Abstract
The knowledge surrounding the application of immune checkpoint inhibitors (ICIs) in the treatment of pediatric cancers is continuously expanding and evolving. These therapies work by enhancing the body’s natural immune response against tumors, which may have been suppressed by certain pathways. The effectiveness of ICIs in treating adult cancers has been widely acknowledged. However, the results of early phase I/II clinical trials that exclusively targeted the use of ICIs for treating different pediatric cancers have been underwhelming. The response rates to ICIs have generally been modest, except for cases of pediatric classic Hodgkin lymphoma. There seems to be a notable disparity in the immunogenicity of childhood cancers compared to adult cancers, potentially accounting for this phenomenon. On average, childhood cancers tend to have significantly fewer neoantigens. In recent times, there has been a renewed sense of optimism regarding the potential benefits of ICI therapies for specific groups of children with cancer. In initial research, individuals diagnosed with pediatric hypermutated and SMARCB1-deficient cancers have shown remarkable positive outcomes when treated with ICI therapies. This is likely due to the underlying biological factors that promote the expression of neoantigens and inflammation within the tumor. Ongoing trials are diligently assessing the effectiveness of ICIs for pediatric cancer patients in these specific subsets. This review aimed to analyze the safety and effectiveness of ICIs in pediatric patients with different types of highly advanced malignancies.
1. Introduction
The main focus of cytotoxic chemotherapy and targeted therapies is to specifically attack cancer cells. However, immune checkpoint inhibitors (ICIs) function by enhancing the body’s immune response against tumors by interfering with co-inhibitory T-cell signaling. Resistance is a common occurrence in patients receiving both conventional cancer treatments and targeted therapies. Nevertheless, a considerable portion of patients who undergo ICI treatment exhibit enduring responses, suggesting the presence of a lasting immunologic memory.
ICIs have completely transformed the way adult cancers are treated [1]. Following its approval in 2011, the cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibody has been utilized for the treatment of metastatic melanoma; eight ICIs targeting the PD-1 pathway have so far received approval. Currently, these ICIs are utilized in the treatment of 18 distinct forms of cancer [2]. Regrettably, the pediatric experience has been notable, as early studies have shown limited effectiveness of ICIs in children. This study emphasizes the distinction in immune response between pediatric and adult cancers, prompting a renewed emphasis on identifying particular subsets of pediatric tumors that may exhibit a greater level of responsiveness to ICI therapies [3]. We thoroughly explore the mechanisms of ICIs, analyze their utilization in both adult and pediatric cancers, and investigate two potential avenues for advancing the use of ICIs in pediatric patients: hypermutated cancers and SMARCB1-deficient tumors.
One example of an ICI is ipilimumab, a completely humanized monoclonal antibody. It focuses on CTLA-4, which is responsible for suppressing the immune response. In 2011, the US Food and Drug Administration (FDA) approved the utilization of ipilimumab in the treatment of advanced metastatic melanoma [4]. Treatment was continued until there was radiographic progression, unacceptable toxic effects, a decision made by the investigator, or the patient withdrew their consent [5,6,7,8].
2. Exploring the Biological Role of CTLA-4 and PD-1
The identification of CTLA-4 as part of the immunoglobulin gene superfamily in activated CD8 T cells marked a major advancement in the study of immune co-inhibitory molecules. In 1987, the cloning and identification of it was successfully achieved by Jean-François Brunet et al. [9]. In 1991, Jeffrey Ledbetter and Peter Linsley conducted a study that explored the interaction between CD28 and B7 in BMS. Their research indicated that CTLA-4 exhibits a higher binding preference for B7 in comparison to CD28 [10]. In the following year, the groups led by Jeffrey Bluestone and Peter Linsley separately conducted in vivo studies. The studies demonstrated that CTLA4-Ig, a soluble variant of CTLA-4, can extend the lifespan of islet grafts and inhibit the antibody response that is not dependent on T cells. One possible role for CTLA-4 is to regulate T-cell activity in a negative regulatory manner [11,12]. In 1994, Jeffrey Bluestone conducted research that highlighted the important role of CTLA-4 in suppressing the response of T cells [13]. It was demonstrated that the expression of CTLA-4 by activated T cells resulted in the direct binding to B7 on antigen-presenting cells, leading to a decrease in T-cell proliferation and function. James Allison conducted an independent study on the function of CTLA-4 and published his findings in 1995. He discovered that CD28 and CTLA-4 both interact with the ligand B7, but they have contrasting effects on T-cell functionality [14]. In 1995, two different research teams, the Tak Mak group and the Arlene Sharpe group, successfully created mice with a CTLA-4 gene knockout. The mice displayed a notable lymphoproliferative disorder which resulted in premature mortality, indicating that CTLA-4 plays a crucial role in the activation and proliferation of T cells [15,16].
The cloning and functional analysis of the PD-1 gene followed a similar procedure to that of CTLA-4. In 1992, Tasuku Honjo discovered the PD-1 gene by cloning it from a stimulated mouse T-cell hybridoma. This gene belongs to the immunoglobulin gene superfamily [17]. Later on, the Honjo group successfully created mice with a PD-1 gene knockout. However, in contrast to the mice lacking the CTLA-4 gene, the anticipated alterations did not occur rapidly. After a few months, as they aged, the C57BL/6 mice had glomerulonephritis and arthritis similar to lupus. [18]. When compared to T cells from wild-type mice, those obtained from animals lacking the PD-1 gene showed far more activated behavior. This finding supports the notion that PD-1 functions as a co-inhibitory molecule. In 2001, a study documented the presence of an autoimmune disease, specifically cardiomyopathy, in PD-1 gene knockout mice with BALB/c background [19]. Research findings indicate that PD-1 exerts a suppressive effect on the activation of T cells. It is worth mentioning that CTLA-4 and PD-1 may have varying functions, likely because of their unique mechanisms of action. We proceeded to search for a genuine PD-1 ligand that exhibits both physical interaction and inhibitory properties. The PD-L1 ligand was identified by the Lieping Chen group, while the PD-L2 ligand was identified by the Drew Pardoll group [20,21]. Extensive research conducted by Gordon Freeman and his team has revealed that the interaction between receptors and ligands, specifically PD-1/PD-L1 and PD-1/PD-L2, plays a significant role in the negative regulation of T-cell responses [22,23]. In 2004, two separate teams, the Lieping Chen group and the Arlene Sharpe group, successfully generated PD-L1 gene knockout mice [24,25,26]. Mice lacking the PD-L2 gene were created in 2006 by research groups led by Chen Dong and Arlene Sharpe [27,28,29]. It was shown that both ligands may bind directly to PD-1 and improve tolerance of T-cell activation.
James Allison and Tasuku Honjo made significant contributions to cancer immunotherapy with their research on CTLA-4 and PD-1, which led to them being awarded the esteemed 2018 Nobel Prize in Physiology and Medicine [30]. Figure 1 depicts the timeline and important milestones in the development of ICIs.
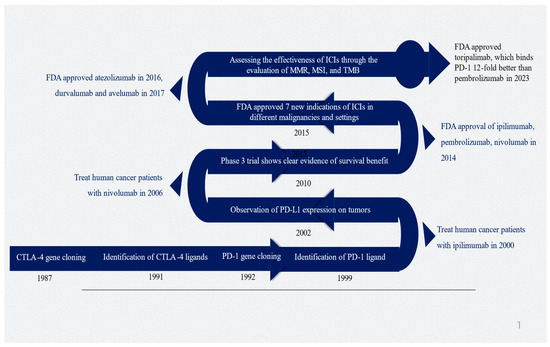
Figure 1.
Timeline of critical milestones for developing immune checkpoint inhibitors. Abbreviations: CTLA-4, cytotoxic T-lymphocyte-associated protein 4; ICI, immune checkpoint inhibitor; MMR, mismatch repair; MSI, microsatellite instability; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; TMD, tumor mutational burden. Figure generated by authors based on the existing literature [9,10,30,31,32,33].
3. Mechanism of Action of Immune Checkpoint Inhibitors
ICIs function by augmenting the body’s inherent T-cell responses against tumors, which might have been compromised by inhibitory pathways. T-cell responses primarily target neoantigens, which are proteins formed due to genetic mutations in cancer cells. This phenomenon occurs as a result of their inherent capacity to elicit an immune response [3]. Neoantigens, like viral antigens, are not found in the normal human proteome, which causes T cells to recognize them as foreign. Upon encountering tumor antigens in the draining lymph nodes, the T cells that target the tumor undergo activation. Migrating to the tumor microenvironment, clonal proliferation, and enhanced effector activity (including cytokine production and cytolytic capability) are all outcomes of the process. When T cells arrive at the tumor site, they come across tumor antigens, which then prompt their activation and subsequent removal of tumor cells. However, once a patient shows signs of disease, their T-cell responses have already been greatly suppressed by different inhibitory pathways. These pathways involve the activation of immune checkpoints, which are types of inhibitory receptors. Immune checkpoints play a crucial role in regulating the activity of tumor-specific T cells, preventing them from effectively eliminating cancer cells. The ICIs work by disrupting the signaling of inhibitory immune checkpoints, which in turn allows tumor-specific T cells to be activated and promotes an immune response against the tumor.
Due to the significant harm caused by conventional therapies, particularly in the developing brains of children, there is a pressing demand for improved treatment strategies. Immunotherapy, which has garnered increasing attention over time, offers a potential solution. Information is scarce regarding the potential application of immunotherapy in brain tumors. While children have generally tolerated ICIs well, their response to these treatments has been extremely limited or nonexistent. Figure 2 depicts the signaling interaction between tumor cells, tumor-associated macrophages, and tumor-infiltrating lymphocytes in the medulloblastoma microenvironment [34]. The initial immune checkpoints that were identified were CTLA-4 and PD-1. Only CTLA-4 and PD-1 have been successfully targeted in clinical settings, despite the discovery and ongoing investigation of several more checkpoints [35]. CTLA-4 is an inhibitory molecule that becomes more active on T cells following the first encounter with an antigen, helping to restrict T-cell activation at this early stage. To achieve successful T-cell activation during priming, two signals are required from antigen-presenting cells. The first signal involves T-cell receptor signaling, which occurs when the T-cell interacts with the antigen in the major histocompatibility complex. The second signal pertains to costimulation via CD28, which occurs when the T-cell engages with B7-1 (CD80) or B7-2 (CD86). Upon receiving these two signals, activated T cells increase the expression of CTLA-4 as a means to regulate excessive activation [36].
