, M.; Wang, J.; et al. Matrix metalloproteinase-2 inducing COL1A1 synthesis via integrin alpha V promotes invasion and metastasis of cholangiocarcinoma cells. Ann. Hepatol. 2024, 29, 101279. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, Y.; Wu, Y.; Gao, Y.; Li, Q.; Abdulrahman, A.A.; Liu, X.F.; Ji, G.Q.; Gao, J.; Li, L.; et al. Identification of COL1A1 as an invasion-related gene in malignant astrocytoma. Int. J. Oncol. 2018, 53, 2542–2554. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Jones, J.L.; O’Byrne, K.J. Matrix metalloproteinase 9 and the epidermal growth factor signal pathway in operable non-small cell lung cancer. Clin. Cancer Res. 2000, 6, 2349–2355. [Google Scholar] [PubMed]
- Sinpitaksakul, S.N.; Pimkhaokham, A.; Sanchavanakit, N.; Pavasant, P. TGF-beta1 induced MMP-9 expression in HNSCC cell lines via Smad/MLCK pathway. Biochem. Biophys. Res. Commun. 2008, 371, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.H.; Du, W.D.; Li, Y.F.; Al-Aroomi, M.A.; Yan, C.; Wang, Y.; Zhang, Z.Y.; Liu, F.Y.; Sun, C.F. The Overexpression of Fibronectin 1 Promotes Cancer Progression and Associated with M2 Macrophages Polarization in Head and Neck Squamous Cell Carcinoma Patients. Int. J. Gen. Med. 2022, 15, 5027–5042. [Google Scholar] [CrossRef]
- Raj, S.; Kesari, K.K.; Kumar, A.; Rathi, B.; Sharma, A.; Gupta, P.K.; Jha, S.K.; Jha, N.K.; Slama, P.; Roychoudhury, S.; et al. Molecular mechanism(s) of regulation(s) of c-MET/HGF signaling in head and neck cancer. Mol. Cancer 2022, 21, 31. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Niklander, S.E.; Murdoch, C.; Hunter, K.D. IL-1/IL-1R Signaling in Head and Neck Cancer. Front. Oral Health 2021, 2, 722676. [Google Scholar] [CrossRef] [PubMed]
- Carla, C.; Daris, F.; Cecilia, B.; Francesca, B.; Francesca, C.; Paolo, F. Angiogenesis in head and neck cancer: A review of the literature. J. Oncol. 2012, 2012, 358472. [Google Scholar] [CrossRef] [PubMed]
- Ilhan-Mutlu, A.; Siehs, C.; Berghoff, A.S.; Ricken, G.; Widhalm, G.; Wagner, L.; Preusser, M. Expression profiling of angiogenesis-related genes in brain metastases of lung cancer and melanoma. Tumour Biol. 2016, 37, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Huang, L.; Lu, Y.G.; Zheng, D.L. Roles of the Wnt Signaling Pathway in Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2020, 7, 590912. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Gehren, A.S.; Rocha, M.R.; de Souza, W.F.; Morgado-Diaz, J.A. Alterations of the apical junctional complex and actin cytoskeleton and their role in colorectal cancer progression. Tissue Barriers 2015, 3, e1017688. [Google Scholar] [CrossRef] [PubMed]
- Egloff, A.M.; Grandis, J.R. Targeting epidermal growth factor receptor and SRC pathways in head and neck cancer. Semin. Oncol. 2008, 35, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Cassell, A.; Grandis, J.R. Investigational EGFR-targeted therapy in head and neck squamous cell carcinoma. Expert Opin. Investig. Drugs 2010, 19, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Swinson, D.E.; Cox, G.; O’Byrne, K.J. Coexpression of epidermal growth factor receptor with related factors is associated with a poor prognosis in non-small-cell lung cancer. Br. J. Cancer 2004, 91, 1301–1307. [Google Scholar] [CrossRef]
- Dai, S.; Li, F.; Xu, S.; Hu, J.; Gao, L. The important role of miR-1-3p in cancers. J. Transl. Med. 2023, 21, 769. [Google Scholar] [CrossRef]
- Thomaidou, A.C.; Batsaki, P.; Adamaki, M.; Goulielmaki, M.; Baxevanis, C.N.; Zoumpourlis, V.; Fortis, S.P. Promising Biomarkers in Head and Neck Cancer: The Most Clinically Important miRNAs. Int. J. Mol. Sci. 2022, 23, 8257. [Google Scholar] [CrossRef] [PubMed]
- Subha, S.T.; Chin, J.W.; Cheah, Y.K.; Mohtarrudin, N.; Saidi, H.I. Multiple microRNA signature panel as promising potential for diagnosis and prognosis of head and neck cancer. Mol. Biol. Rep. 2022, 49, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Kozlowska-Maslon, J.; Guglas, K.; Kolenda, T.; Lamperska, K.; Makalowska, I. miRNA in head and neck squamous cell carcinomas: Promising but still distant future of personalized oncology. Rep. Pract. Oncol. Radiother. 2023, 28, 681–697. [Google Scholar] [CrossRef]
