
Quantitative Proteomics and Molecular Mechanisms of Non-Hodgkin Lymphoma Mice Treated with Incomptine A, Part II

 ,
,  , , ,
, , ,  , and
, and 
Abstract
:
1. Introduction
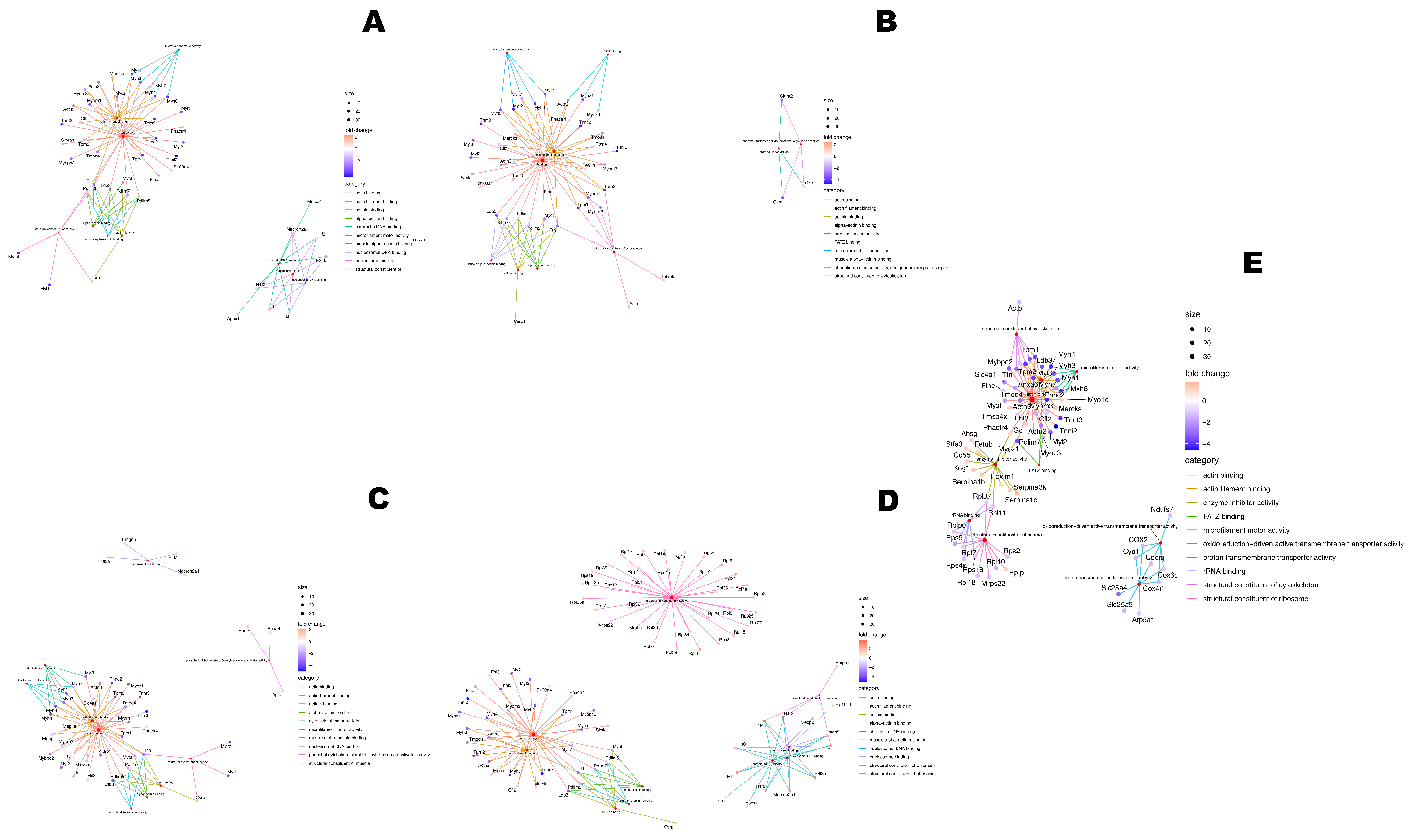
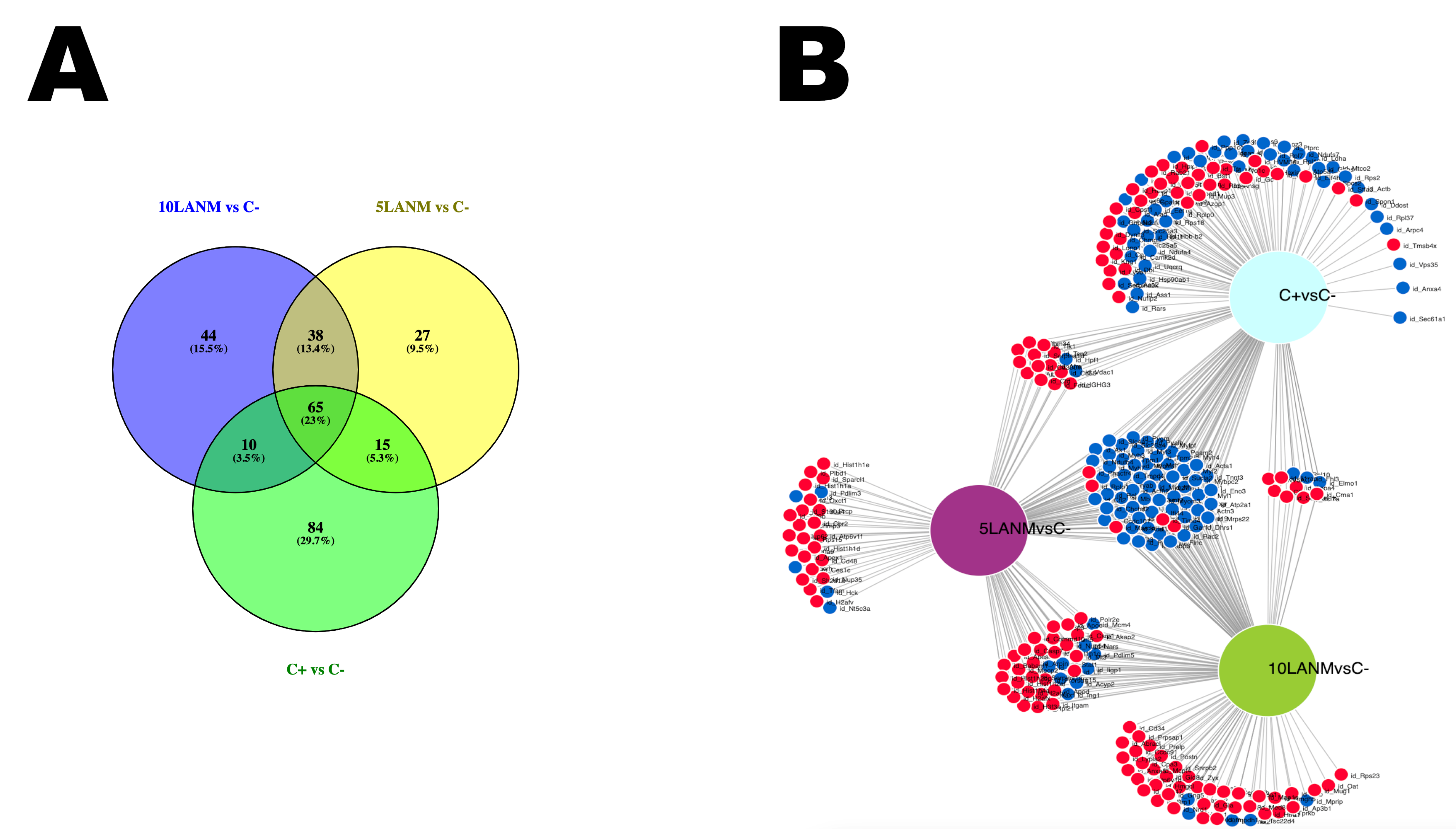
2. Results
3. Discussion
4. Materials and Methods
4.1. Incomptine A (IA) Isolation
4.2. Chemicals and Instrumentation
4.3. Cell Culture Conditions
4.4. Animals
4.4.1. Anti-Lymphoma Treatments
4.4.2. In Vivo Lymphoma Male Balb/c Mice Model
4.5. Non-Hodgkin’s Lymphoma Protein Expression Induced Through IA
Sample Preparation for TMT-Based Proteomic Analysis
4.6. Nano LC-MS/MS Analyses
4.7. Data Analysis
4.8. Differential Protein Analysis
4.9. Bioinformatic Methodology
4.10. In Silico Studies, Molecular Docking
In Silico Physicochemical, Pharmacokinetic and Toxicological Properties
4.11. Comparison of Shared Processes and Molecules
5. Conclusions
Strengths and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Palacio-Mejia, L.S.; Hernandez-Avila, J.E.; Hernandez-Avila, M.; Dyer-Leal, D.; Barranco, A.; Quezada-Sanchez, A.D.; Alvarez-Aceves, M.; Cortes-Alcala, R.; Fernandez-Wheatley, J.L.; Ordonez-Hernandez, I.; et al. Leading causes of excess mortality in Mexico during the COVID-19 pandemic 2020-2021: A death certificates study in a middle-income country. Lancet Reg. Health Am. 2022, 13, 100303. [Google Scholar] [CrossRef] [PubMed]
- INEGI. Estadísticas de Mortalidad/Defunciones Registradas (Mortalidad General). Available online: https://www.inegi.org.mx/programas/mortalidad/#Tabulados (accessed on 24 March 2023).