CTLA-4 is structurally similar to CD28, but it binds B7-1 and B7-2 ligands more strongly [37]. CTLA-4 expression efficiently outperforms CD28 binding to its ligands and controls the costimulatory signal. This passage discusses how monoclonal antibodies can inhibit the interaction between CTLA-4 and CD28 ligands, resulting in increased effectiveness and duration of T-cell activation. The tumor microenvironment displays increased clonal diversity of tumor-specific T cells, enhanced T-cell migration to tumors, and a higher ratio of effector-to-regulatory T cells. CTLA-4 is an immunological regulator. It interacts with the B7 ligand on antigen-presenting cells, which plays a role in T-cell suppression. The evasion of the immune response against tumor cells can be achieved by inhibiting the activity of cytotoxic T-cells through the CTLA-4/B7 checkpoint. Research suggests that CTLA-4 plays a role in suppressing T-cell activation by affecting T-cell receptor (TCR) signaling, altering CD28 localization, and sequestering B7 ligands from antigen-presenting cells (APCs). CTLA-4 inhibitors function by blocking T-cell inhibitory signals, resulting in heightened T-cell activity and proliferation. This ultimately strengthens the immune response against cancer cells [38].
Activated T cells, B cells, and monocytes are among the immune cells that contain PD-1. It has a significant impact on suppressing the immune system’s response. PD-L1 and PD-L2 ligands are detectable on both antigen-presenting cells (APCs) and tumor cells. The interaction between PD-L1 and PD-1 on T cells leads to the suppression of their function, enabling tumor cells to evade detection by the immune system. The inhibitors impede the interaction, thus promoting the activation of cytotoxic T-cells against the tumor cells. Tumors with heightened PD-L1 expression can effectively evade the immune system and hinder the body’s ability to respond to T cells that combat cancer. One way in which PD-1/PD-L1 inhibitors work is by preventing the immune system from tolerating tumor cells [39].
PD-1 hinders T-cell activity in peripheral tissues following prolonged exposure to antigens, in contrast to CTLA-4 which primarily suppresses T cells during the initial activation process. Prolonged exposure to antigens in the tumor microenvironment leads to T-cell fatigue. This process results in the reprogramming of T cells and a gradual decrease in their effector functioning as time goes on. PD-1 expression is responsible for the development of T-cell exhaustion. This protein helps bring inhibitory phosphatases to the immune synapse by binding to its ligands PD-L1 and PD-L2. This changes the communication between T-cell receptors. The endothelium is one of several tissues that often contain PD-1 ligands. However, in certain instances, cancers can also display elevated levels of PD-L1 [40]. The recruitment of suppressive myeloid cells expressing PD-L1 to the tumor microenvironment can occur, effectively exploiting a pathway that typically aids in preventing immunopathology in healthy tissue. This passage provides a clear and scholarly explanation of how monoclonal antibodies can improve T-cell function and empower them to fight against tumors by inhibiting the PD-1/PD-L1 interaction. It is important to highlight that PD-1 functions differently from CTLA-4, which offers a scientific justification for using ICIs that target both pathways concurrently. Significant progress is being made in the field of brain cancer treatment, both in preclinical and clinical environments. New therapies targeting emerging factors like T-cell immunoglobulin, mucin domain 3 (TIM-3), and indoleamine 2,3-dioxygenase-1 (IDO1) are showing promising results. In addition, there is promising potential in agonistic therapies that focus on the positive stimulatory immune checkpoint CD40 [41,42].
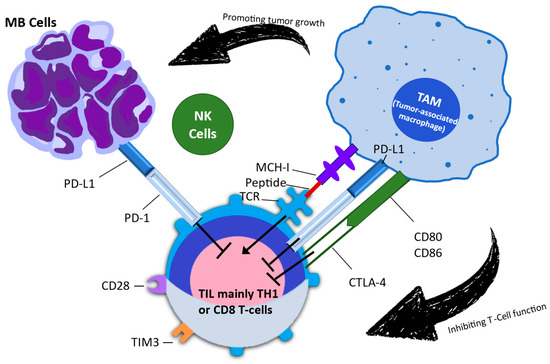
Figure 2.
This figure illustrates the intricate signaling interactions between tumor cells, tumor-associated macrophages, and tumor-infiltrating lymphocytes within the medulloblastoma microenvironment. This figure is based on the research conducted by Kurdi et al. (2023) [43]. MB: medulloblastoma; NK: natural killer; TAM: tumor-associated macrophage; TIL: tumor-infiltrating lymphocyte; TIM-3: T-cell immunoglobulin and mucin domain 3.
Figure 2.
This figure illustrates the intricate signaling interactions between tumor cells, tumor-associated macrophages, and tumor-infiltrating lymphocytes within the medulloblastoma microenvironment. This figure is based on the research conducted by Kurdi et al. (2023) [43]. MB: medulloblastoma; NK: natural killer; TAM: tumor-associated macrophage; TIL: tumor-infiltrating lymphocyte; TIM-3: T-cell immunoglobulin and mucin domain 3.
3.1. Experience with Immune Checkpoint Inhibitors in Pediatric Cancers
The most extensive research with immune checkpoint inhibitors in pediatric cancers has focused on the PD-1 axis. Childhood cancers are often classified as “cold” tumors because of their low tumor mutational burden (TMB), PD-1 expression, and T-cell infiltrates. Four studies conducted between 2020 and 2022 present the most data on the effectiveness of different immunotherapy drugs as monotherapies for recurrent and refractory pediatric tumors [44]. All four studies found that the ICIs were well tolerated by children when administered at weight-based dosing that aligned with the approved adult doses. The dosages used were 3 mg/kg of nivolumab administered every 2 weeks, 2 mg/kg of pembrolizumab given every 3 weeks, 15 mg/kg of atezolizumab administered every 3 weeks, and 20 mg/kg of avelumab given every 2 weeks [45,46]. The adverse events observed in pediatric patients were similar to those seen in adults, except for a higher incidence of cytopenia, including grades 3–4. This could be attributed to the higher level of pretreatment in pediatric patients compared to adults. Furthermore, the most common adverse events observed were constitutional symptoms such as fatigue and fever, in grades 1 and 2. The most prevalent adverse event connected to the immune system was shown to be liver toxicity, most especially increased transaminases grades 1 and 2. In addition, the following immune-related side effects have been reported on rare occasions: colitis, pleural and pericardial effusions, thyroiditis, pancreatitis/elevated lipase, and thyroiditis. Out of the 350 pediatric patients examined in these studies, it is important to highlight that two individuals experienced notable toxicities that may be linked to the treatment.
Immunotherapy drugs targeting PD-1/PD-L1 have demonstrated notable benefits in treating pediatric classic Hodgkin lymphoma, with the disease’s biology playing a crucial role in determining treatment response. Reed–Sternberg cells often display pathological instances of chromosome 9p24.1 mutations, resulting in high PD-L1 and PD-L2 levels [47]. Hodgkin lymphoma’s genetic susceptibility makes it highly responsive to checkpoint blockade, enhancing its potential for treatment. In pediatric patients with relapsed Hodgkin lymphoma, PD-1/PD-L1-targeting ICIs demonstrated an objective response rate of 30% to 60%, similar to that observed in adults. The current FDA approval for pembrolizumab includes this indication.
Despite the various obstacles, there is cause for optimism regarding the potential benefits that children with cancer may experience as a result of advancements in oncology, particularly in the areas of immune amplifiers and ICIs. At the outset, it is observed that pediatric tumors have relatively low mutation burdens upon initial diagnosis. However, it has been noted that the frequency of mutations tends to rise with the administration of chemoradiotherapy [48,49]. Neoantigens can arise from either direct production or mutations in mismatch repair genes. Exploring the combination of checkpoint inhibition and agents that promote non-synonymous somatic mutations (NSSMs) shows great potential in the study of adult cancers. The potential of these NSSMs is significant, as they can facilitate the development of focused therapeutic approaches. Through the identification of precise mutations in cancer cells, researchers can develop drugs that specifically target these mutations. This advancement enables the creation of treatment options that are both more effective and tailored to individual patients [50,51,52,53]. This strategy holds potential for children as well, with a planned study in neuroblastoma (Clinicaltrials.Gov: NCT02914405). Research has shown that radiation can affect the growth of cancers, leading to the emergence of new targets for immune cells involved in tumor suppression [54,55]. In addition, radiation has the potential to improve immune activation and enhance the response to checkpoint blockade. This can be achieved through various mechanisms, including the augmentation of neoantigens. In some intriguing situations, a metastatic location outside of the radiation field was able to regress due to an abscopal effect caused by a combination of targeted radiation and checkpoint blockage [56]. In their study, Leruste and colleagues made an interesting discovery regarding rhabdoid tumors. It was discovered that these tumors have a notable abundance of T cells, including worn-out effector and expanded memory CD8 populations [57]. In addition, there is a high presence of immunosuppressive myeloid-derived cells in these tumors [58].
There is limited experience in targeting CTLA-4 with ipilimumab in pediatrics. Ipilimumab was considered safe, but it appeared to result in more immune-related adverse events compared to PD-1/PD-L1 ICIs, especially at higher doses, similar to those in adults [3]. The FDA has authorized the use of ipilimumab to treat metastatic or unresectable melanoma in children and adolescents over the age of 12, based on these findings and by extending adult data to pediatric patients with comparable tumor characteristics [59]. On the other hand, nivolumab has been approved for a wider range of cancers compared to ipilimumab [60].
Recent studies have shifted their focus to combination ICIs, highlighting the additional benefits observed in adult patients and the biological rationale behind the synergistic effects of CTLA-4 and PD-1 blockade [61]. Regrettably, up to now, the ipilimumab/nivolumab combination does not appear to offer enhanced effectiveness against pediatric solid tumors [62]. When it comes to children, why do ICIs not work as well? Although PD-L1 expression is still rare in pediatric malignancies, there is minimal indication that it is a crucial factor. The apparent lack of effectiveness indicates a fundamental distinction in the immunobiology of cancers in children and adults. Childhood cancers are often categorized as “cold tumors”, known for having low mutational burdens and fewer infiltrating T cells [63]. Ongoing research into the immunobiology of pediatric cancers aims to improve the utilization of immunotherapies for these patients. While ICIs may not be beneficial for most pediatric cancer patients, recent data have identified specific patient groups that may respond well to ICIs. Patients with distinct tumor biology often exhibit increased neoantigen burdens or greater numbers of infiltrating immune cells. Interest has been sparked in tumors with MMRD and SMARCB-1 deficiencies, reigniting enthusiasm for the potential use of ICIs in these patients (Figure 3). Patients with hypermutated tumors, including those with alterations in SMARCB1, may have a more favorable immune response to certain treatments like ICIs if they have neoantigen-reactive T cells [64]. Tumors that have undergone hypermutation may have a higher chance of responding to these therapies [3]. This is because they have a wide range of neoantigens that can be targeted by the activated T cells.
