


Do Molecular Fingerprints Identify Diverse Active Drugs in Large-Scale Virtual Screening? (No)

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
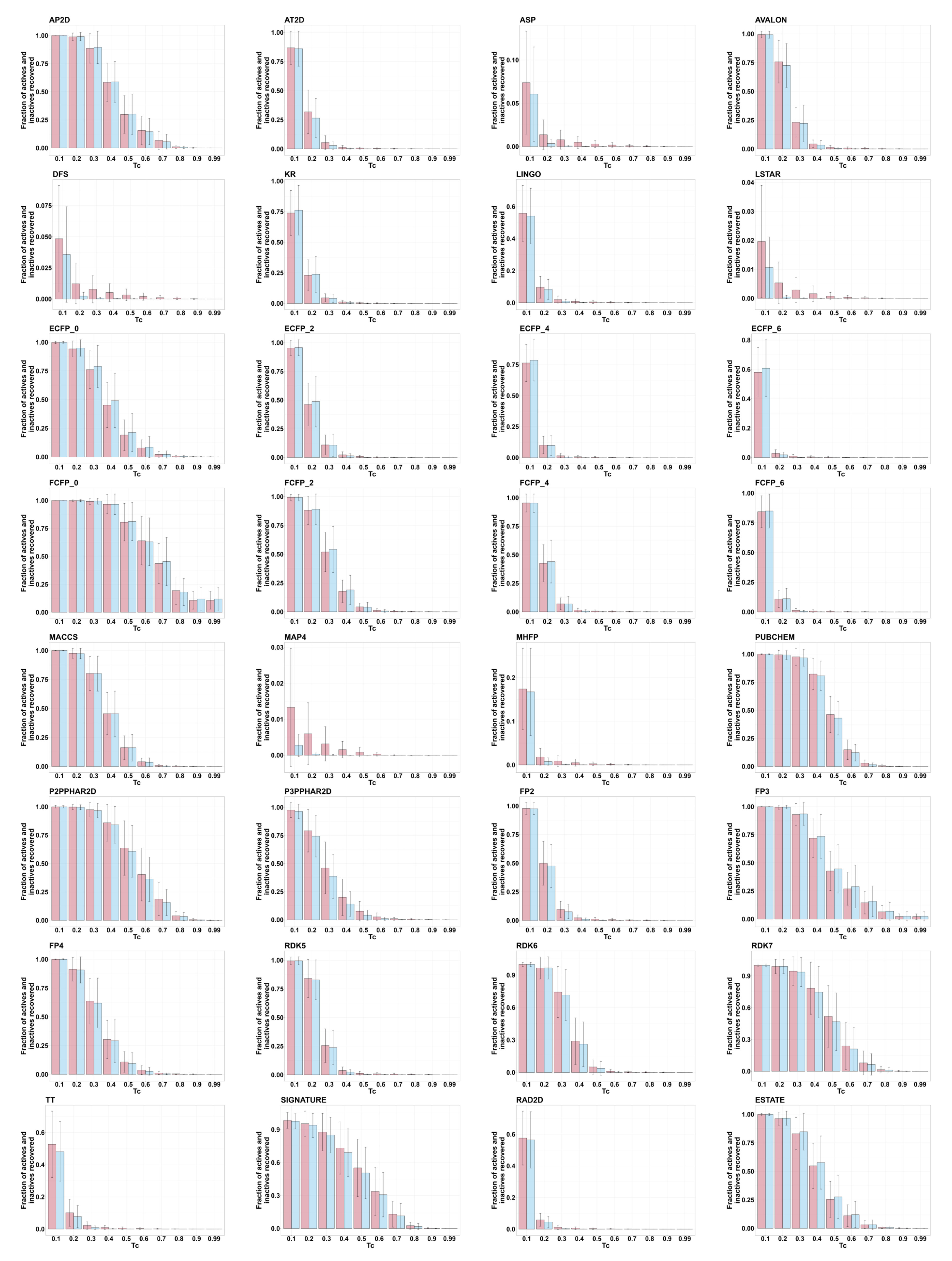
2.1. Enrichment for Active Molecules
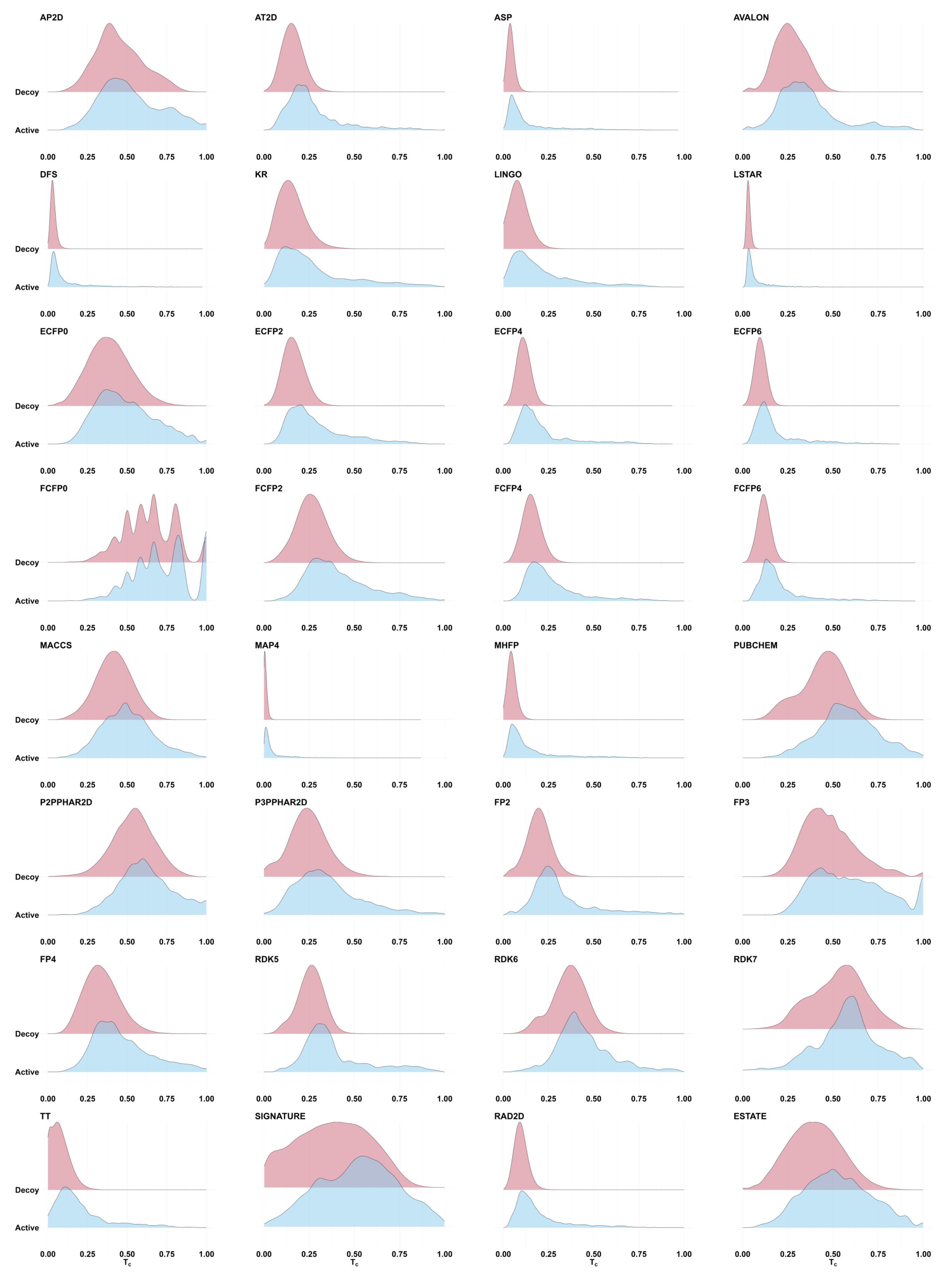
2.2. Tanimoto Similarity Distributions Are Generally Indistinguishable
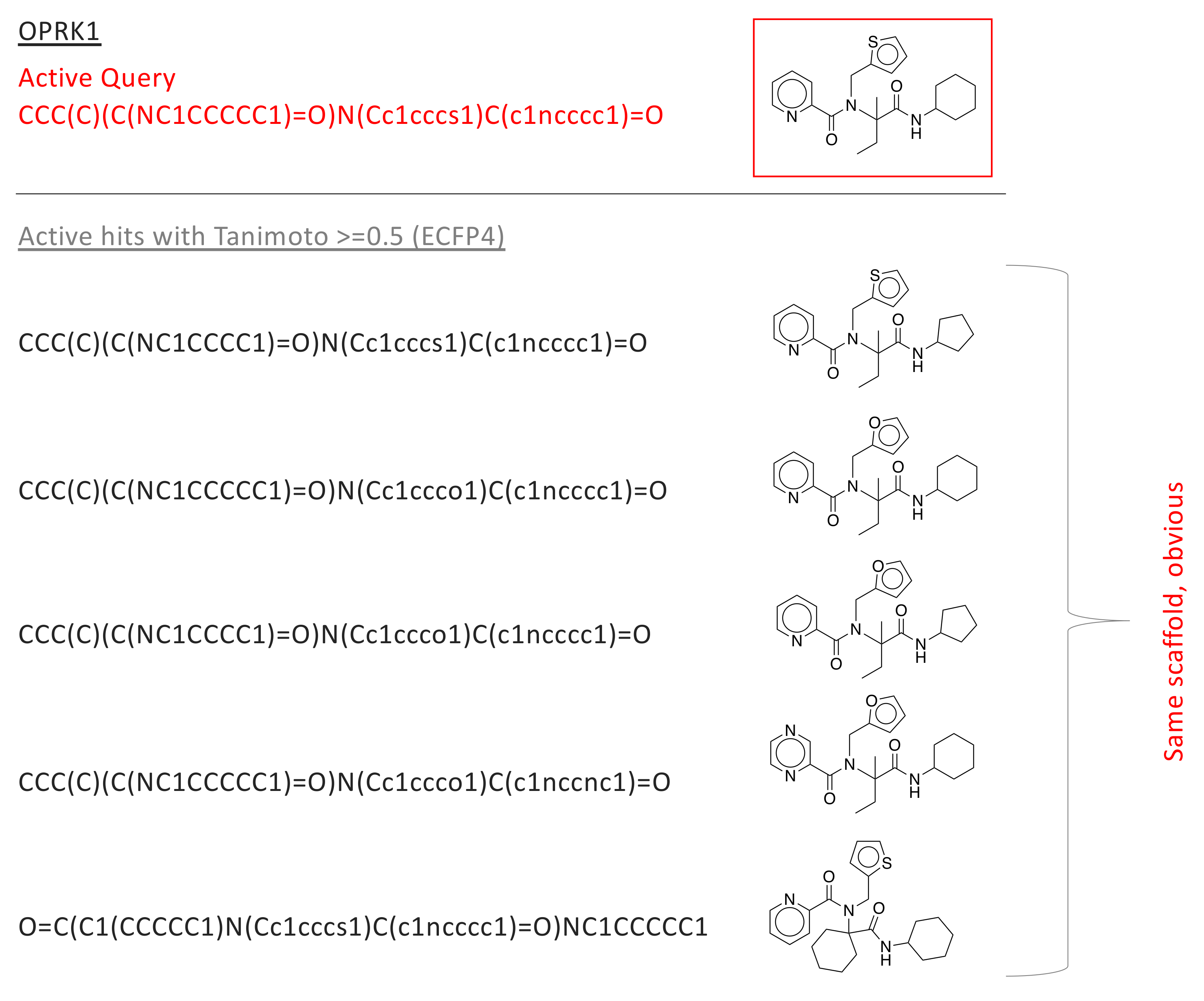
2.3. Tanimoto Score Distributions, and Their Utility in Scaffold Hopping
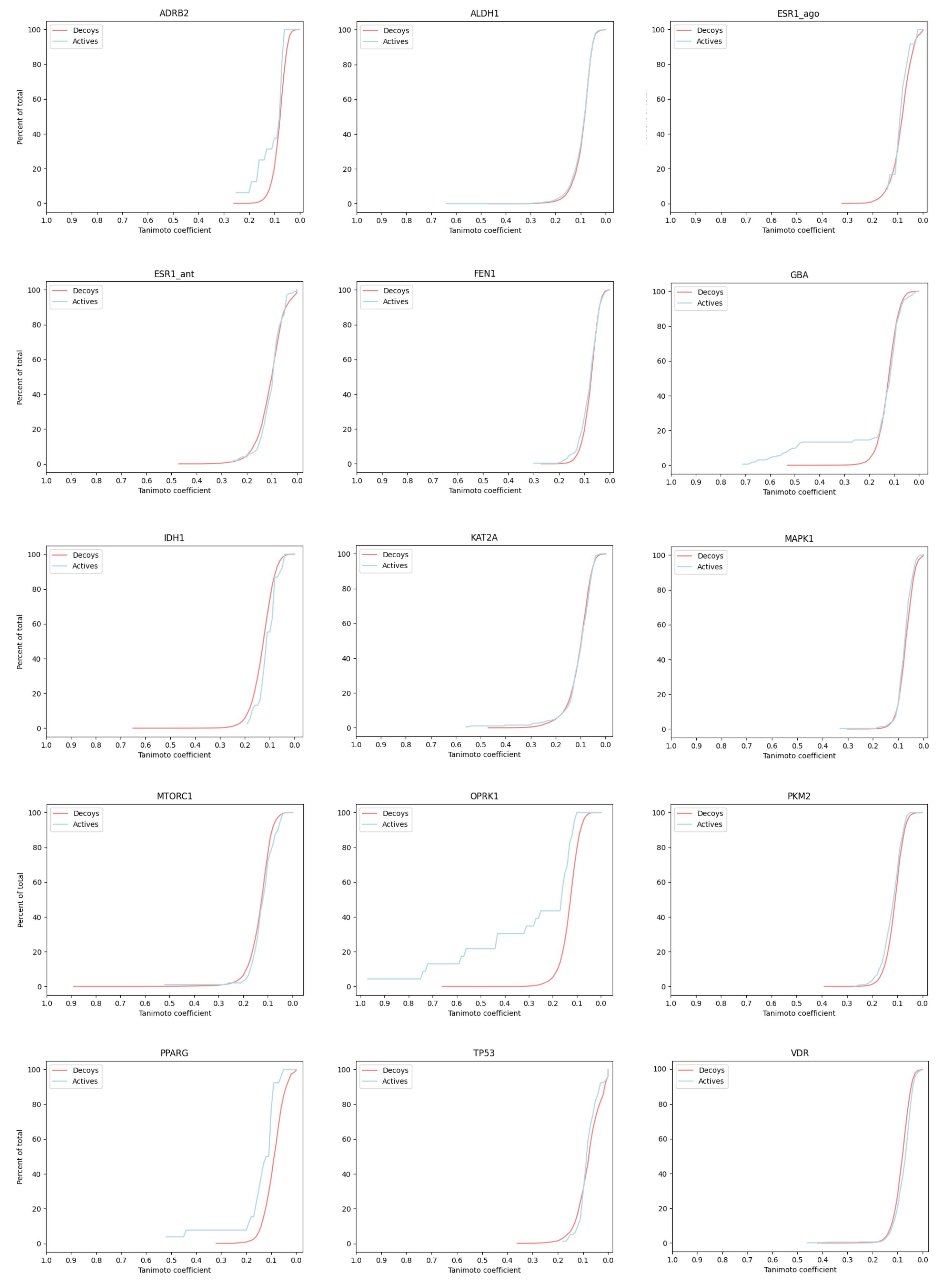
2.4. Evaluation on a Target with Many Validated Inactive Molecules
2.5. Fingerprint Similarity Values Do Not Correlate with Compound Potency
3. Discussion
4. Materials and Methods
4.1. Fingerprint Representations
4.2. Benchmarking Data Sets
- DUD-E: Directory of Useful Decoys, Enhanced [89]: DUD-E is a widely used data set for VS benchmarking, containing data for 102 protein targets. Each target is represented by an average of ∼224 active ligands, ∼90 experimental decoys, and ∼14,000 computational decoys. Compounds are considered active based on a 1 M experimental affinity cutoff, and experimental decoys are ligands with no measurable affinity up to 30 M. Computational decoy ligands are selected from ZINC [83] to have 50 physical properties (rotatable bonds, hydrogen bond acceptors/donors, molecular weight, logP, net charge) similar to the actives, but with low fingerprint (Daylight [90]) Tanimoto coefficient . (Note: this means that computational decoys are, by construction, expected to have low fingerprint similarity).
