

Immune Checkpoint Inhibitors for Pediatric Cancers: Is It Still a Stalemate?



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Exploring the Biological Role of CTLA-4 and PD-1
3. Mechanism of Action of Immune Checkpoint Inhibitors
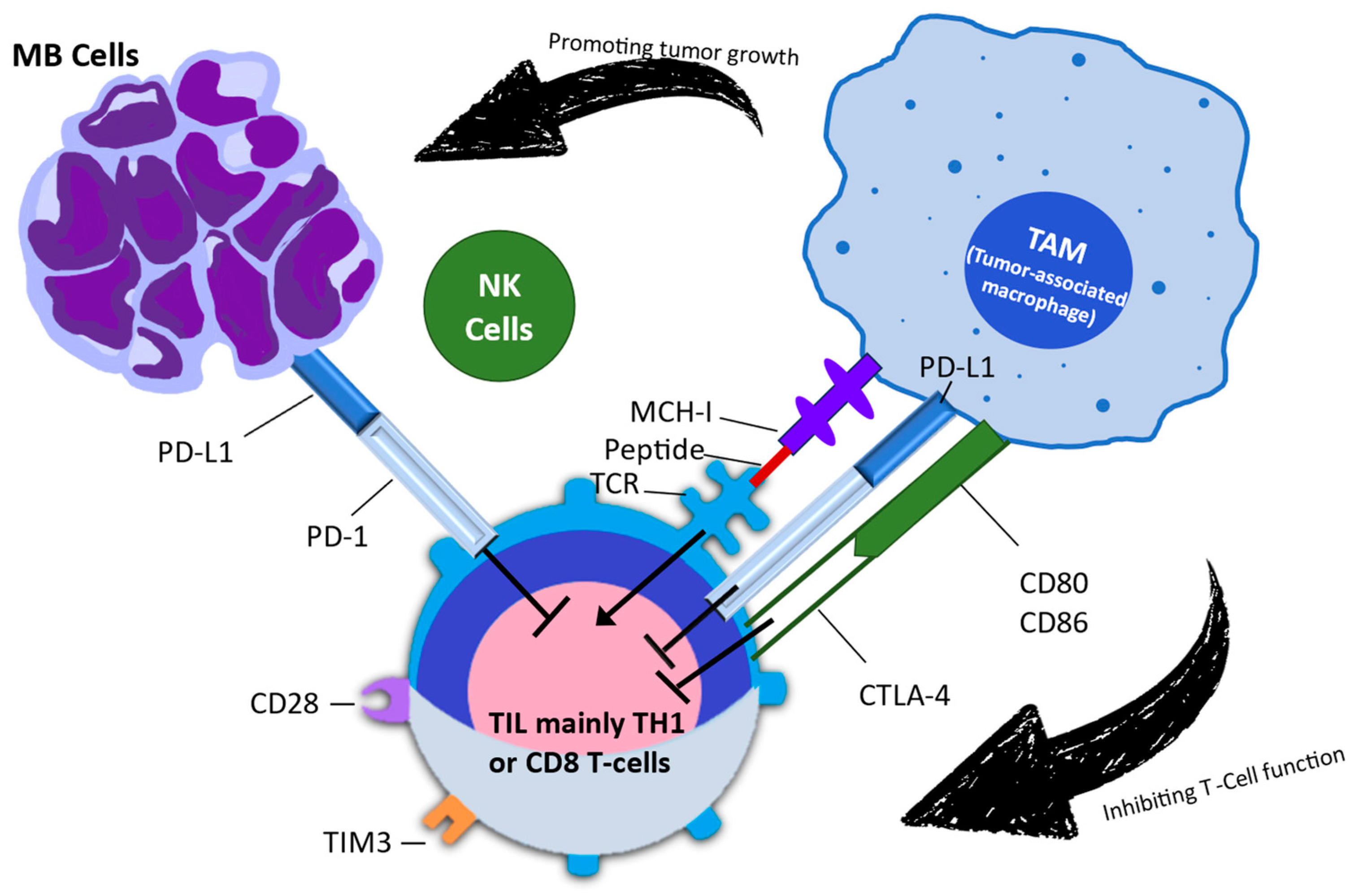
3.1. Experience with Immune Checkpoint Inhibitors in Pediatric Cancers
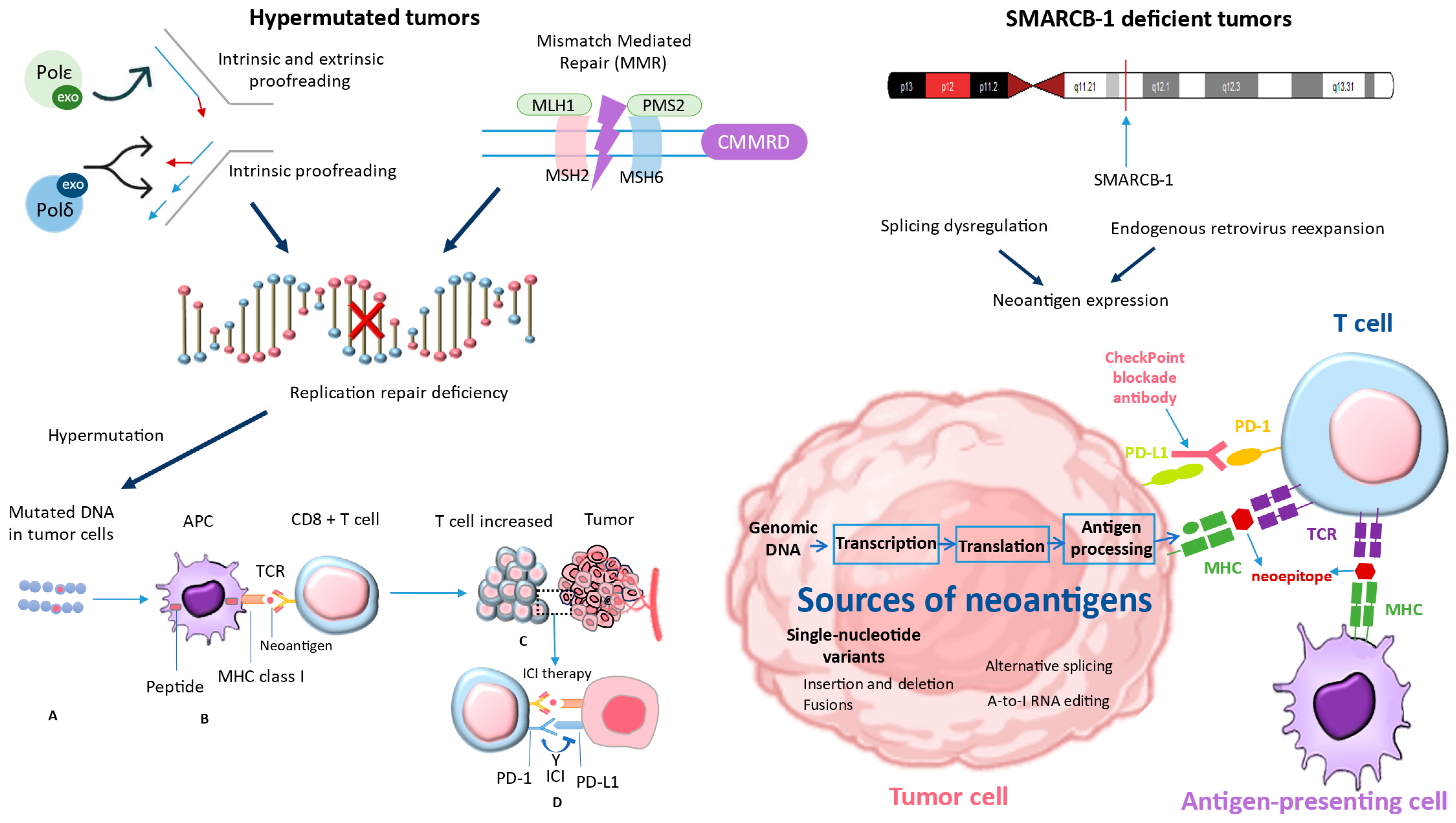
3.2. Exploring Immune Checkpoint Inhibition in Hypermutated Cancers
3.3. Normal and Malignant Human Cells Have Different Microsatellite Instability Signals from DNA Polymerase and Mismatch Repair
3.4. Immune Checkpoint Inhibition for Hypermutant Tumors
4. How the Body Develops Intolerance to ICI Treatment
5. Adverse Events of Immune Checkpoint Inhibitors
5.1. Pulmonary Toxicity of Immune Checkpoint Immunotherapy
5.2. Cellular Autoimmunity/Higher T-Cell Activity
5.3. Genetic Predisposition
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ebrahimi, S.; Habibzadeh, A.; Khojasteh-Kaffash, S.; Valizadeh, P.; Samieefar, N.; Rezaei, N. Immune Checkpoint Inhibitors Therapy as the Game-Changing Approach for Pediatric Lymphoma: A Brief Landscape. Crit. Rev. Oncol. Hematol. 2023, 193, 104225. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Kim, H.R.; Ha, S.J. Immune Checkpoint Inhibitors in 10 Years: Contribution of Basic Research and Clinical Application in Cancer Immunotherapy. Immune Netw. 2022, 22, e2. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Morgenstern, D.A.; Leruste, A.; Bourdeaut, F.; Davis, K.L. Checkpoint Immunotherapy in Pediatrics: Here, Gone, and Back Again. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Tentori, L.; Navarra, P. Ipilimumab: A novel immunostimulatory monoclonal antibody for the treatment of cancer. Pharmacol. Res. 2012, 65, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhou, Q.; Gao, Y.; Hu, W.; Lou, G.; Sun, H.; Zhu, J.; Shu, J.; Zhou, X.; Sun, R.; et al. A multicenter phase 2 trial of camrelizumab plus famitinib for women with recurrent or metastatic cervical squamous cell carcinoma. Nat. Commun. 2022, 13, 7581. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos Usta, E.; Yanez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Lu, C.; Mao, L.; Zhu, Y.; Kong, W.; Shen, S.; Tang, M.; Bao, S.; Cheng, H.; Li, G.; et al. PD-1 blockade plus chemoradiotherapy as preoperative therapy for patients with BRPC/LAPC: A biomolecular exploratory, phase II trial. Cell Rep. Med. 2023, 4, 100972. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily-CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Zeng, Y.; Thistlethwaite, J.R.; Montag, A.; Brady, W.; Gibson, M.G.; Linsley, P.S.; Bluestone, J.A. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science 1992, 257, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Wallace, P.M.; Johnson, J.; Gibson, M.G.; Greene, J.L.; Ledbetter, J.A.; Singh, C.; Tepper, M.A. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science 1992, 257, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Tseng, S.Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Flies, D.B.; van Deursen, J.M.; Chen, L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity 2004, 20, 327–336. [Google Scholar] [CrossRef]