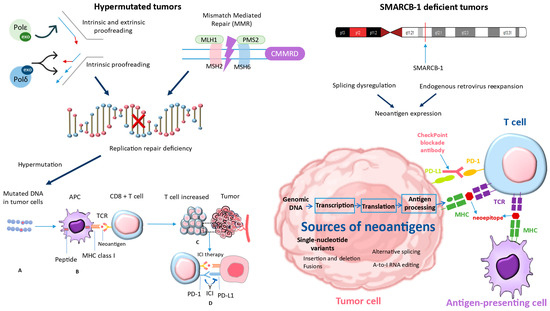
Figure 3.
The studies conducted by Davis et al. (2021) [64] were used to derive the bottom-right corner of Figure 3, while the left part of Figure 3 was derived from Long et al. (2022) [3]. These studies demonstrated that tumors with deficiencies in MMRD and SMARCB-1 have reignited interest in the potential use of ICIs in these patients. MMRD: mismatch repair deficiency; SMARCB-1: SWI/SNF-Related, Matrix-Associated, Actin-Dependent Regulator Of Chromatin, Subfamily B, Member 1.
The mutational burden of a tumor is an inherent characteristic that is associated with the immune response against the tumor and its response to ICI. This correlation is likely due to the increased formation of neoantigens resulting from a higher number of non-synonymous single-nucleotide variants [65]. Microsatellite instability due to a lack of DNA mismatch repair and an improved response to PD-1 inhibition have been linked in studies [66].
ICIs may not be very effective in the general pediatric cancer population, except for Hodgkin lymphoma. The overexpression of PD-L1 due to the amplification of a specific region on chromosome 9p24.1, which includes PD-L1, PD-L2, and JAK2, has been observed in Hodgkin lymphoma. This overexpression is believed to be responsible for the significant clinical response to PD-1 blockade [67]. However, there is a possibility of higher response rates in specific subgroups. This subgroup includes tumors that have a mismatch repair deficiency caused by mutations or epigenetic silencing of certain genes, as well as a polymerase-proofreading deficiency caused by mutations in other genes. The identified deficiencies result in hypermutation and the subsequent generation of neoantigens. These neoantigens have the potential to elicit a response from T cells. One possible subgroup comprises tumors that lack the SMARCB1 gene. Various mechanisms have been proposed, such as dysregulation of splicing, re-expression of endogenous retroviruses leading to neoantigen expression, and enhanced infiltration of tumor immune cells. These processes may be interconnected or influenced by other yet unknown factors [3]. Immunotherapy medications, such as anti-PD-1, enhance the body’s defenses against malignancies by blocking the PD-1–ligand interaction. Consequently, the tumor’s T cells become more activated.
3.2. Exploring Immune Checkpoint Inhibition in Hypermutated Cancers
Pediatric cancers generally exhibit a lower mutational burden in comparison to adult cancers, particularly carcinomas. This difference is likely due to the distinct processes of carcinogenesis [68]. Certain types of adult cancers, such as melanoma, non-small cell lung cancer, and bladder cancer, are particularly responsive to ICI therapies. These cancers typically have a significant number of genetic mutations. Multiple studies have found a correlation between higher TMB and positive outcomes in adult patients with melanoma, non-small cell lung cancer, and colorectal cancer. These outcomes include sustained clinical improvement, favorable radiologic response, and extended periods without disease progression [69,70]. On the other hand, the relatively small number of mutations found in pediatric cancers indicates a reduced presence of neoantigens. As a result, there is a higher chance that the immune system will not identify the tumor as foreign. The relatively low TMB observed in most pediatric cancers may explain the limited efficacy of ICIs in the overall pediatric cancer population [71].
It is crucial to note that the majority of pediatric cancers exhibit a low TMB. However, there is a small subset that shows a significantly higher mutational burden. Among the nearly 3000 pediatric cancers that were examined, a small fraction (5%) displayed a significant TMB (tumor mutational burden) exceeding 10 mutations per megabase [3]. A minuscule fraction (1%) fell under the category of ultra-hypermutated, displaying a TMB surpassing 100 mutations per megabase. Replication repair deficiency can lead to high mutational burdens. This occurs when crucial genes responsible for maintaining the integrity of DNA replication undergo mutations [72]. Replication repair deficiency arises from deficiencies in two key mechanisms: DNA MMR and DNA polymerase proofreading [73]. These mechanisms play a crucial role in ensuring precise replication, either individually or in conjunction. Hypermutated cancers often exhibit a deficiency in DNA MMR, a crucial surveillance system that corrects errors in base-pairing during the process of DNA replication. MMRD is the result of genetic mutations in the MMR genes, namely MLH1, MSH2, MSH6, and PMS2 [74]. In addition, it is worth noting that ultra-hypermutated tumors frequently exhibit a combination of MMRD and polymerase-proofreading deficiency. It is usual to perform this by introducing extra mutations into polymerases ε or δ (POLE or POLD1) [75]. Without proper proofreading and surveillance systems in place, there is a rapid accumulation of additional mutations, resulting in a substantial increase in TMB. Ultimately, this leads to the emergence of numerous new neoantigens capable of eliciting an immune response.
3.3. Normal and Malignant Human Cells Have Different Microsatellite Instability Signals from DNA Polymerase and Mismatch Repair
Both somatic and germline mutations can give rise to tumor MMRD and polymerase-proofreading deficiency. Certain types of cancers that affect the gastrointestinal and genitourinary systems can be attributed to specific genetic mutations. These mutations can occur in genes like POLE and POLD1, as well as in MMR genes, which are associated with Lynch syndrome [76]. Moreover, the lack of MMR function in the germline results in a notable cancer predisposition syndrome known as constitutional MMRD [77]. This syndrome is characterized by the early onset of various types of cancers, including high-grade gliomas, gastrointestinal malignancies, and hematologic malignancies. Cancers often develop in individuals with constitutional MMRD as a result of early somatic-driver mutations in POLE or POLD1 [78]. Hence, mutations can accumulate quickly, with some cases even reaching up to 600 mutations per cell division. Managing patients with these conditions can pose significant challenges due to the aggressive nature of their malignancies, limited response to chemotherapy, and the increased likelihood of concurrent cancer diagnoses. Immunohistochemistry plays a crucial role in the examination of suspected cases. Patients with constitutional MMRD demonstrate a deficiency in MMR protein expression in both tumor and normal tissues. Nevertheless, it is worth noting that individuals with Lynch syndrome will only exhibit a deficiency in MMR expression within tumor cells [79].
An effective way to assess MMRD is by analyzing its influence on MSI, rather than focusing on the broader measurement of TMB [80]. MSI assessment has predominantly been utilized in the examination of adult cancers. Typical panels utilized for MSI testing focus on a restricted set of microsatellites, which are short tandem repetitive DNA sequences. If multiple loci exhibit changes, the tumor is categorized as MSI-high. If a single sequence undergoes mutation, it is categorized as MSI-low [81]. Based on the current circumstances, the tumor is categorized as microsatellite-stable. Nevertheless, the reliability of panel testing might be compromised when conducted on pediatric patients with constitutional MMRD. Panel testing was employed to assess MSI and revealed that a minority of constitutional MMRD cancers displayed MSI-high features. Further investigation of MSI in childhood constitutional MMRD cancers has uncovered the occurrence of microsatellite insertions or deletions throughout the entire genome, rather than being limited to the typical five loci. It has been established that these types of cancers exhibit MSI accumulation, albeit at varying locations. It is crucial to acknowledge that variations in loci can result in a false-negative outcome on panel testing. Caution should be exercised when using standard MSI methodologies to assess the occurrence of MMRD in pediatric cancers.
3.4. Immune Checkpoint Inhibition for Hypermutant Tumors
A report published in 2016 examined the use of ICIs in the treatment of pediatric patients with refractory hypermutated cancers [82]. The case study focused on two brothers who were diagnosed with recurrent multifocal glioblastoma with constitutional MMRD. It also mentioned that they had positive responses to nivolumab. Many children worldwide with constitutional MMRD have received ICIs as a treatment for recurring illnesses when other options are not feasible. The International Replication Repair Deficiency Consortium recently collected data from these patients, which have confirmed significant response rates. The study included a total of 38 patients who were given either nivolumab or pembrolizumab as treatment [83]. Of the 45 cancers examined, 25 tumors, or 55.5% of the cases, showed a positive response in the form of objective responses or stable disease. In patients who underwent a central radiology review, the objective response rate was 42%. Out of this, 17% achieved a complete response, while 25% achieved a partial response. Patients with progressive or recurrent malignancies of the central nervous system had a somewhat worse 3-year survival rate of 39.3%, compared to the total group survival rate of 41.4%. This represents a substantial improvement compared to previous projections. An investigation was carried out to explore the relationship between genomic biomarkers and outcomes following ICI therapy [76]. The study enrolled patients who had germline replication repair deficiency, such as Lynch syndrome, constitutional MMRD, or POLE mutation. These patients also had confirmed tumor MMRD and elevated TMB. The study uncovered a connection between these biomarkers and the effectiveness of treatments. The study revealed a connection between a higher TMB (based on single-nucleotide variants) and enhanced tumor response and overall survival. The study found that the presence of both tumor MMRD and polymerase-proofreading deficiency had a significant impact on the outcomes, as opposed to MMRD alone. Furthermore, it is important to highlight that a strong correlation was found between a higher occurrence of microsatellite insertions or deletions and a favorable response, as well as enhanced overall survival. It is worth noting that this correlation was only observed in tumors with MMRD alone, which also showed a slightly lower TMB. In contrast, there was no observed correlation in tumors that exhibited both MMRD and polymerase-proofreading deficiency. A prediction model study for both replication repair-deficient cancer types showed a good prognosis associated with total microsatellite insertions or deletions and a high number of single-nucleotide variations.