- MUV: Maximum Unbiased Validation [91]: MUV data sets are based on bioactivity data available in PubChem [48]. This benchmark consists of sets of 30 actives (taken from confirmation assays) and 15,000 decoys (drawn from corresponding primary screens) for each of the 17 targets. The goal of the experimental design is to obtain an optimal spread of actives in the chemical space of the decoys. Since the data are taken from high-throughput screening assays that can be affected by experimental noise and artifacts (caused by unspecific activity of chemical compounds), an assay filter is applied to remove compounds interfering with optical detection methods (autofluorescence and luciferase inhibition) and potential aggregators.
- DEKOIS: The Demanding Evaluation Kits for Objective In silico Screening (DEKOIS) [42] benchmark is based on BindingDB [92] bioactivity data (, , or values). The DEKOIS data set is derived from a set of 15 million molecules randomly selected from ZINC, which are divided into 10,752 bins based on their molecular weight (12 bins), octanol–water partition coefficient (8 bins), number of hydrogen bond acceptors (4 bins), number of hydrogen bond donors (4 bins), and number of rotatable bonds (7 bins). Active ligands are also placed into these pre-defined bins. For each active ligand, 1500 decoys are sampled from the active’s bin (or neighboring bins, if necessary). These are further refined to a final set of 30 structurally diverse decoys per active. The DEKOIS data set includes 81 protein targets found in the DUD-E data set.
- LIT-PCBA: The LIT-PCBA benchmark [43] is a curated subset of the PubChem BioAssay database [48], containing data from experiments where more than 10,000 chemicals were screened against a single protein target, and dose–response curves identified at least 50 actives. Active ligands identified in a bioassay experiment are not guaranteed to bind to the same pocket of the target protein; to overcome this concern, LIT-PCBA includes only targets with representative ligand-bound structures present in the PDB, such that the PDB ligands share the same phenotype or function as the true active ligands from the bioassay experiments. The LIT-PCBA data set was further refined to contain only targets for which at least one of the VS methods (2D fingerprint similarity, 3D shape similarity, and molecular docking) achieved an enrichment in true positives (i.e., the most challenging protein targets have been removed, so that enrichment results are, by design, expected to show good results). Targets in the LIT-PCBA have a variable active to decoy ratio that ranges from as low as 1:20 to 1:19,000.