- Latchman, Y.E.; Liang, S.C.; Wu, Y.; Chernova, T.; Sobel, R.A.; Klemm, M.; Kuchroo, V.K.; Freeman, G.J.; Sharpe, A.H. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10691–10696. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Park, J.S.; Jeong, Y.H.; Son, J.; Ban, Y.H.; Lee, B.H.; Chen, L.; Chang, J.; Chung, D.H.; Choi, I.; et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J. Immunol. 2015, 194, 5801–5811. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Zhang, Y.; Chung, Y.; Bishop, C.; Daugherty, B.; Chute, H.; Holst, P.; Kurahara, C.; Lott, F.; Sun, N.; Welcher, A.A.; et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl. Acad. Sci. USA 2006, 103, 11695–11700. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Greenwald, R.J.; Latchman, Y.E.; Rosas, L.; Satoskar, A.; Freeman, G.J.; Sharpe, A.H. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur. J. Immunol. 2006, 36, 58–64. [Google Scholar] [CrossRef]
- Guo, Z.S. The 2018 Nobel Prize in medicine goes to cancer immunotherapy (editorial for BMC Cancer). BMC Cancer 2018, 18, 1086. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Mittra, A.; Naqash, A.R.; Takebe, N. A review of mechanisms of resistance to immune checkpoint inhibitors and potential strategies for therapy. Cancer Drug Resist. 2020, 18, 252–275. [Google Scholar] [CrossRef] [PubMed]
- Rechberger, J.S.; Toll, S.A.; Vanbilloen, W.J.F.; Daniels, D.J.; Khatua, S. Exploring the Molecular Complexity of Medulloblastoma: Implications for Diagnosis and Treatment. Diagnostics 2023, 13, 2398. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, D.C.; Theocharopoulos, C.; Lialios, P.P.; Foteinou, D.; Koumprentziotis, I.A.; Xynos, G.; Gogas, H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment. Cancers 2023, 15, 2718. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Voskamp, M.J.; Li, S.; van Daalen, K.R.; Crnko, S.; Ten Broeke, T.; Bovenschen, N. Immunotherapy in Medulloblastoma: Current State of Research, Challenges, and Future Perspectives. Cancers 2021, 13, 5387. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Mulla, N.; Malibary, H.; Bamaga, A.K.; Fadul, M.M.; Faizo, E.; Hakamy, S.; Baeesa, S. Immune microenvironment of medulloblastoma: The association between its molecular subgroups and potential targeted immunotherapeutic receptors. World J. Clin. Oncol. 2023, 14, 117–130. [Google Scholar] [CrossRef]