The FDA has approved the use of nivolumab and the combination of nivolumab/ipilimumab in the treatment of recurrent metastatic colorectal cancer. This approval is specifically for patients aged 12 and above who have been designated as having MMRD or MSI-high. For the treatment of recurrent advanced solid tumors with MSI-high designation or MMRD, pembrolizumab received expedited approval by the FDA in 2017, regardless of histology. This approval was extended in June 2020 to include advanced solid cancers with a high TMB of above 10 mutations per megabase. This milestone marked the initial FDA approval predicated on TMB. The approvals were given after the data from the adult phase II KEYNOTE-158 study were released. The study revealed a 34.3% objective response rate in non-colorectal solid tumors classified as MSI-high or with MMRD. Furthermore, the rate of response stood at 29% among patients with hypermutated tumors, characterized by a TMB exceeding 10 mutations per megabase.
In general, there are promising data that support the utilization of ICIs in children who have hypermutated cancers associated with MMRD [84]. The response rates in patients with ultra-hypermutated cancers are particularly noteworthy. These cancers are often linked to constitutional MMRD and the subsequent acquisition of a somatic POLE/POLD1 mutation, which results in the rapid accumulation of mutations. It remains uncertain how tumors with a lower number of mutations (10–100 or even 5–10 mutations per megabase) respond, and whether therapy-induced hypermutation can predict response in the same manner as hypermutation caused by MMRD. It should be noted that in the field of adult malignancies, there seems to be a correlation between the precise underlying cause of high TMB, such as MMRD or polymerase-proofreading deficiency mutation, and better treatment results after ICI therapy. Nevertheless, the increased TMB does not appear to have a direct impact on the enhanced outcome [81]. Additional analysis reveals that the correlation between higher TMB and the efficacy of ICIs is absent in cancer types where TMB does not correspond to neoantigen load or CD8 T-cell infiltration. Hence, while TMB can provide valuable insights into patient eligibility, it is crucial to take into account additional factors, particularly those related to the tumor immune environment, when evaluating the possible effectiveness of ICI therapy. Currently, it is not advisable to suggest routine MSI or TMB testing for all pediatric patients with cancer. If a patient is going through panel, whole exome, or genome sequencing and has a recurrence, it is essential to assess TMB. Patients with constitutional MMRD-associated malignancies or Lynch syndrome must also have TMB testing, even though these cases are very uncommon.
4. How the Body Develops Intolerance to ICI Treatment
Our understanding of the mechanisms behind innate and acquired resistance to ICI therapy is still incomplete. One contributing factor to this is the lack of comprehensive understanding regarding the various clinical, molecular, and immunologic factors associated with the clinical response and long-term advantages of ICI therapy. It is commonly recognized that a successful immune response against tumors after PD-1/PD-L1 blockade requires the reactivation and clonal proliferation of T cells with previous antigen exposure in the tumor microenvironment (TME) [85]. To ensure durable immunologic memory and sustain efficient disease control, a particular subset of effector T cells must undergo differentiation into effector memory T cells. The process is facilitated by the help of CD4+ helper T cells and dendritic cells. The organisms remain viable throughout their entire lifespan and demonstrate a reaction when exposed to antigens [86]. Tumors employ multiple mechanisms within their structure to evade the immune system. The mechanisms encompass genetic and epigenetic alterations that impact the creation, presentation, and processing of neoantigens. In addition, changes in cellular signaling pathways can interfere with the function of cytotoxic T cells. Non-cancerous stromal or immune cells, as well as other systemic effects, may collaborate with cancer cells to enhance tumor development and resistance to ICI; this process is known as creating tumor-extrinsic pathways.
Failure of ICI therapy can occur due to deficiencies in any of the mentioned steps, which can be categorized into three distinct groups: (1) insufficient generation of anti-tumor T cells, (2) inadequate functioning of tumor-specific T cells, and (3) issues with the formation of T-cell memory are potential factors to consider (Figure 4) [33]. Tumor-reactive T-cell formation can be impeded by multiple factors, including inadequate or inappropriate neoantigens, compromised neoantigen processing, and impaired neoantigen presentation [87]. Various methods can be employed to promote immunogenic cell death, such as chemotherapy and radiation. Additionally, activating innate immune responses and enhancing dendritic cell function can help increase antigen presentation. These approaches have the potential to facilitate the development or display of suitable antigens in tumors that lack immune cells and have a non-inflamed tumor microenvironment. In addition, inhibiting immunosuppressive factors could enhance dendritic cell migration, maturation, and function. This could potentially improve T-cell priming and collaboration with ICIs.
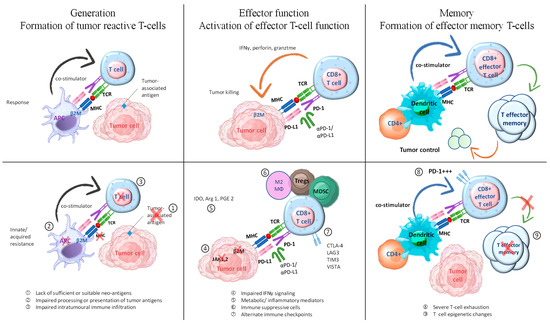
Figure 4.
Exploring combination therapies to address resistance in ICI therapy. The process of generating an effective tumor-directed T-cell response involves several steps. These include the formation of tumor-specific T cells, the activation of effector T-cell function, and the development of effector memory T cells. This figure is derived from the study of Fujiwara et al. (2020) [33].
Checkpoints or co-inhibitory receptors on the immune system, cytokines that suppress the immune system, metabolites that inhibit the immunological system, and cells that suppress the immune system are all components of immune evasion. Numerous combination therapies are currently under development to tackle innate resistance by focusing on potential immune evasion mechanisms within the TME.
5. Adverse Events of Immune Checkpoint Inhibitors
ICIs can lead to immune system overstimulation, which in turn raises the potential for immune-mediated toxicity. Although the exact physiopathology is still unclear, there exist several hypotheses regarding the underlying mechanism responsible for the development of immune-related adverse events (irAEs) [88]. ICIs are widely regarded as a safer alternative to chemotherapy due to their lower incidence of severe side effects. However, it is important to note that ICIs still carry some risks, including the possibility of serious immune-related adverse events. Studies estimate that around 2% of treatment-related deaths can be attributed to ICIs [89]. The pathophysiology of irAEs may differ based on the specific organ involved. Neurological adverse events can be attributed to the release of onconeural peptides that occur when cancer cells die. They initiate the production of onconeural autoantibodies, which can potentially result in harm to the organs [90].
Patients with a variety of advanced malignancies are now being treated with ICIs, which has led to remarkable improvements in their overall health. Nevertheless, when ICIs stimulate the immune system, they also inadvertently disrupt the immune balance in non-tumor tissues, causing significant immune and inflammatory responses. These encompass clinical immune-related adverse events. ICIs are effective in treating tumors, but it is worth noting that a significant percentage of patients, ranging from 30% to 60%, may experience irAEs. Immune checkpoint inhibitors have shown effectiveness in treating different types of cancer, but they can cause specific adverse events related to the immune system in different organs [91]. Immune checkpoints are essential for regulating the T-cell immune response by functioning as negative regulators. The immune system relies on these chemicals to keep tolerance levels high and stop autoimmunity in its tracks. In contrast to the cytotoxic effects of traditional chemotherapy, which occur in cycles and primarily impact rapidly dividing cells, irAEs present as sporadic and unpredictable manifestations that often target specific organs. The skin, colon, liver, endocrine glands, and lungs are frequently involved in this condition [92]. Typically, reactions of grade 3 or higher are deemed sufficient to warrant the permanent discontinuation of ICIs [93].
5.1. Pulmonary Toxicity of Immune Checkpoint Immunotherapy
Tumors can evade the immune system through different mechanisms. These include modifying or eliminating antigens, increasing the expression of immune checkpoint molecules, and manipulating cytokines and oncogenic signaling to establish an immune-suppressive environment within the tumor. There have been associations found between ICI therapy and various lung complications. Checkpoint inhibitor-related pneumonitis (CIP) is one of many possible side effects, along with sarcoid-like granulomatosis, pleural effusion, and worsening of obstructive lung disease. The development of CIP is characterized by increased immune activation mediated by T cells, imbalances in cytokine levels, the upregulation of autoantibodies, and genetic predispositions. These factors collectively contribute to inflammation in the lungs [94]. However, diagnosing CIP solely based on radiographic features is not possible. Several alterations have been detected by CT scans; as many as half of the cases documented in the literature show either ground-glass opacities or consolidative lesions. Research has explored genetic variations as a potential contributor to the occurrence of irAEs, which are frequently associated with autoimmune conditions.
5.2. Cellular Autoimmunity/Higher T-Cell Activity
There is a growing body of evidence suggesting that the upregulation of T cells may be involved in the development of CIP, as illustrated in Figure 5. A thorough assessment of clinical symptoms and radiological findings is the main basis for the diagnosis of CIP. There are a wide variety of radiographic patterns that may be seen in chronic interstitial pneumonia (CIP), including cryptogenic organizing pneumonia (COP), hypersensitivity pneumonitis (HP), non-specific interstitial pneumonia, and acute interstitial pneumonia [95]. Treatment for CIP that exceeds grade 2 typically involves the use of high-dose corticosteroids. Recurrent CIP can occur following steroid treatment, regardless of the status of ICI administration.
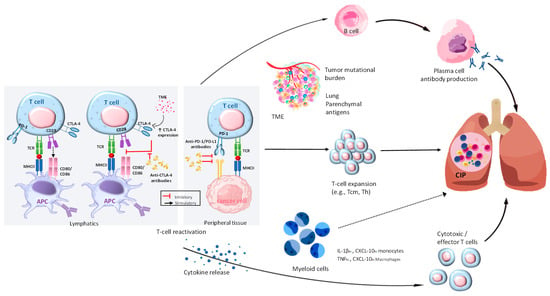
Figure 5.