4.3. Virtual Screening Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| VS | Virtual Screening |
| SPP | Similar Property Principle |
| LBVS | Ligand-Based Virtual Screening |
| DRF | Decoy Retention Factor |
| ROC | Receiver Operating Characteristic |
| AUC | Area Under the Curve |
References
- Sadybekov, A.A.; Sadybekov, A.V.; Liu, Y.; Iliopoulos-Tsoutsouvas, C.; Huang, X.P.; Pickett, J.; Houser, B.; Patel, N.; Tran, N.K.; Tong, F.; et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 2022, 601, 452–459. [Google Scholar] [CrossRef]
- Venkatraman, V.; Colligan, T.H.; Lesica, G.T.; Olson, D.R.; Gaiser, J.; Copeland, C.J.; Wheeler, T.J.; Roy, A. Drugsniffer: An Open Source Workflow for Virtually Screening Billions of Molecules for Binding Affinity to Protein Targets. Front. Pharmacol. 2022, 13, 874746. [Google Scholar] [CrossRef]
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; et al. Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. [Google Scholar] [CrossRef]
- Warr, W.A.; Nicklaus, M.C.; Nicolaou, C.A.; Rarey, M. Exploration of ultralarge compound collections for drug discovery. J. Chem. Inf. Model. 2022, 62, 2021–2034. [Google Scholar] [CrossRef]
- Walters, W.P. Virtual Chemical Libraries. J. Med. Chem. 2018, 62, 1116–1124. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Johnson, M.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2013, 57, 3186–3204. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Škuta, C.; Bender, A.; Svozil, D. QSAR-derived affinity fingerprints (part 2): Modeling performance for potency prediction. J. Cheminf. 2020, 12, 41. [Google Scholar] [CrossRef]
- Venkatraman, V. FP-MAP: An extensive library of fingerprint-based molecular activity prediction tools. Front. Chem. 2023, 11, 1239467. [Google Scholar] [CrossRef]
- Tetko, I.V.; Bruneau, P.; Mewes, H.W.; Rohrer, D.C.; Poda, G.I. Can we estimate the accuracy of ADME–Tox predictions? Drug Discov. Today 2006, 11, 700–707. [Google Scholar] [CrossRef]
- Mellor, C.; Robinson, R.M.; Benigni, R.; Ebbrell, D.; Enoch, S.; Firman, J.; Madden, J.; Pawar, G.; Yang, C.; Cronin, M. Molecular fingerprint-derived similarity measures for toxicological read-across: Recommendations for optimal use. Regul. Toxicol. Pharmacol. 2019, 101, 121–134. [Google Scholar] [CrossRef]
- Venkatraman, V. FP-ADMET: A compendium of fingerprint-based ADMET prediction models. J. Cheminf. 2021, 13, 75. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Falcao, A.O. Structural Similarity Based Kriging for Quantitative Structure Activity and Property Relationship Modeling. J. Chem. Inf. Model. 2014, 54, 1833–1849. [Google Scholar] [CrossRef]
- Vidal, D.; Thormann, M.; Pons, M. LINGO, an Efficient Holographic Text Based Method To Calculate Biophysical Properties and Intermolecular Similarities. J. Chem. Inf. Model. 2005, 45, 386–393. [Google Scholar] [CrossRef]
- Kubinyi, H. Similarity and dissimilarity: A medicinal chemist’s view. Perspect. Drug Discov. Des. 1998, 9, 225–252. [Google Scholar] [CrossRef]
- Maggiora, G.M. On Outliers and Activity CliffsWhy QSAR Often Disappoints. J. Chem. Inf. Model. 2006, 46, 1535. [Google Scholar] [CrossRef]
- Stumpfe, D.; Hu, H.; Bajorath, J. Evolving Concept of Activity Cliffs. ACS Omega 2019, 4, 14360–14368. [Google Scholar] [CrossRef]
- Van Tilborg, D.; Alenicheva, A.; Grisoni, F. Exposing the limitations of molecular machine learning with activity cliffs. J. Chem. Inf. Model. 2022, 62, 5938–5951. [Google Scholar] [CrossRef]
- Barbosa, F.; Horvath, D. Molecular Similarity and Property Similarity. Curr. Top. Med. Chem. 2004, 4, 589–600. [Google Scholar] [CrossRef]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity Searching Using 2D Structural Fingerprints. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; pp. 133–158. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Similarity searching. WIRES Comput. Mol. Sci. 2011, 1, 260–282. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Gao, K.; Nguyen, D.D.; Sresht, V.; Mathiowetz, A.M.; Tu, M.; Wei, G.W. Are 2D fingerprints still valuable for drug discovery? Phys. Chem. Chem. Phys. 2020, 22, 8373–8390. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do Structurally Similar Molecules Have Similar Biological Activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Oprea, T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Venkatraman, V.; Pérez-Nueno, V.I.; Mavridis, L.; Ritchie, D.W. Comprehensive Comparison of Ligand-Based Virtual Screening Tools Against the DUD Data set Reveals Limitations of Current 3D Methods. J. Chem. Inf. Model. 2010, 50, 2079–2093. [Google Scholar] [CrossRef]
- Sciabola, S.; Torella, R.; Nagata, A.; Boehm, M. Critical Assessment of State-of-the-Art Ligand-Based Virtual Screening Methods. Mol. Inf. 2022, 41, 2200103. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Berdini, V.; Hartshorn, M.J.; Mooij, W.T.M.; Murray, C.W.; Taylor, R.D.; Watson, P. Virtual Screening Using Protein-Ligand Docking: Avoiding Artificial Enrichment. J. Chem. Inf. Model. 2004, 44, 793–806. [Google Scholar] [CrossRef]
- Sieg, J.; Flachsenberg, F.; Rarey, M. In Need of Bias Control: Evaluating Chemical Data for Machine Learning in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2019, 59, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sze, K.H.; Lu, G.; Ballester, P.J. Machine-learning scoring functions for structure-based drug lead optimization. WIRES Comput. Mol. Sci. 2020, 10, e1465. [Google Scholar] [CrossRef]
- Imrie, F.; Bradley, A.R.; Deane, C.M. Generating property-matched decoy molecules using deep learning. Bioinformatics 2021, 37, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.M.; Yang, Y.; Balius, T.E.; O’Meara, M.J.; Lyu, J.; Young, J.; Tang, K.; Shoichet, B.K.; Irwin, J.J. Property-Unmatched Decoys in Docking Benchmarks. J. Chem. Inf. Model. 2021, 61, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Réau, M.; Langenfeld, F.; Zagury, J.F.; Lagarde, N.; Montes, M. Decoys Selection in Benchmarking Datasets: Overview and Perspectives. Front. Pharmacol. 2018, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cruz, A.; Ramsey, S.; Dickson, C.J.; Duca, J.S.; Hornak, V.; Koes, D.R.; Kurtzman, T. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS ONE 2019, 14, e0220113. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Sayle, R.A. Comparing structural fingerprints using a literature-based similarity benchmark. J. Cheminf. 2016, 8, 36. [Google Scholar] [CrossRef]
- Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M.; Boeckler, F.M. Evaluation and Optimization of Virtual Screening Workflows with DEKOIS 2.0—A Public Library of Challenging Docking Benchmark Sets. J. Chem. Inf. Model. 2013, 53, 1447–1462. [Google Scholar] [CrossRef]
- Tran-Nguyen, V.K.; Jacquemard, C.; Rognan, D. LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4263–4273. [Google Scholar] [CrossRef]
- Baldi, P.; Nasr, R. When is chemical similarity significant? The statistical distribution of chemical similarity scores and its extreme values. J. Chem. Inf. Model. 2010, 50, 1205–1222. [Google Scholar] [CrossRef]
- O Hagan, S.; Swainston, N.; Handl, J.; Kell, D.B. A rule of 0.5 for the metabolite-likeness of approved pharmaceutical drugs. Metabolomics 2015, 11, 323–339. [Google Scholar] [CrossRef]
- Vogt, M.; Bajorath, J. Modeling tanimoto similarity value distributions and predicting search results. Mol. Inform. 2017, 36, 1600131. [Google Scholar] [CrossRef]