- Liu, C.; Yang, M.; Zhang, D.; Chen, M.; Zhu, D. Clinical cancer immunotherapy: Current progress and prospects. Front. Immunol. 2022, 13, 961805. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.C.; Joubert, A.M.; Visagie, M.H. Molecular Farming of Pembrolizumab and Nivolumab. Int. J. Mol. Sci. 2023, 24, 10045. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, J.J.; Xing, R.; Zeng, Y.C. Combination therapy: Future directions of immunotherapy in small cell lung cancer. Transl. Oncol. 2021, 14, 100889. [Google Scholar] [CrossRef]
- Sadaf, H.; Ambroziak, M.; Binkowski, R.; Kluebsoongnoen, J.; Paszkiewicz-Kozik, E.; Steciuk, J.; Markowicz, S.; Walewski, J.; Sarnowska, E.; Sarnowski, T.J.; et al. New molecular targets in Hodgkin and Reed-Sternberg cells. Front. Immunol. 2023, 14, 1155468. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef]
- Schramm, A.; Koster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef]
- Haymaker, C.L.; Kim, D.; Uemura, M.; Vence, L.M.; Phillip, A.; McQuail, N.; Brown, P.D.; Fernandez, I.; Hudgens, C.W.; Creasy, C.; et al. Metastatic Melanoma Patient Had a Complete Response with Clonal Expansion after Whole Brain Radiation and PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 100–105. [Google Scholar] [CrossRef]
- Herter-Sprie, G.S.; Koyama, S.; Korideck, H.; Hai, J.; Deng, J.; Li, Y.Y.; Buczkowski, K.A.; Grant, A.K.; Ullas, S.; Rhee, K.; et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 2016, 1, e87415. [Google Scholar] [CrossRef]
- Koller, K.M.; Mackley, H.B.; Liu, J.; Wagner, H.; Talamo, G.; Schell, T.D.; Pameijer, C.; Neves, R.I.; Anderson, B.; Kokolus, K.M.; et al. Improved survival and complete response rates in patients with advanced melanoma treated with concurrent ipilimumab and radiotherapy versus ipilimumab alone. Cancer Biol. Ther. 2017, 18, 36–42. [Google Scholar] [CrossRef]
- Liniker, E.; Menzies, A.M.; Kong, B.Y.; Cooper, A.; Ramanujam, S.; Lo, S.; Kefford, R.F.; Fogarty, G.B.; Guminski, A.; Wang, T.W.; et al. Activity and safety of radiotherapy with anti-PD-1 drug therapy in patients with metastatic melanoma. Oncoimmunology 2016, 5, e1214788. [Google Scholar] [CrossRef]
- Findlay, J.M.; Castro-Giner, F.; Makino, S.; Rayner, E.; Kartsonaki, C.; Cross, W.; Kovac, M.; Ulahannan, D.; Palles, C.; Gillies, R.S.; et al. Differential clonal evolution in oesophageal cancers in response to neoadjuvant chemotherapy. Nat. Commun. 2016, 7, 11111. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Cleary, J.M.; Reardon, B.; Pike, J.; Braunstein, L.Z.; Kim, J.; Amin-Mansour, A.; Miao, D.; Damish, A.; Chin, J.; et al. Genomic Evolution after Chemoradiotherapy in Anal Squamous Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Mazeron, R.; Dercle, L.; Ammari, S.; Canova, C.; Marabelle, A.; Rose, S.; Rubin, E.; Deutsch, E.; Soria, J.C.; et al. Abscopal effect in a Hodgkin lymphoma patient treated by an anti-programmed death 1 antibody. Eur. J. Cancer 2016, 66, 91–94. [Google Scholar] [CrossRef]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J.; et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Yarmarkovich, M.; Maris, J.M. When Cold Is Hot: Immune Checkpoint Inhibition Therapy for Rhabdoid Tumors. Cancer Cell 2019, 36, 575–576. [Google Scholar] [CrossRef]