Exploring the pathophysiological mechanisms in CIP. By utilizing ICI, T cells can overcome the immunosuppression caused by cancer. The activation of different pathways leads to the proliferation of B and plasma cells, which in turn produce autoimmune antibodies like anti-CD74. In addition, they trigger the release of inflammatory cytokines such as IL-1β, TNF-α, and CXCL-10, which can affect various cell types. In addition, T cells, such as Tcm, Th, and clonal T cells, undergo expansion in response to various factors within the tumor microenvironment, the tumor’s mutational burden, and self-antigens located in the lung tissue. The combination of these various pathways can cause inflammation in the lungs, leading to CIP. The role of myeloid cells in CIP is clear, although the exact mechanisms are still not fully understood. Their role in T-cell activation and expansion can be influenced by the T-cell and cytokine environment, potentially leading to pulmonary injury. The presence of clear lines in CIP signifies the existence of established mechanisms, whereas the presence of dotted lines suggests potential mechanisms. This figure is derived from the study of Ghanbar et al. (2024) [94].
5.3. Genetic Predisposition
Genetic variations have been extensively studied as a possible factor in the development of irAEs, which are frequently linked to autoimmunity. Multiple pathways may be involved in the complex interaction of various irAEs, as indicated by the association of several genes with single-nucleotide polymorphisms [96,97,98]. The variations in human leukocyte antigens hold immense importance due to their critical involvement in the interaction of immune cells.
6. Discussion
Autoimmune events, also known as irAEs, are commonly seen as complications of ICIs. Immune-related adverse events (IrAEs) arise from the activation of effector T cells and the suppression of the Treg function, leading to an inflammatory condition. Auto-reactivity in healthy organ tissue occurs as a result [99,100,101]. Checkpoint inhibitors are responsible for the majority of immune-related adverse events (irAEs) in cancer treatment [102].
A study revealed that a notable proportion of individuals who are prescribed ICIs experience adverse events. Furthermore, a notable proportion of individuals, ranging from 17 to 59%, experience severe grade 3 or 4 side effects, as described in the Common Terminology Criteria for Adverse Events (CTCAE). In this study, it was found that around 17% of patients who were given anti-PD-1 monotherapy experienced notable side effects. On the other hand, more than 59% of individuals who underwent ipilimumab and nivolumab therapy encountered grade 3 or 4 toxicities [103].
Monitoring guidelines are necessary because of the notable prevalence of immune side effects. In 2017, the Toxicity Management Working Group was established by the Society for Immunotherapy of Cancer (SITC). This group later published guidelines for monitoring adverse immune events. The group recommends conducting a variety of medical tests before treatment. These tests include blood count, metabolic panel, thyroid studies, cortisol and adreno-corticotropic hormone levels, hemoglobin A1c, electrocardiogram, and various other tests [104]. It is important to establish a foundation by repeating these steps before moving on to future cycles. The assessment of potential adverse events differs depending on the specific organ system involved. Colitis is frequently observed in individuals with cancer and can arise from either infectious or autoimmune causes when undergoing checkpoint inhibitor treatment. Crucial to a thorough evaluation is the use of stool samples for culture, parasites, C. difficile antigens, CMV polymerase chain reaction, inflammatory markers, CT scans, and colonoscopy, if necessary. Pneumonitis is a less common but important adverse event that is associated with higher mortality rates. It is recommended to include chest CT, pulmonary function testing, and a six-minute walking test in the diagnostic evaluation. The development of arthropathy in patients may warrant a comprehensive evaluation to investigate potential rheumatologic causes. Some of the tests that may be included in this examination are MRI, erythrocyte sedimentation rate, C-reactive protein, cyclic citrullinated peptide antibody, rheumatoid factor, and anti-nuclear antibody. Although uncommon, therapy might cause neurologic side effects including neuropathy, transverse myelitis, and aseptic meningitis. Brain MRI and lumbar puncture should be considered as part of the work-up [105].
The management of irAEs involves the use of corticosteroids to suppress the immune response. Recommended for more severe events, high-dose oral corticosteroids (0.5–2 mg/kg) have shown effectiveness as a treatment [99]. If steroids and withholding the checkpoint inhibitor do not yield desired results, more potent immunosuppressants may be considered. For instance, significant progress has been made in the management of irAEs through the utilization of TNF-alpha and calcineurin inhibitors [106,107,108].
Restarting ICIs in patients who have experienced an irAE carries both advantages and potential drawbacks. According to a retrospective study, a significant number of patients experienced a recurrence of the same adverse reaction (24%), or developed a new reaction in a different organ system (26%), upon restarting the same drug [109]. It is worth noting that the administration of a different class of checkpoint inhibitors resulted in a relatively low rate of recurring irAE. In a study conducted by Menzies et al. in 2017, it was found that among 67 patients who experienced irAE while taking ipilimumab, only 2 individuals experienced a recurrence of the same irAE after initiating anti-PD-1 inhibition [110,111]. However, a significant portion of the subjects experienced the emergence of a new irAE as a result of the treatment. However, the authors assert that the reactions were mild and that switching drug classes is a feasible and secure alternative. It is still unclear whether the same applies when moving from PD-1 to CTLA-4 inhibition. While only 70% of PD-1/PD-L1 patients have irAEs, data show that as many as 90% of patients on anti-CTLA-4 treatment do [112].
7. Conclusions