- Verras, A.; Waller, C.L.; Gedeck, P.; Green, D.V.S.; Kogej, T.; Raichurkar, A.; Panda, M.; Shelat, A.A.; Clark, J.; Guy, R.K.; et al. Shared Consensus Machine Learning Models for Predicting Blood Stage Malaria Inhibition. J. Chem. Inf. Model. 2017, 57, 445–453. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Kendall, M.G. A new measure of rank correlation. Biometrika 1938, 30, 81–93. [Google Scholar] [CrossRef]
- Vogt, M.; Bajorath, J. Similarity Searching for Potent Compounds Using Feature Selection. J. Chem. Inf. Model. 2013, 53, 1613–1619. [Google Scholar] [CrossRef]
- Mathea, M.; Klingspohn, W.; Baumann, K. Chemoinformatic classification methods and their applicability domain. Mol. Inform. 2016, 35, 160–180. [Google Scholar] [CrossRef]
- Bender, A. How similar are those molecules after all? Use two descriptors and you will have three different answers. Expert Opin. Drug Discov. 2010, 5, 1141–1151. [Google Scholar] [CrossRef]
- Kilchmann, F.; Marcaida, M.J.; Kotak, S.; Schick, T.; Boss, S.D.; Awale, M.; Gonczy, P.; Reymond, J.L. Discovery of a selective aurora a kinase inhibitor by virtual screening. J. Med. Chem. 2016, 59, 7188–7211. [Google Scholar] [CrossRef]
- Ozhathil, L.C.; Delalande, C.; Bianchi, B.; Nemeth, G.; Kappel, S.; Thomet, U.; Ross-Kaschitza, D.; Simonin, C.; Rubin, M.; Gertsch, J.; et al. Identification of potent and selective small molecule inhibitors of the cation channel TRPM4. Br. J. Pharmacol. 2018, 175, 2504–2519. [Google Scholar] [CrossRef]
- Zhu, D.; Johannsen, S.; Masini, T.; Simonin, C.; Haupenthal, J.; Illarionov, B.; Andreas, A.; Awale, M.; Gierse, R.M.; van der Laan, T.; et al. Discovery of novel drug-like antitubercular hits targeting the MEP pathway enzyme DXPS by strategic application of ligand-based virtual screening. Chem. Sci. 2022, 13, 10686–10698. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, K.; Repasky, M.P.; Leswing, K.; Abel, R.; Shoichet, B.K.; Jerome, S.V. Efficient exploration of chemical space with docking and deep learning. J. Chem. Theory Comput. 2021, 17, 7106–7119. [Google Scholar] [CrossRef]
- Gorantla, R.; Kubincova, A.; Weiße, A.Y.; Mey, A.S. From Proteins to Ligands: Decoding Deep Learning Methods for Binding Affinity Prediction. J. Chem. Inf. Model. 2023, 64, 2496–2507. [Google Scholar] [CrossRef]
- Gentile, F.; Yaacoub, J.C.; Gleave, J.; Fernandez, M.; Ton, A.T.; Ban, F.; Stern, A.; Cherkasov, A. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 2022, 17, 672–697. [Google Scholar] [CrossRef]
- Pérez-Nueno, V.I.; Venkatraman, V.; Mavridis, L.; Clark, T.; Ritchie, D.W. Using Spherical Harmonic Surface Property Representations for Ligand-Based Virtual Screening. Mol. Inform. 2010, 30, 151–159. [Google Scholar] [CrossRef]
- Pérez-Nueno, V.I.; Venkatraman, V.; Mavridis, L.; Ritchie, D.W. Detecting Drug Promiscuity Using Gaussian Ensemble Screening. J. Chem. Inf. Model. 2012, 52, 1948–1961. [Google Scholar] [CrossRef]
- Hofbauer, C.; Lohninger, H.; Aszódi, A. SURFCOMP: A Novel Graph-Based Approach to Molecular Surface Comparison. J. Chem. Inf. Model. 2004, 44, 837–847. [Google Scholar] [CrossRef]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodolà, E.; Boscaini, D.; Bronstein, M.M.; Correia, B.E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2019, 17, 184–192. [Google Scholar] [CrossRef]
- Douguet, D.; Payan, F. sensaas: Shape-based Alignment by Registration of Colored Point-based Surfaces. Mol. Inf. 2020, 39, 2000081. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Jenkins, J.L.; Scheiber, J.; Sukuru, S.C.K.; Glick, M.; Davies, J.W. How Similar Are Similarity Searching Methods? A Principal Component Analysis of Molecular Descriptor Space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Faulon, J.L.; Visco, D.P.; Pophale, R.S. The Signature Molecular Descriptor. 1. Using Extended Valence Sequences in QSAR and QSPR Studies. J. Chem. Inf. Model. 2003, 43, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom pairs as molecular features in structure-activity studies: Definition and applications. J. Chem. Inf. Model. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Hinselmann, G.; Rosenbaum, L.; Jahn, A.; Fechner, N.; Zell, A. jCompoundMapper: An open source Java library and command-line tool for chemical fingerprints. J. Cheminf. 2011, 3, 3. [Google Scholar] [CrossRef]
- Ralaivola, L.; Swamidass, S.J.; Saigo, H.; Baldi, P. Graph kernels for chemical informatics. Neural Netw. 2005, 18, 1093–1110. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Chertó, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2.0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminf. 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.H.; Kier, L.B. Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information. J. Chem. Inf. Model. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Klekota, J.; Roth, F.P. Chemical substructures that enrich for biological activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Model. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, A.; Probst, D.; Reymond, J.L. One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome. J. Cheminf. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Probst, D.; Reymond, J.L. A probabilistic molecular fingerprint for big data settings. J. Cheminf. 2018, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Mahé, P.; Ralaivola, L.; Stoven, V.; Vert, J.P. The Pharmacophore Kernel for Virtual Screening with Support Vector Machines. J. Chem. Inf. Model. 2006, 46, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- PubChem Substructure Fingerprint, Version: 1.3. 2022. Available online: ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/ (accessed on 1 March 2024).
- Landrum, G. RDKit: Open-Source Cheminformatics, 2022. Release: 2022.03.5. Available online: https://www.rdkit.org (accessed on 1 March 2024).
- Raymond, J.W.; Willett, P. Effectiveness of graph-based and fingerprint-based similarity measures for virtual screening of 2D chemical structure databases. J. Comput. Aided Mol. Des. 2002, 16, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminf. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, N.; Zagury, J.F.; Montes, M. Benchmarking Data Sets for the Evaluation of Virtual Ligand Screening Methods: Review and Perspectives. J. Chem. Inf. Model. 2015, 55, 1297–1307. [Google Scholar] [CrossRef]
- Irwin, J.J. Community benchmarks for virtual screening. J. Comput. Aided Mol. Des. 2008, 22, 193–199. [Google Scholar] [CrossRef]
- Tran-Nguyen, V.K.; Rognan, D. Benchmarking Data Sets from PubChem BioAssay Data: Current Scenario and Room for Improvement. Int. J. Mol. Sci. 2020, 21, 4380. [Google Scholar] [CrossRef]
- Nisius, B.; Bajorath, J. Rendering Conventional Molecular Fingerprints for Virtual Screening Independent of Molecular Complexity and Size Effects. ChemMedChem 2010, 5, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Chaput, L.; Martinez-Sanz, J.; Saettel, N.; Mouawad, L. Benchmark of four popular virtual screening programs: Construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminf. 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Shelat, A.; Doman, T.N.; Guy, R.K.; Shoichet, B.K. High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol. 2005, 1, 146–148. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Daylight Chemical Information Systems, I. Daylight Theory Manual, Version 4.9. 2011. Available online: https://www.daylight.com/dayhtml/doc/theory (accessed on 1 March 2024).