- Tao, S.; Li, H.; Ma, X.; Ma, Y.; He, J.; Gao, Y.; Li, J. Elevating microRNA-1-3p shuttled by cancer-associated fibroblasts-derived extracellular vesicles suppresses breast cancer progression and metastasis by inhibiting GLIS1. Cancer Gene Ther. 2021, 28, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, M.; Jin, H.; Peng, B.; Dai, L.; Wang, S.; Xing, H.; Wang, B.; Wu, Z. Synthetic Evaluation of MicroRNA-1-3p Expression in Head and Neck Squamous Cell Carcinoma Based on Microarray Chips and MicroRNA Sequencing. BioMed Res. Int. 2021, 2021, 6529255. [Google Scholar] [CrossRef]
- Ye, J.; Liao, Q.; Zeng, X.; Liu, C.; Ding, Y.; Liu, X.; Zeng, L.; Guan, T.; Yuan, Y. MicroRNA-124-3p inhibited progression of nasopharyngeal carcinoma by interaction with PCDH8 and the inactivation of PI3K/AKT/mTOR pathway. J. Cancer 2021, 12, 4933–4944. [Google Scholar] [CrossRef]
- Shibata, T.; Cao, D.Y.; Dar, T.B.; Ahmed, F.; Bhat, S.A.; Veiras, L.C.; Bernstein, E.A.; Khan, A.A.; Chaum, M.; Shiao, S.L.; et al. miR766-3p and miR124-3p Dictate Drug Resistance and Clinical Outcome in HNSCC. Cancers 2022, 14, 5273. [Google Scholar] [CrossRef]
- Hauser, B.; Zhao, Y.; Pang, X.; Ling, Z.; Myers, E.; Wang, P.; Califano, J.; Gu, X. Functions of MiRNA-128 on the regulation of head and neck squamous cell carcinoma growth and apoptosis. PLoS ONE 2015, 10, e0116321. [Google Scholar] [CrossRef]
- Jiang, G.; Li, R.H.; Sun, C.; Liu, Y.Q.; Zheng, J.N. Dacarbazine combined targeted therapy versus dacarbazine alone in patients with malignant melanoma: A meta-analysis. PLoS ONE 2014, 9, e111920. [Google Scholar] [CrossRef]
- Tomasz, M. Mitomycin C: Small, fast and deadly (but very selective). Chem. Biol. 1995, 2, 575–579. [Google Scholar] [CrossRef]
- Shah, M.A.; Bodoky, G.; Starodub, A.; Cunningham, D.; Yip, D.; Wainberg, Z.A.; Bendell, J.; Thai, D.; He, J.; Bhargava, P.; et al. Phase III Study to Evaluate Efficacy and Safety of Andecaliximab With mFOLFOX6 as First-Line Treatment in Patients With Advanced Gastric or GEJ Adenocarcinoma (GAMMA-1). J. Clin. Oncol. 2021, 39, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Taverna, F.; Goveia, J.; Karakach, T.K.; Khan, S.; Rohlenova, K.; Treps, L.; Subramanian, A.; Schoonjans, L.; Dewerchin, M.; Eelen, G.; et al. BIOMEX: An interactive workflow for (single cell) omics data interpretation and visualization. Nucleic Acids Res. 2020, 48, W385–W394. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramirez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Gursoy, A.; Keskin, O.; Nussinov, R. Topological properties of protein interaction networks from a structural perspective. Biochem. Soc. Trans. 2008, 36, 1398–1403. [Google Scholar] [CrossRef]
- Newman, M.E.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004, 69, 026113. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Han, K.; Suh, Y.J.; Jung, I. Stability selection for LASSO with weights based on AUC. Sci. Rep. 2023, 13, 5207. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Lin, Y.C.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. miRDB: A microRNA target prediction and functional annotation database with a wiki interface. RNA 2008, 14, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Cannon, M.; Stevenson, J.; Stahl, K.; Basu, R.; Coffman, A.; Kiwala, S.; McMichael, J.F.; Kuzma, K.; Morrissey, D.; Cotto, K.; et al. DGIdb 5.0: Rebuilding the drug–gene interaction database for precision medicine and drug discovery platforms. Nucleic Acids Res. 2023, 52, D1227–D1235. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danishuddin; Haque, M.A.; Malik, M.Z.; Arya, R.; Singh, P.; Lee, J.-S.; Kim, J.-J.; Lee, K.-W.; Jung, T.-S. Unveiling the Mechanisms Underlying the Immunotherapeutic Potential of Gene–miRNA and Drugs in Head and Neck Cancer. Pharmaceuticals 2024, 17, 921. https://doi.org/10.3390/ph17070921
Danishuddin, Haque MA, Malik MZ, Arya R, Singh P, Lee J-S, Kim J-J, Lee K-W, Jung T-S. Unveiling the Mechanisms Underlying the Immunotherapeutic Potential of Gene–miRNA and Drugs in Head and Neck Cancer. Pharmaceuticals. 2024; 17(7):921. https://doi.org/10.3390/ph17070921
Chicago/Turabian StyleDanishuddin, Md Azizul Haque, Md. Zubbair Malik, Rakesh Arya, Pooja Singh, Jeong-Sang Lee, Jong-Joo Kim, Keun-Woo Lee, and Tae-Sung Jung. 2024. "Unveiling the Mechanisms Underlying the Immunotherapeutic Potential of Gene–miRNA and Drugs in Head and Neck Cancer" Pharmaceuticals 17, no. 7: 921. https://doi.org/10.3390/ph17070921
APA StyleDanishuddin, Haque, M. A., Malik, M. Z., Arya, R., Singh, P., Lee, J.-S., Kim, J.-J., Lee, K.-W., & Jung, T.-S. (2024). Unveiling the Mechanisms Underlying the Immunotherapeutic Potential of Gene–miRNA and Drugs in Head and Neck Cancer. Pharmaceuticals, 17(7), 921. https://doi.org/10.3390/ph17070921