Until now, precision medicine has primarily focused on utilizing the molecular and genomic characteristics of tumors to prescribe targeted small molecules and biologics. However, emerging precision medicine platforms may present new possibilities for customizing treatments for individual patients or specific patient groups. As we continue to uncover the intricacies of how ICIs are responded to and resisted, we must incorporate molecular and functional technologies to create innovative precision immune-oncology platforms. The justification for using ICIs in the adjuvant setting is to eliminate micro-metastases and extend survival. However, it is crucial to not put the cart before the horse.
Author Contributions
Conceptualization, T.-H.J.; methodology, T.-H.J. and Y.-L.W.; writing—original draft preparation, T.-H.J.; writing—review and editing, C.-C.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
No conflicts of interest are declared by the authors.
References
- Ebrahimi, S.; Habibzadeh, A.; Khojasteh-Kaffash, S.; Valizadeh, P.; Samieefar, N.; Rezaei, N. Immune Checkpoint Inhibitors Therapy as the Game-Changing Approach for Pediatric Lymphoma: A Brief Landscape. Crit. Rev. Oncol. Hematol. 2023, 193, 104225. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Kim, H.R.; Ha, S.J. Immune Checkpoint Inhibitors in 10 Years: Contribution of Basic Research and Clinical Application in Cancer Immunotherapy. Immune Netw. 2022, 22, e2. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Morgenstern, D.A.; Leruste, A.; Bourdeaut, F.; Davis, K.L. Checkpoint Immunotherapy in Pediatrics: Here, Gone, and Back Again. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Tentori, L.; Navarra, P. Ipilimumab: A novel immunostimulatory monoclonal antibody for the treatment of cancer. Pharmacol. Res. 2012, 65, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhou, Q.; Gao, Y.; Hu, W.; Lou, G.; Sun, H.; Zhu, J.; Shu, J.; Zhou, X.; Sun, R.; et al. A multicenter phase 2 trial of camrelizumab plus famitinib for women with recurrent or metastatic cervical squamous cell carcinoma. Nat. Commun. 2022, 13, 7581. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos Usta, E.; Yanez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Lu, C.; Mao, L.; Zhu, Y.; Kong, W.; Shen, S.; Tang, M.; Bao, S.; Cheng, H.; Li, G.; et al. PD-1 blockade plus chemoradiotherapy as preoperative therapy for patients with BRPC/LAPC: A biomolecular exploratory, phase II trial. Cell Rep. Med. 2023, 4, 100972. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily-CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Zeng, Y.; Thistlethwaite, J.R.; Montag, A.; Brady, W.; Gibson, M.G.; Linsley, P.S.; Bluestone, J.A. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science 1992, 257, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Wallace, P.M.; Johnson, J.; Gibson, M.G.; Greene, J.L.; Ledbetter, J.A.; Singh, C.; Tepper, M.A. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science 1992, 257, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Tseng, S.Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Flies, D.B.; van Deursen, J.M.; Chen, L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity 2004, 20, 327–336. [Google Scholar] [CrossRef]
- Latchman, Y.E.; Liang, S.C.; Wu, Y.; Chernova, T.; Sobel, R.A.; Klemm, M.; Kuchroo, V.K.; Freeman, G.J.; Sharpe, A.H. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10691–10696. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Park, J.S.; Jeong, Y.H.; Son, J.; Ban, Y.H.; Lee, B.H.; Chen, L.; Chang, J.; Chung, D.H.; Choi, I.; et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J. Immunol. 2015, 194, 5801–5811. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Zhang, Y.; Chung, Y.; Bishop, C.; Daugherty, B.; Chute, H.; Holst, P.; Kurahara, C.; Lott, F.; Sun, N.; Welcher, A.A.; et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl. Acad. Sci. USA 2006, 103, 11695–11700. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Greenwald, R.J.; Latchman, Y.E.; Rosas, L.; Satoskar, A.; Freeman, G.J.; Sharpe, A.H. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur. J. Immunol. 2006, 36, 58–64. [Google Scholar] [CrossRef]
- Guo, Z.S. The 2018 Nobel Prize in medicine goes to cancer immunotherapy (editorial for BMC Cancer). BMC Cancer 2018, 18, 1086. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Mittra, A.; Naqash, A.R.; Takebe, N. A review of mechanisms of resistance to immune checkpoint inhibitors and potential strategies for therapy. Cancer Drug Resist. 2020, 18, 252–275. [Google Scholar] [CrossRef] [PubMed]
- Rechberger, J.S.; Toll, S.A.; Vanbilloen, W.J.F.; Daniels, D.J.; Khatua, S. Exploring the Molecular Complexity of Medulloblastoma: Implications for Diagnosis and Treatment. Diagnostics 2023, 13, 2398. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, D.C.; Theocharopoulos, C.; Lialios, P.P.; Foteinou, D.; Koumprentziotis, I.A.; Xynos, G.; Gogas, H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment. Cancers 2023, 15, 2718. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Voskamp, M.J.; Li, S.; van Daalen, K.R.; Crnko, S.; Ten Broeke, T.; Bovenschen, N. Immunotherapy in Medulloblastoma: Current State of Research, Challenges, and Future Perspectives. Cancers 2021, 13, 5387. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Mulla, N.; Malibary, H.; Bamaga, A.K.; Fadul, M.M.; Faizo, E.; Hakamy, S.; Baeesa, S. Immune microenvironment of medulloblastoma: The association between its molecular subgroups and potential targeted immunotherapeutic receptors. World J. Clin. Oncol. 2023, 14, 117–130. [Google Scholar] [CrossRef]
- Liu, C.; Yang, M.; Zhang, D.; Chen, M.; Zhu, D. Clinical cancer immunotherapy: Current progress and prospects. Front. Immunol. 2022, 13, 961805. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.C.; Joubert, A.M.; Visagie, M.H. Molecular Farming of Pembrolizumab and Nivolumab. Int. J. Mol. Sci. 2023, 24, 10045. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, J.J.; Xing, R.; Zeng, Y.C. Combination therapy: Future directions of immunotherapy in small cell lung cancer. Transl. Oncol. 2021, 14, 100889. [Google Scholar] [CrossRef]
- Sadaf, H.; Ambroziak, M.; Binkowski, R.; Kluebsoongnoen, J.; Paszkiewicz-Kozik, E.; Steciuk, J.; Markowicz, S.; Walewski, J.; Sarnowska, E.; Sarnowski, T.J.; et al. New molecular targets in Hodgkin and Reed-Sternberg cells. Front. Immunol. 2023, 14, 1155468. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef]
- Schramm, A.; Koster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef]