- Rohrer, S.G.; Baumann, K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2015, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Truchon, J.F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Clark, R.D.; Webster-Clark, D.J. Managing bias in ROC curves. J. Comput. Aided Mol. Des. 2008, 22, 141–146. [Google Scholar] [CrossRef]
- Lopes, J.C.D.; dos Santos, F.M.; Martins-José, A.; Augustyns, K.; Winter, H.D. The power metric: A new statistically robust enrichment-type metric for virtual screening applications with early recovery capability. J. Cheminf. 2017, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hevener, K.E.; White, S.W.; Lee, R.E.; Boyett, J.M. A statistical framework to evaluate virtual screening. BMC Bioinf. 2009, 10, 225. [Google Scholar] [CrossRef] [PubMed]
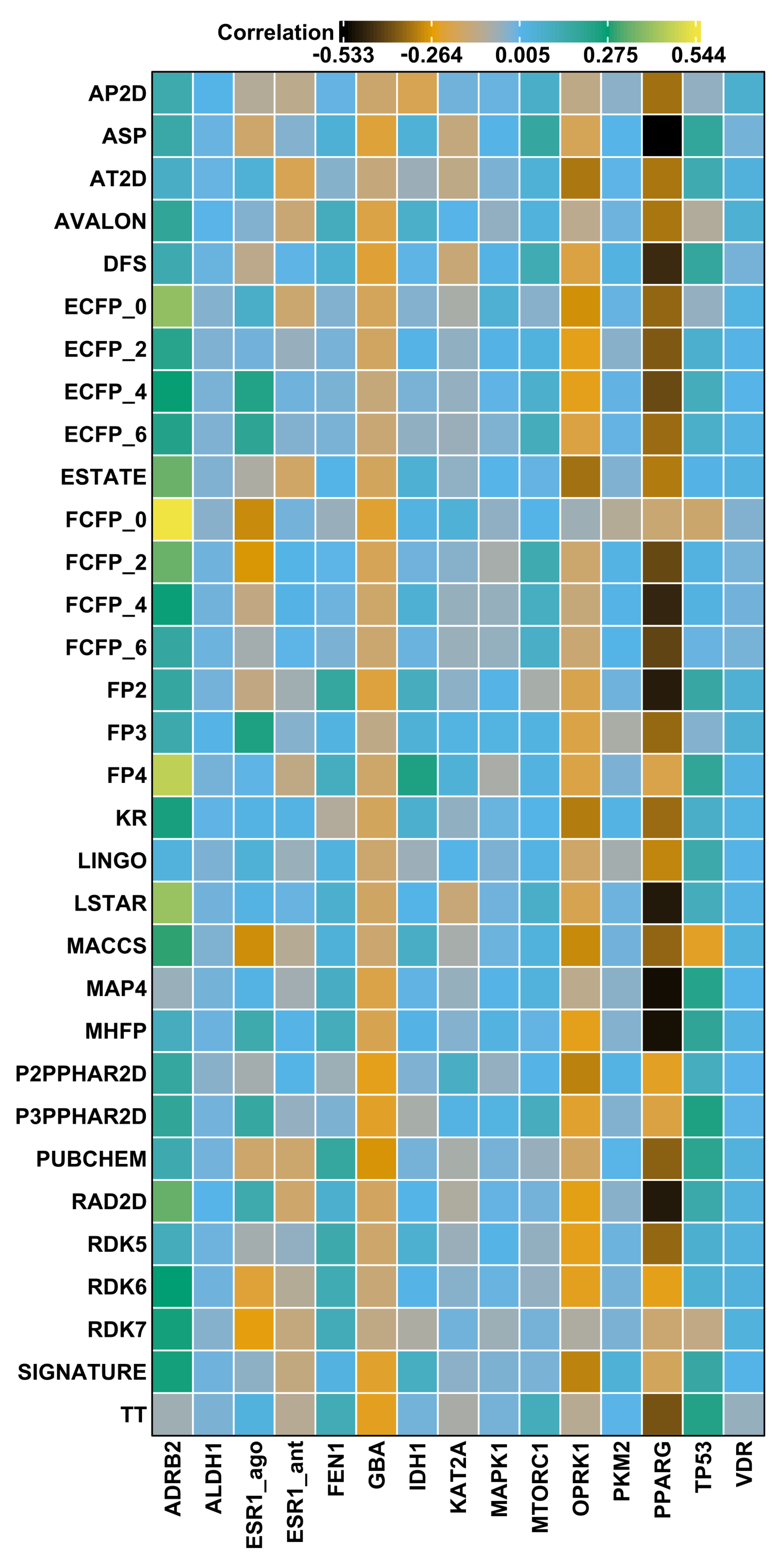
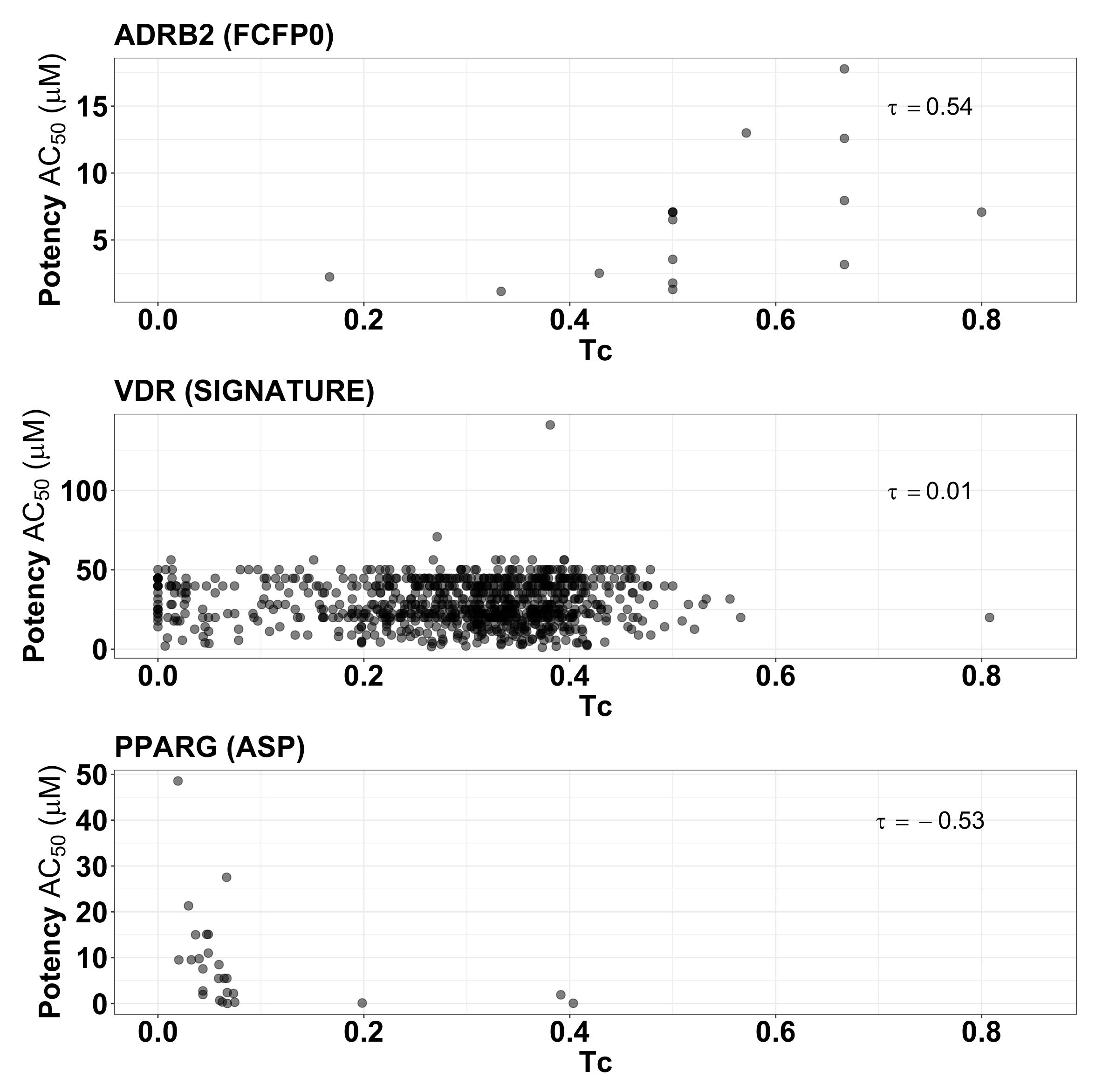

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FP | AUC | DRF | ||||||
|---|---|---|---|---|---|---|---|---|
| DEKOIS | DUDE | MUV | LIT-PCBA | DEKOIS | DUDE | MUV | LIT-PCBA | |
| AP2D | 0.64 | 0.66 | 0.49 | 0.51 | 0.55 | 0.39 | 1.24 | 1.00 |
| AT2D | 0.78 | 0.79 | 0.58 | 0.55 | 0.20 | 0.12 | 0.86 | 0.83 |
| AVALON | 0.72 | 0.73 | 0.60 | 0.55 | 0.30 | 0.18 | 0.85 | 0.97 |
| ESTATE | 0.71 | 0.75 | 0.53 | 0.50 | 0.31 | 0.17 | 0.98 | 0.94 |
| FP3 | 0.68 | 0.77 | 0.51 | 0.52 | 0.45 | 0.20 | 0.89 | 0.83 |
| FP4 | 0.74 | 0.80 | 0.58 | 0.54 | 0.30 | 0.12 | 0.89 | 0.91 |
| MACCS | 0.71 | 0.75 | 0.55 | 0.54 | 0.33 | 0.18 | 0.99 | 0.93 |
| PUBCHEM | 0.76 | 0.76 | 0.55 | 0.54 | 0.28 | 0.20 | 1.06 | 0.97 |
| RDK5 | 0.76 | 0.75 | 0.58 | 0.56 | 0.24 | 0.16 | 0.90 | 0.89 |
| RDK6 | 0.70 | 0.70 | 0.59 | 0.58 | 0.38 | 0.29 | 0.86 | 0.82 |
| RDK7 | 0.62 | 0.63 | 0.58 | 0.59 | 0.74 | 0.70 | 0.98 | 0.85 |
| KR | 0.72 | 0.74 | 0.54 | 0.51 | 0.31 | 0.19 | 1.05 | 0.96 |
| SIGNATURE | 0.72 | 0.72 | 0.55 | 0.54 | 0.33 | 0.23 | 0.97 | 0.99 |
| SUBSTRUCTURE | 0.71 | 0.73 | 0.56 | 0.54 | 0.36 | 0.24 | 0.96 | 0.91 |
| ASP | 0.80 | 0.79 | 0.58 | 0.53 | 0.17 | 0.12 | 0.89 | 0.99 |
| DFS | 0.79 | 0.78 | 0.55 | 0.52 | 0.18 | 0.14 | 0.98 | 1.01 |
| FP2 | 0.79 | 0.78 | 0.55 | 0.54 | 0.20 | 0.14 | 1.01 | 0.90 |
| LSTAR | 0.78 | 0.78 | 0.54 | 0.51 | 0.19 | 0.13 | 0.97 | 1.03 |
| TT | 0.80 | 0.80 | 0.61 | 0.56 | 0.15 | 0.10 | 0.72 | 0.85 |
| PATH | 0.75 | 0.75 | 0.57 | 0.54 | 0.18 | 0.13 | 0.91 | 0.96 |
| ECFP0 | 0.70 | 0.77 | 0.53 | 0.50 | 0.33 | 0.13 | 0.97 | 0.96 |
| ECFP2 | 0.77 | 0.81 | 0.54 | 0.51 | 0.19 | 0.09 | 0.99 | 1.01 |
| ECFP4 | 0.76 | 0.80 | 0.54 | 0.51 | 0.19 | 0.09 | 0.99 | 1.00 |
| ECFP6 | 0.75 | 0.78 | 0.54 | 0.52 | 0.20 | 0.10 | 0.99 | 0.98 |
| FCFP0 | 0.66 | 0.69 | 0.54 | 0.52 | 0.35 | 0.23 | 0.41 | 0.42 |
| FCFP2 | 0.76 | 0.75 | 0.55 | 0.52 | 0.24 | 0.19 | 0.93 | 1.01 |
| FCFP4 | 0.78 | 0.76 | 0.54 | 0.52 | 0.20 | 0.15 | 0.93 | 1.01 |