- Rose, K.; Walson, P.D. Are Regulatory Age Limits in Pediatric Melanoma Justified? Curr. Ther. Res. Clin. Exp. 2019, 90, 113–118. [Google Scholar] [CrossRef]
- Haanen, J.B.; Robert, C. Immune Checkpoint Inhibitors. Prog. Tumor Res. 2015, 42, 55–66. [Google Scholar]
- Liu, W.; Zhang, L.; Xiu, Z.; Guo, J.; Wang, L.; Zhou, Y.; Jiao, Y.; Sun, M.; Cai, J. Combination of Immune Checkpoint Inhibitors with Chemotherapy in Lung Cancer. Onco. Targets Ther. 2020, 13, 7229–7241. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Fox, E.; Isikwei, E.; Reid, J.M.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; Mackall, C.L. A Phase I/II Trial of Nivolumab plus Ipilimumab in Children and Young Adults with Relapsed/Refractory Solid Tumors: A Children’s Oncology Group Study ADVL1412. Clin. Cancer Res. 2022, 28, 5088–5097. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Tarduno, A.; Lu, Y.C. Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy. Cancers 2021, 13, 6061. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Dupain, C.; Harttrampf, A.C.; Urbinati, G.; Geoerger, B.; Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Mol. Ther. Nucleic Acids. 2017, 6, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Valero, C.; Lee, M.; Hoen, D.; Wang, J.; Nadeem, Z.; Patel, N.; Postow, M.A.; Shoushtari, A.N.; Plitas, G.; Balachandran, V.P.; et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat. Genet. 2021, 53, 11–15. [Google Scholar] [CrossRef]
- Gridelli, C.; Peters, S.; Mok, T.; Forde, P.M.; Reck, M.; Attili, I.; de Marinis, F. First-line immunotherapy in advanced non-small-cell lung cancer patients with ECOG performance status 2: Results of an International Expert Panel Meeting by the Italian Association of Thoracic Oncology. ESMO Open 2022, 7, 100355. [Google Scholar] [CrossRef]
- Li, L.; Bai, L.; Lin, H.; Dong, L.; Zhang, R.; Cheng, X.; Liu, Z.; Ouyang, Y.; Ding, K. Multiomics analysis of tumor mutational burden across cancer types. Comput. Struct. Biotechnol. J. 2021, 19, 5637–5646. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Maruvka, Y.E.; Sudhaman, S.; Kelly, J.; Haradhvala, N.J.; Bianchi, V.; Edwards, M.; Forster, V.J.; Nunes, N.M.; Galati, M.A.; et al. DNA Polymerase and Mismatch Repair Exert Distinct Microsatellite Instability Signatures in Normal and Malignant Human Cells. Cancer Discov. 2021, 11, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, G.M. DNA mismatch repair in the context of chromatin. Cells Biosci. 2020, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Sudhaman, S.; Morgenstern, D.; Coblentz, A.; Chung, J.; Stone, S.C.; Alsafwani, N.; Liu, Z.A.; Karsaneh, O.A.A.; Soleimani, S.; et al. Genomic predictors of response to PD-1 inhibition in children with germline DNA replication repair deficiency. Nat. Med. 2022, 28, 125–135. [Google Scholar] [CrossRef]