- Haymaker, C.L.; Kim, D.; Uemura, M.; Vence, L.M.; Phillip, A.; McQuail, N.; Brown, P.D.; Fernandez, I.; Hudgens, C.W.; Creasy, C.; et al. Metastatic Melanoma Patient Had a Complete Response with Clonal Expansion after Whole Brain Radiation and PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 100–105. [Google Scholar] [CrossRef]
- Herter-Sprie, G.S.; Koyama, S.; Korideck, H.; Hai, J.; Deng, J.; Li, Y.Y.; Buczkowski, K.A.; Grant, A.K.; Ullas, S.; Rhee, K.; et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 2016, 1, e87415. [Google Scholar] [CrossRef]
- Koller, K.M.; Mackley, H.B.; Liu, J.; Wagner, H.; Talamo, G.; Schell, T.D.; Pameijer, C.; Neves, R.I.; Anderson, B.; Kokolus, K.M.; et al. Improved survival and complete response rates in patients with advanced melanoma treated with concurrent ipilimumab and radiotherapy versus ipilimumab alone. Cancer Biol. Ther. 2017, 18, 36–42. [Google Scholar] [CrossRef]
- Liniker, E.; Menzies, A.M.; Kong, B.Y.; Cooper, A.; Ramanujam, S.; Lo, S.; Kefford, R.F.; Fogarty, G.B.; Guminski, A.; Wang, T.W.; et al. Activity and safety of radiotherapy with anti-PD-1 drug therapy in patients with metastatic melanoma. Oncoimmunology 2016, 5, e1214788. [Google Scholar] [CrossRef]
- Findlay, J.M.; Castro-Giner, F.; Makino, S.; Rayner, E.; Kartsonaki, C.; Cross, W.; Kovac, M.; Ulahannan, D.; Palles, C.; Gillies, R.S.; et al. Differential clonal evolution in oesophageal cancers in response to neoadjuvant chemotherapy. Nat. Commun. 2016, 7, 11111. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Cleary, J.M.; Reardon, B.; Pike, J.; Braunstein, L.Z.; Kim, J.; Amin-Mansour, A.; Miao, D.; Damish, A.; Chin, J.; et al. Genomic Evolution after Chemoradiotherapy in Anal Squamous Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Mazeron, R.; Dercle, L.; Ammari, S.; Canova, C.; Marabelle, A.; Rose, S.; Rubin, E.; Deutsch, E.; Soria, J.C.; et al. Abscopal effect in a Hodgkin lymphoma patient treated by an anti-programmed death 1 antibody. Eur. J. Cancer 2016, 66, 91–94. [Google Scholar] [CrossRef]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J.; et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Yarmarkovich, M.; Maris, J.M. When Cold Is Hot: Immune Checkpoint Inhibition Therapy for Rhabdoid Tumors. Cancer Cell 2019, 36, 575–576. [Google Scholar] [CrossRef]
- Rose, K.; Walson, P.D. Are Regulatory Age Limits in Pediatric Melanoma Justified? Curr. Ther. Res. Clin. Exp. 2019, 90, 113–118. [Google Scholar] [CrossRef]
- Haanen, J.B.; Robert, C. Immune Checkpoint Inhibitors. Prog. Tumor Res. 2015, 42, 55–66. [Google Scholar]
- Liu, W.; Zhang, L.; Xiu, Z.; Guo, J.; Wang, L.; Zhou, Y.; Jiao, Y.; Sun, M.; Cai, J. Combination of Immune Checkpoint Inhibitors with Chemotherapy in Lung Cancer. Onco. Targets Ther. 2020, 13, 7229–7241. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Fox, E.; Isikwei, E.; Reid, J.M.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; Mackall, C.L. A Phase I/II Trial of Nivolumab plus Ipilimumab in Children and Young Adults with Relapsed/Refractory Solid Tumors: A Children’s Oncology Group Study ADVL1412. Clin. Cancer Res. 2022, 28, 5088–5097. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Tarduno, A.; Lu, Y.C. Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy. Cancers 2021, 13, 6061. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Dupain, C.; Harttrampf, A.C.; Urbinati, G.; Geoerger, B.; Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Mol. Ther. Nucleic Acids. 2017, 6, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Valero, C.; Lee, M.; Hoen, D.; Wang, J.; Nadeem, Z.; Patel, N.; Postow, M.A.; Shoushtari, A.N.; Plitas, G.; Balachandran, V.P.; et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat. Genet. 2021, 53, 11–15. [Google Scholar] [CrossRef]
- Gridelli, C.; Peters, S.; Mok, T.; Forde, P.M.; Reck, M.; Attili, I.; de Marinis, F. First-line immunotherapy in advanced non-small-cell lung cancer patients with ECOG performance status 2: Results of an International Expert Panel Meeting by the Italian Association of Thoracic Oncology. ESMO Open 2022, 7, 100355. [Google Scholar] [CrossRef]
- Li, L.; Bai, L.; Lin, H.; Dong, L.; Zhang, R.; Cheng, X.; Liu, Z.; Ouyang, Y.; Ding, K. Multiomics analysis of tumor mutational burden across cancer types. Comput. Struct. Biotechnol. J. 2021, 19, 5637–5646. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Maruvka, Y.E.; Sudhaman, S.; Kelly, J.; Haradhvala, N.J.; Bianchi, V.; Edwards, M.; Forster, V.J.; Nunes, N.M.; Galati, M.A.; et al. DNA Polymerase and Mismatch Repair Exert Distinct Microsatellite Instability Signatures in Normal and Malignant Human Cells. Cancer Discov. 2021, 11, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, G.M. DNA mismatch repair in the context of chromatin. Cells Biosci. 2020, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Sudhaman, S.; Morgenstern, D.; Coblentz, A.; Chung, J.; Stone, S.C.; Alsafwani, N.; Liu, Z.A.; Karsaneh, O.A.A.; Soleimani, S.; et al. Genomic predictors of response to PD-1 inhibition in children with germline DNA replication repair deficiency. Nat. Med. 2022, 28, 125–135. [Google Scholar] [CrossRef]
- Weber, C.A.M.; Kronke, N.; Volk, V.; Auber, B.; Forster, A.; Trost, D.; Geffers, R.; Esmaeilzadeh, M.; Lalk, M.; Nabavi, A.; et al. Rare germline variants in POLE and POLD1 encoding the catalytic subunits of DNA polymerases epsilon and delta in glioma families. Acta Neuropathol. Commun. 2023, 11, 184. [Google Scholar] [CrossRef]
- Gola, M.; Stefaniak, P.; Godlewski, J.; Jereczek-Fossa, B.A.; Starzynska, A. Prospects of POLD1 in Human Cancers: A Review. Cancers 2023, 15, 1905. [Google Scholar] [CrossRef]
- Dencic, T.; Petrovic, A.; Jovicic Milentijevic, M.; Radenkovic, G.; Jovic, M.; Zivkovic, N.; Salinger, S.; Brankovic, B.; Velickov, A.; Ilic, I. The Importance of Immunohistochemical Heterogeneous Expression of MMR Protein in Patients with Colorectal Cancer in Stage II and III of the Disease. Medicina 2023, 59, 489. [Google Scholar] [CrossRef]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cells Int. 2020, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Ciurej, A.; Lewis, E.; Gupte, A.; Al-Antary, E. Checkpoint Immunotherapy in Pediatric Oncology: Will We Say Checkmate Soon? Vaccines 2023, 11, 1843. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, D.; Giannatempo, P.; Grazia, G.; Aiello, M.M.; Bertolini, F.; Mirabile, A.; Buti, S.; Vasile, E.; Scotti, V.; Pisapia, P.; et al. Patients Selection for Immunotherapy in Solid Tumors: Overcome the Naive Vision of a Single Biomarker. Biomed. Res. Int. 2019, 2019, 9056417. [Google Scholar] [CrossRef] [PubMed]