| FCFP6 | 0.78 | 0.75 | 0.54 | 0.52 | 0.20 | 0.15 | 0.96 | 0.98 |
| MAP4 | 0.81 | 0.83 | 0.56 | 0.54 | 0.14 | 0.07 | 0.91 | 0.85 |
| MHFP | 0.81 | 0.81 | 0.54 | 0.53 | 0.17 | 0.10 | 0.97 | 0.94 |
| RAD2D | 0.76 | 0.77 | 0.53 | 0.53 | 0.23 | 0.14 | 0.99 | 0.93 |
| CIRCULAR | 0.76 | 0.77 | 0.54 | 0.52 | 0.26 | 0.11 | 0.98 | 0.99 |
| P2PPHAR2D | 0.66 | 0.74 | 0.51 | 0.54 | 0.50 | 0.25 | 1.20 | 0.94 |
| P3PPHAR2D | 0.71 | 0.76 | 0.52 | 0.55 | 0.33 | 0.16 | 1.17 | 0.93 |
| PHARMACOPHORE | 0.75 | 0.76 | 0.54 | 0.53 | 0.42 | 0.21 | 1.19 | 0.94 |
| LINGO | 0.77 | 0.79 | 0.54 | 0.54 | 0.21 | 0.10 | 1.02 | 0.91 |
| Protein | Actives | Inactives |
|---|---|---|
| ADRB2 | 17 | 311,748 |
| ALDH1 | 5363 | 101,874 |
| ESR_ago | 13 | 4,378 |
| ESR_antago | 88 | 3,820 |
| FEN1 | 360 | 350,718 |
| GBA | 163 | 291,241 |
| IDH1 | 39 | 358,757 |
| KAT2A | 194 | 342,729 |
| MAPK1 | 308 | 61,567 |
| MTORC1 | 97 | 32,972 |
| OPRK1 | 24 | 269,475 |
| PKM2 | 546 | 244,679 |
| PPARG | 24 | 4,071 |
| TP53 | 64 | 3,345 |
| VDR | 655 | 262,648 |
| TYPE | FAMILY | DESCRIPTION | SIZE (bits) |
|---|---|---|---|
| AP2D [66] | SUBSTRUCTURE | Topological Atom Pairs | 4096 |
| ASP [67] | PATH | All-Shortest Path encoding | 4096 |
| AT2D [67] | SUBSTRUCTURE | Topological Atom Triplets | 4096 |
| AVALON | SUBSTRUCTURE | Enumerates paths and feature classes | 1024 |
| DFS [68] | PATH | All-path encodings | 4096 |
| ECFP_0 [69,70] | CIRCULAR | Extended-connectivity fingerprint of diameter 0 | 1024 |
| ECFP_2 [69,70] | CIRCULAR | Extended-connectivity fingerprint of diameter 2 | 1024 |
| ECFP_4 [69,70] | CIRCULAR | Extended-connectivity fingerprint of diameter 4 | 1024 |
| ECFP_6 [69,70] | CIRCULAR | Extended-connectivity fingerprint of diameter 6 | 1024 |
| ESTATE [70,71] | SUBSTRUCTURE | Fingerprint based on E-State fragments | 79 |
| FCFP_0 [69,70] | CIRCULAR | Feature-class fingerprint of diameter 0 | 1024 |
| FCFP_2 [69,70] | CIRCULAR | Feature-class fingerprint of diameter 2 | 1024 |
| FCFP_4 [69,70] | CIRCULAR | Feature-class fingerprint of diameter 4 | 1024 |
| FCFP_6 [69,70] | CIRCULAR | Feature-class fingerprint of diameter 6 | 1024 |
| FP2 [72] | PATH | Indexes linear fragments up to 7 atoms in length | – |
| FP3 [72] | SUBSTRUCTURE | Based on 55 SMARTS patterns defining functional groups | – |
| FP4 [72] | SUBSTRUCTURE | Based on SMARTS patterns defining functional groups | – |
| KR [70,73] | SUBSTRUCTURE | Klekota–Roth SMARTS-based fingerprint | 4860 |
| LINGO [16,70] | TEXT | Fragmentation of SMILES strings | – |
| LSTAR [67] | PATH | Local Path Environments | 4096 |
| MACCS [74] | SUBSTRUCTURE | Molecular ACCess System structural keys | 166 |
| MAP4 [75] | CIRCULAR | Combines substructure and atom-pair concepts | 2048 |
| MHFP [76] | CIRCULAR | Encodes circular substructures | 2048 |
| P2PPHAR2D [77] | PHARMACOPHORE | Pharmacophore pair encoding | 4096 |