- Weber, C.A.M.; Kronke, N.; Volk, V.; Auber, B.; Forster, A.; Trost, D.; Geffers, R.; Esmaeilzadeh, M.; Lalk, M.; Nabavi, A.; et al. Rare germline variants in POLE and POLD1 encoding the catalytic subunits of DNA polymerases epsilon and delta in glioma families. Acta Neuropathol. Commun. 2023, 11, 184. [Google Scholar] [CrossRef]
- Gola, M.; Stefaniak, P.; Godlewski, J.; Jereczek-Fossa, B.A.; Starzynska, A. Prospects of POLD1 in Human Cancers: A Review. Cancers 2023, 15, 1905. [Google Scholar] [CrossRef]
- Dencic, T.; Petrovic, A.; Jovicic Milentijevic, M.; Radenkovic, G.; Jovic, M.; Zivkovic, N.; Salinger, S.; Brankovic, B.; Velickov, A.; Ilic, I. The Importance of Immunohistochemical Heterogeneous Expression of MMR Protein in Patients with Colorectal Cancer in Stage II and III of the Disease. Medicina 2023, 59, 489. [Google Scholar] [CrossRef]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cells Int. 2020, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Ciurej, A.; Lewis, E.; Gupte, A.; Al-Antary, E. Checkpoint Immunotherapy in Pediatric Oncology: Will We Say Checkmate Soon? Vaccines 2023, 11, 1843. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, D.; Giannatempo, P.; Grazia, G.; Aiello, M.M.; Bertolini, F.; Mirabile, A.; Buti, S.; Vasile, E.; Scotti, V.; Pisapia, P.; et al. Patients Selection for Immunotherapy in Solid Tumors: Overcome the Naive Vision of a Single Biomarker. Biomed. Res. Int. 2019, 2019, 9056417. [Google Scholar] [CrossRef] [PubMed]
- Westcott, P.M.K.; Muyas, F.; Hauck, H.; Smith, O.C.; Sacks, N.J.; Ely, Z.A.; Jaeger, A.M.; Rideout, W.M., 3rd; Zhang, D.; Bhutkar, A.; et al. Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity. Nat. Genet. 2023, 55, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Koh, C.H.; Lee, S.; Kwak, M.; Kim, B.S.; Chung, Y. CD8 T-cell subsets: Heterogeneity, functions, and therapeutic potential. Exp. Mol. Med. 2023, 55, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of Resistance to Immune Checkpoint Blockade. Am. J. Clin. Dermatol. 2019, 20, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Thouvenin, L.; Olivier, T.; Banna, G.; Addeo, A.; Friedlaender, A. Immune checkpoint inhibitor-induced aseptic meningitis and encephalitis: A case series and narrative review. Ther. Adv. Drug. Saf. 2021, 12, 20420986211004745. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Mohn, N.; Beutel, G.; Gutzmer, R.; Ivanyi, P.; Satzger, I.; Skripuletz, T. Neurological Immune-Related Adverse Events Associated with Nivolumab, Ipilimumab, and Pembrolizumab Therapy-Review of the Literature and Future Outlook. J. Clin. Med. 2019, 8, 1777. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Ibis, B.; Aliazis, K.; Cao, C.; Yenyuwadee, S.; Boussiotis, V.A. Immune-related adverse effects of checkpoint immunotherapy and implications for the treatment of patients with cancer and autoimmune diseases. Front. Immunol. 2023, 14, 1197364. [Google Scholar] [CrossRef]