- Westcott, P.M.K.; Muyas, F.; Hauck, H.; Smith, O.C.; Sacks, N.J.; Ely, Z.A.; Jaeger, A.M.; Rideout, W.M., 3rd; Zhang, D.; Bhutkar, A.; et al. Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity. Nat. Genet. 2023, 55, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Koh, C.H.; Lee, S.; Kwak, M.; Kim, B.S.; Chung, Y. CD8 T-cell subsets: Heterogeneity, functions, and therapeutic potential. Exp. Mol. Med. 2023, 55, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of Resistance to Immune Checkpoint Blockade. Am. J. Clin. Dermatol. 2019, 20, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Thouvenin, L.; Olivier, T.; Banna, G.; Addeo, A.; Friedlaender, A. Immune checkpoint inhibitor-induced aseptic meningitis and encephalitis: A case series and narrative review. Ther. Adv. Drug. Saf. 2021, 12, 20420986211004745. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Mohn, N.; Beutel, G.; Gutzmer, R.; Ivanyi, P.; Satzger, I.; Skripuletz, T. Neurological Immune-Related Adverse Events Associated with Nivolumab, Ipilimumab, and Pembrolizumab Therapy-Review of the Literature and Future Outlook. J. Clin. Med. 2019, 8, 1777. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Ibis, B.; Aliazis, K.; Cao, C.; Yenyuwadee, S.; Boussiotis, V.A. Immune-related adverse effects of checkpoint immunotherapy and implications for the treatment of patients with cancer and autoimmune diseases. Front. Immunol. 2023, 14, 1197364. [Google Scholar] [CrossRef]
- Nagpal, C.; Rastogi, S.; Shamim, S.A.; Prakash, S. Re-challenge of immune checkpoint inhibitor pembrolizumab with concurrent tocilizumab after prior grade 3 pneumonitis. Ecancermedicalscience 2023, 17, 1644. [Google Scholar] [CrossRef] [PubMed]
- Ghanbar, M.I.; Suresh, K.; Ghanbar, M.I.; Suresh, K. Pulmonary toxicity of immune checkpoint immunotherapy. J. Clin. Investig. 2024, 134, e170503. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.X.; Huang, W.; Liu, P.P.; Pan, Y.; Cui, Y.H. Dynamic changes in the radiologic manifestation of a recurrent checkpoint inhibitor-related pneumonitis in a non-small cell lung cancer patient: A case report. World J. Clin. Cases 2021, 9, 9108–9113. [Google Scholar] [CrossRef] [PubMed]
- Bins, S.; Basak, E.A.; El Bouazzaoui, S.; Koolen, S.L.W.; Oomen-de Hoop, E.; van der Leest, C.H.; van der Veldt, A.A.M.; Sleijfer, S.; Debets, R.; van Schaik, R.H.N.; et al. Association between single-nucleotide polymorphisms and adverse events in nivolumab-treated non-small cell lung cancer patients. Br. J. Cancer 2018, 118, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.C.; Dorak, M.T.; Bettinotti, M.P.; Bingham, C.O.; Shah, A.A. Association of HLA-DRB1 shared epitope alleles and immune checkpoint inhibitor-induced inflammatory arthritis. Rheumatology 2019, 58, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, N.; Diab, A.; Yu, R.K.; Futreal, A.; Criswell, L.A.; Tayar, J.H.; Dadu, R.; Shannon, V.; Shete, S.S.; Suarez-Almazor, M.E. Genetic determinants of immune-related adverse events in patients with melanoma receiving immune checkpoint inhibitors. Cancer Immunol. Immunother. 2021, 70, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Shojaie, L.; Ali, M.; Iorga, A.; Dara, L. Mechanisms of immune checkpoint inhibitor-mediated liver injury. Acta Pharm. Sin. B 2021, 11, 3727–3739. [Google Scholar] [CrossRef] [PubMed]
- Vilarino, N.; Bruna, J.; Kalofonou, F.; Anastopoulou, G.G.; Argyriou, A.A. Immune-Driven Pathogenesis of Neurotoxicity after Exposure of Cancer Patients to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 5774. [Google Scholar] [CrossRef]
- Teng, Y.S.; Yu, S. Molecular Mechanisms of Cutaneous Immune-Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitors. Curr. Oncol. 2023, 30, 6805–6819. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chavez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suarez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Tomsitz, D.; Ruf, T.; Zierold, S.; French, L.E.; Heinzerling, L. Steroid-Refractory Immune-Related Adverse Events Induced by Checkpoint Inhibitors. Cancers 2023, 15, 2538. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Abu-Sbeih, H.; Ascierto, P.A.; Brufsky, J.; Cappelli, L.C.; Cortazar, F.B.; Gerber, D.E.; Hamad, L.; Hansen, E.; Johnson, D.B.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 2021, 9, e002435. [Google Scholar] [CrossRef]
- Alsalem, A.N.; Scarffe, L.A.; Briemberg, H.R.; Aaroe, A.E.; Harrison, R.A. Neurologic Complications of Cancer Immunotherapy. Curr. Oncol. 2023, 30, 5876–5897. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.; Obeid, M.; Spain, L.; Carbonnel, F.; Wang, Y.; Robert, C.; Lyon, A.R.; Wick, W.; Kostine, M.; Peters, S.; et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 1217–1238. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K.; Committee, E.G. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv264–iv266. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Wolchok, J.D.; Bass, A.R. TNF in the era of immune checkpoint inhibitors: Friend or foe? Nat. Rev. Rheumatol. 2021, 17, 213–223. [Google Scholar] [CrossRef]
- Bylsma, S.; Yun, K.; Patel, S.; Dennis, M.J. Immune Checkpoint Inhibitor Rechallenge After Prior Immune Toxicity. Curr. Treat. Options Oncol. 2022, 23, 1153–1168. [Google Scholar] [CrossRef]
- Menzies, A.M.; Johnson, D.B.; Ramanujam, S.; Atkinson, V.G.; Wong, A.N.M.; Park, J.J.; McQuade, J.L.; Shoushtari, A.N.; Tsai, K.K.; Eroglu, Z.; et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann. Oncol. 2017, 28, 368–376. [Google Scholar] [CrossRef]
- Haanen, J.; Ernstoff, M.; Wang, Y.; Menzies, A.; Puzanov, I.; Grivas, P.; Larkin, J.; Peters, S.; Thompson, J.; Obeid, M. Rechallenge patients with immune checkpoint inhibitors following severe immune-related adverse events: Review of the literature and suggested prophylactic strategy. J. Immunother. Cancer 2020, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Gorska, M.; Politynska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar] [CrossRef] [PubMed]
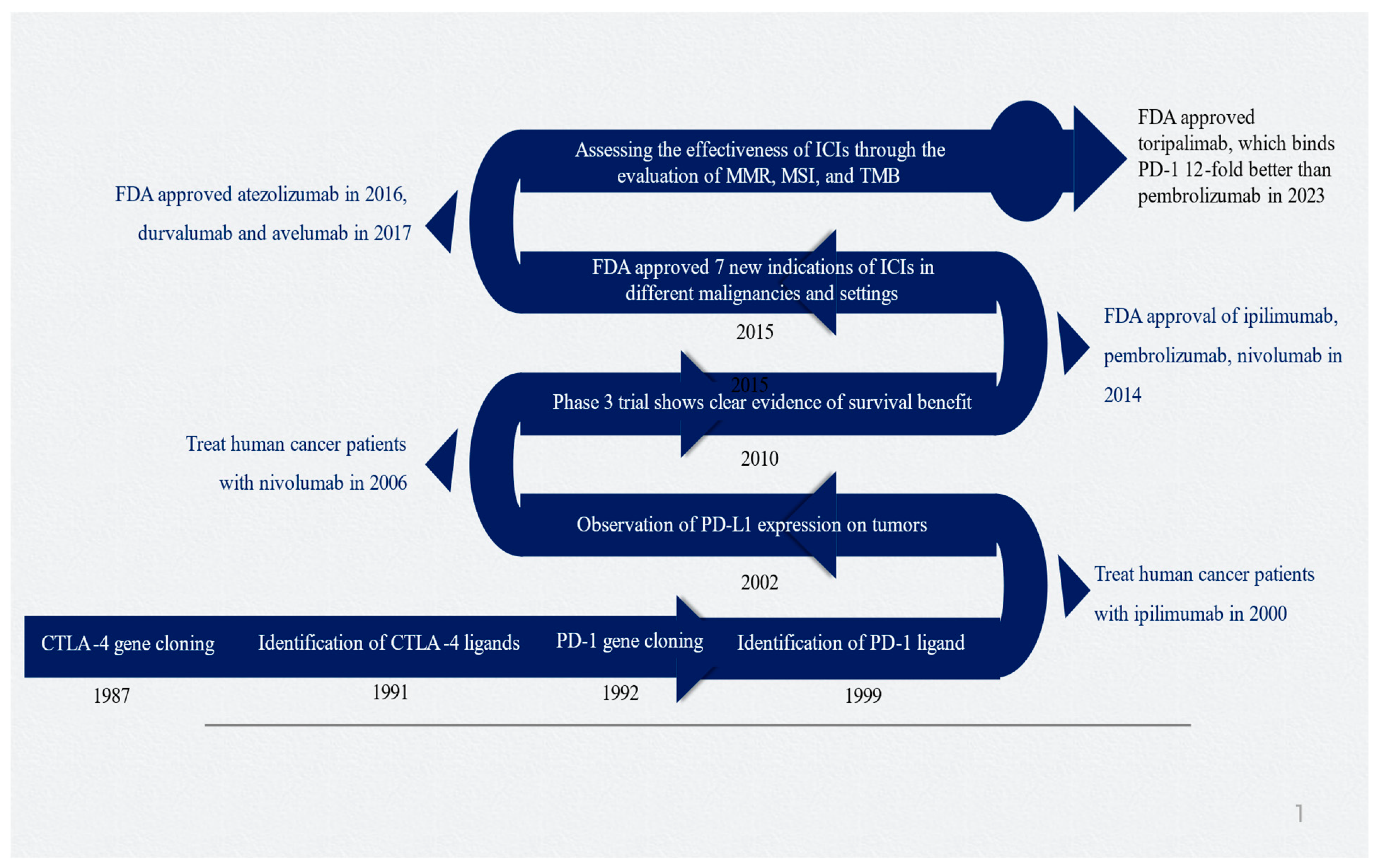
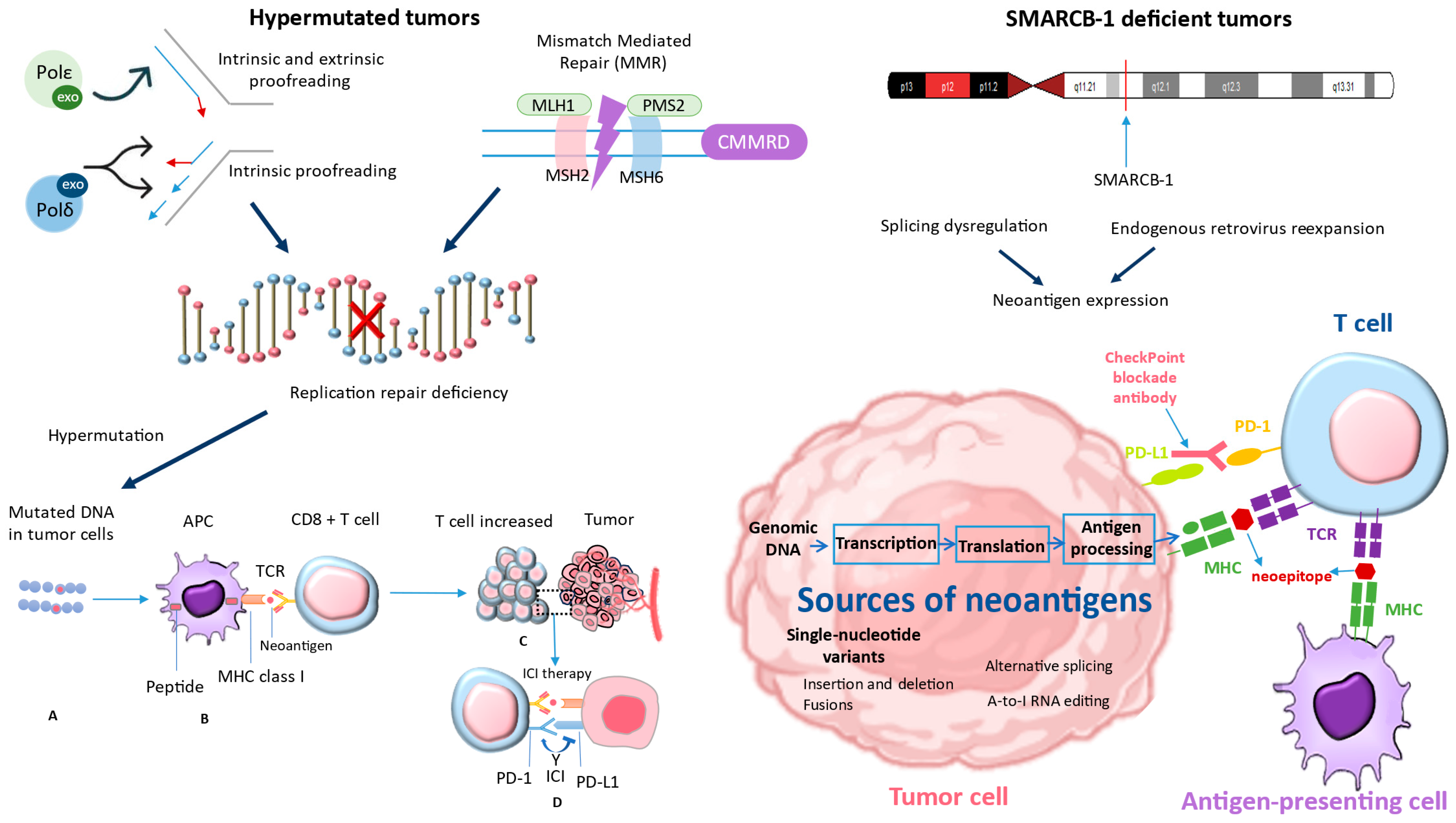
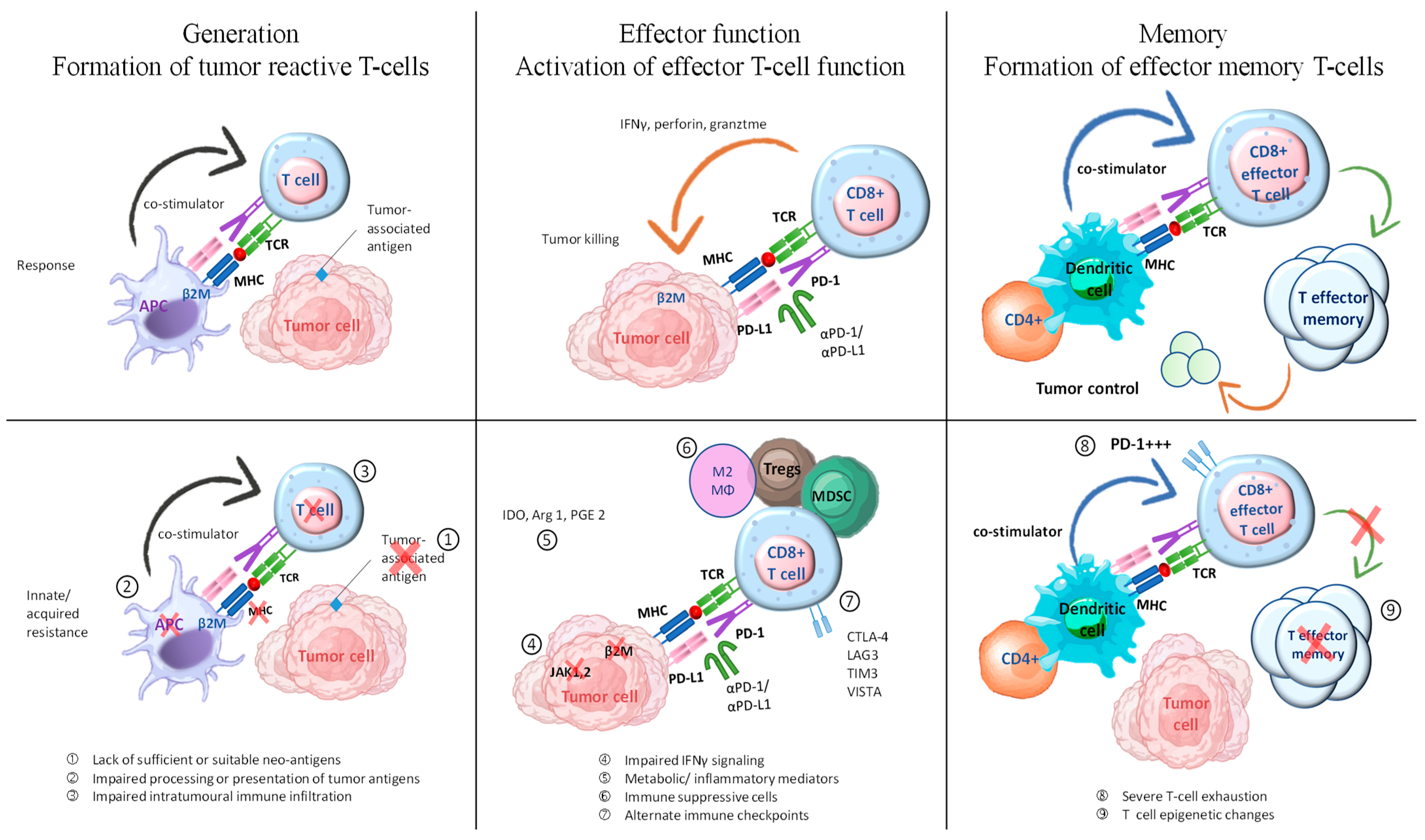
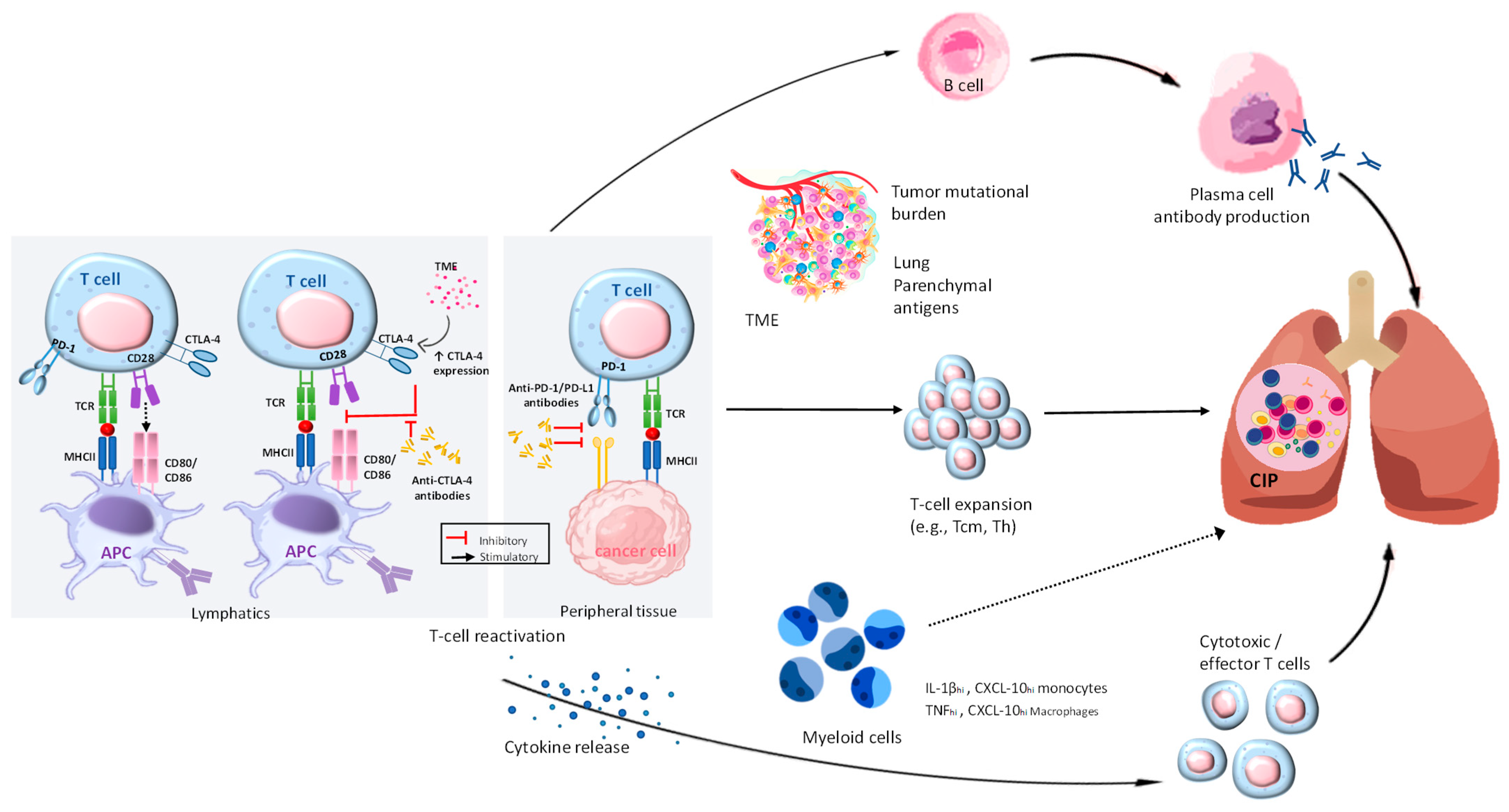
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).