| P3PPHAR2D [77] | PHARMACOPHORE | Pharmacophore triplet encoding | 4096 |
| PUBCHEM [70,78] | SUBSTRUCTURE | Substructure fingerprint | 881 |
| RAD2D [22] | CIRCULAR | Topological Molprint-like fingerprints | 4096 |
| RDK5 [79] | SUBSTRUCTURE | Encodes substructures at most 5 bonds long | 1024 |
| RDK6 [79] | SUBSTRUCTURE | Encodes substructures at most 6 bonds long | 1024 |
| RDK7 [79] | SUBSTRUCTURE | Encodes substructures at most 7 bonds long | 1024 |
| SIGNATURE [65,70] | SUBSTRUCTURE | Based on an array of atom signatures | – |
| TT [66] | PATH | Based on bond paths of four non-hydrogen atoms | – |
| Data Set | Active Source | Decoy Generation | Comments |
|---|---|---|---|
| DUD-E | ChEMBL09 | Total of 0.65% from experiments. Total of 99.35% generated choosing different topologies with similar chemical properties using 2D similarity methods. | No rigorous method to remove false positives. Decoys biased towards 2D similarity methods. |
| DEKOIS | DUD (from literature) [87] | Decoys generated choosing different topologies with similar chemical properties using 2D similarity methods. | Low active to decoy ratio. Decoys biased towards 2D similarity methods |
| MUV | PubChem BioAssay | Unbiased distribution of decoys from experimentally available data. | Data processed to remove false positives and assay artifacts [88]. Low active-to-decoy ratio. |
| LIT-PCBA | PubChem BioAssay | Decoys were chosen from experimentally available data and pruned to have chemical properties similar to actives. | High active to decoy ratio. Actives may not bind to the same pocket of a target. Variable performance in 2D and 3D similarity search and docking across different target sets. Data processed to remove false positives and assay artifacts [88]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatraman, V.; Gaiser, J.; Demekas, D.; Roy, A.; Xiong, R.; Wheeler, T.J. Do Molecular Fingerprints Identify Diverse Active Drugs in Large-Scale Virtual Screening? (No). Pharmaceuticals 2024, 17, 992. https://doi.org/10.3390/ph17080992
Venkatraman V, Gaiser J, Demekas D, Roy A, Xiong R, Wheeler TJ. Do Molecular Fingerprints Identify Diverse Active Drugs in Large-Scale Virtual Screening? (No). Pharmaceuticals. 2024; 17(8):992. https://doi.org/10.3390/ph17080992
Chicago/Turabian StyleVenkatraman, Vishwesh, Jeremiah Gaiser, Daphne Demekas, Amitava Roy, Rui Xiong, and Travis J. Wheeler. 2024. "Do Molecular Fingerprints Identify Diverse Active Drugs in Large-Scale Virtual Screening? (No)" Pharmaceuticals 17, no. 8: 992. https://doi.org/10.3390/ph17080992
APA StyleVenkatraman, V., Gaiser, J., Demekas, D., Roy, A., Xiong, R., & Wheeler, T. J. (2024). Do Molecular Fingerprints Identify Diverse Active Drugs in Large-Scale Virtual Screening? (No). Pharmaceuticals, 17(8), 992. https://doi.org/10.3390/ph17080992