- Nagpal, C.; Rastogi, S.; Shamim, S.A.; Prakash, S. Re-challenge of immune checkpoint inhibitor pembrolizumab with concurrent tocilizumab after prior grade 3 pneumonitis. Ecancermedicalscience 2023, 17, 1644. [Google Scholar] [CrossRef] [PubMed]
- Ghanbar, M.I.; Suresh, K.; Ghanbar, M.I.; Suresh, K. Pulmonary toxicity of immune checkpoint immunotherapy. J. Clin. Investig. 2024, 134, e170503. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.X.; Huang, W.; Liu, P.P.; Pan, Y.; Cui, Y.H. Dynamic changes in the radiologic manifestation of a recurrent checkpoint inhibitor-related pneumonitis in a non-small cell lung cancer patient: A case report. World J. Clin. Cases 2021, 9, 9108–9113. [Google Scholar] [CrossRef] [PubMed]
- Bins, S.; Basak, E.A.; El Bouazzaoui, S.; Koolen, S.L.W.; Oomen-de Hoop, E.; van der Leest, C.H.; van der Veldt, A.A.M.; Sleijfer, S.; Debets, R.; van Schaik, R.H.N.; et al. Association between single-nucleotide polymorphisms and adverse events in nivolumab-treated non-small cell lung cancer patients. Br. J. Cancer 2018, 118, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.C.; Dorak, M.T.; Bettinotti, M.P.; Bingham, C.O.; Shah, A.A. Association of HLA-DRB1 shared epitope alleles and immune checkpoint inhibitor-induced inflammatory arthritis. Rheumatology 2019, 58, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, N.; Diab, A.; Yu, R.K.; Futreal, A.; Criswell, L.A.; Tayar, J.H.; Dadu, R.; Shannon, V.; Shete, S.S.; Suarez-Almazor, M.E. Genetic determinants of immune-related adverse events in patients with melanoma receiving immune checkpoint inhibitors. Cancer Immunol. Immunother. 2021, 70, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Shojaie, L.; Ali, M.; Iorga, A.; Dara, L. Mechanisms of immune checkpoint inhibitor-mediated liver injury. Acta Pharm. Sin. B 2021, 11, 3727–3739. [Google Scholar] [CrossRef] [PubMed]
- Vilarino, N.; Bruna, J.; Kalofonou, F.; Anastopoulou, G.G.; Argyriou, A.A. Immune-Driven Pathogenesis of Neurotoxicity after Exposure of Cancer Patients to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 5774. [Google Scholar] [CrossRef]
- Teng, Y.S.; Yu, S. Molecular Mechanisms of Cutaneous Immune-Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitors. Curr. Oncol. 2023, 30, 6805–6819. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chavez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suarez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Tomsitz, D.; Ruf, T.; Zierold, S.; French, L.E.; Heinzerling, L. Steroid-Refractory Immune-Related Adverse Events Induced by Checkpoint Inhibitors. Cancers 2023, 15, 2538. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Abu-Sbeih, H.; Ascierto, P.A.; Brufsky, J.; Cappelli, L.C.; Cortazar, F.B.; Gerber, D.E.; Hamad, L.; Hansen, E.; Johnson, D.B.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 2021, 9, e002435. [Google Scholar] [CrossRef]
- Alsalem, A.N.; Scarffe, L.A.; Briemberg, H.R.; Aaroe, A.E.; Harrison, R.A. Neurologic Complications of Cancer Immunotherapy. Curr. Oncol. 2023, 30, 5876–5897. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.; Obeid, M.; Spain, L.; Carbonnel, F.; Wang, Y.; Robert, C.; Lyon, A.R.; Wick, W.; Kostine, M.; Peters, S.; et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 1217–1238. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K.; Committee, E.G. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv264–iv266. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Wolchok, J.D.; Bass, A.R. TNF in the era of immune checkpoint inhibitors: Friend or foe? Nat. Rev. Rheumatol. 2021, 17, 213–223. [Google Scholar] [CrossRef]
- Bylsma, S.; Yun, K.; Patel, S.; Dennis, M.J. Immune Checkpoint Inhibitor Rechallenge After Prior Immune Toxicity. Curr. Treat. Options Oncol. 2022, 23, 1153–1168. [Google Scholar] [CrossRef]
- Menzies, A.M.; Johnson, D.B.; Ramanujam, S.; Atkinson, V.G.; Wong, A.N.M.; Park, J.J.; McQuade, J.L.; Shoushtari, A.N.; Tsai, K.K.; Eroglu, Z.; et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann. Oncol. 2017, 28, 368–376. [Google Scholar] [CrossRef]
- Haanen, J.; Ernstoff, M.; Wang, Y.; Menzies, A.; Puzanov, I.; Grivas, P.; Larkin, J.; Peters, S.; Thompson, J.; Obeid, M. Rechallenge patients with immune checkpoint inhibitors following severe immune-related adverse events: Review of the literature and suggested prophylactic strategy. J. Immunother. Cancer 2020, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Gorska, M.; Politynska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaing, T.-H.; Wang, Y.-L.; Chiu, C.-C. Immune Checkpoint Inhibitors for Pediatric Cancers: Is It Still a Stalemate? Pharmaceuticals 2024, 17, 991. https://doi.org/10.3390/ph17080991
Jaing T-H, Wang Y-L, Chiu C-C. Immune Checkpoint Inhibitors for Pediatric Cancers: Is It Still a Stalemate? Pharmaceuticals. 2024; 17(8):991. https://doi.org/10.3390/ph17080991
Chicago/Turabian StyleJaing, Tang-Her, Yi-Lun Wang, and Chia-Chi Chiu. 2024. "Immune Checkpoint Inhibitors for Pediatric Cancers: Is It Still a Stalemate?" Pharmaceuticals 17, no. 8: 991. https://doi.org/10.3390/ph17080991
APA StyleJaing, T.-H., Wang, Y.-L., & Chiu, C.-C. (2024). Immune Checkpoint Inhibitors for Pediatric Cancers: Is It Still a Stalemate? Pharmaceuticals, 17(8), 991. https://doi.org/10.3390/ph17080991