- Calzada, F.; Bautista, E.; Hidalgo-Figueroa, S.; Garcia-Hernandez, N.; Barbosa, E.; Velazquez, C.; Ordonez-Razo, R.M.; Arietta-Garcia, A.G. Antilymphoma Effect of Incomptine A: In Vivo, In Silico, and Toxicological Studies. Molecules 2021, 26, 6646. [Google Scholar] [CrossRef]
- Hernandez-Ruiz, E.; Alvarado-Ibarra, M.; Juan Lien-Chang, L.E.; Banda-Garcia, L.; Aquino-Salgado, J.L.; Barragan-Ibanez, G.; Ramirez-Romero, E.F.; Nolasco-Cancino, C.; Herrera-Olivares, W.; Morales-Adrian, J.J.; et al. Epidemiology and Clinical Characteristics of Non-Hodgkin Lymphoma in Mexico. World J. Oncol. 2021, 12, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Calzada, F.; Ramirez-Santos, J.; Valdes, M.; Garcia-Hernandez, N.; Pina-Jiménez, E.; Ordoñez-Razo, R.M. Evaluation of Acute Oral Toxicity, Brine Shrimp Lethality, and Antilymphoma Activity of Geranylgeraniol and Annona macroprophyllata Leaf Extracts. Rev. Bras. Farmacogn. 2020, 30, 301–304. [Google Scholar] [CrossRef]
- Howard, S.C.; McCormick, J.; Pui, C.H.; Buddington, R.K.; Harvey, R.D. Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 2016, 21, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Dominguez, J.; Marchat, L.A.; Lopez-Camarillo, C.; Mendoza-Hernandez, G.; Sanchez-Espindola, E.; Calzada, F.; Ortega-Hernandez, A.; Sanchez-Monroy, V.; Ramirez-Moreno, E. Effect of the sesquiterpene lactone incomptine A in the energy metabolism of Entamoeba histolytica. Exp. Parasitol. 2013, 135, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, M.; Yazdy, M.S.; Cheson, B.D. Targeting Biology in Non-Hodgkin Lymphoma. Hematol. Oncol. Clin. N. Am. 2019, 33, 727–738. [Google Scholar] [CrossRef]
- Valla, K.; Flowers, C.R.; Koff, J.L. Targeting the B cell receptor pathway in non-Hodgkin lymphoma. Expert Opin. Investig. Drugs 2018, 27, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar] [CrossRef]
- Pina-Jimenez, E.; Calzada, F.; Bautista, E.; Ordonez-Razo, R.M.; Velazquez, C.; Barbosa, E.; Garcia-Hernandez, N. Incomptine A Induces Apoptosis, ROS Production and a Differential Protein Expression on Non-Hodgkin’s Lymphoma Cells. Int. J. Mol. Sci. 2021, 22, 10516. [Google Scholar] [CrossRef]
- Calzada, F.; Garcia-Hernandez, N.; Hidalgo-Figueroa, S.; Bautista, E.; Barbosa, E.; Velazquez, C.; Hernandez-Caballero, M.E. Expanding the Study of the Cytotoxicity of Incomptines A and B against Leukemia Cells. Molecules 2022, 27, 1687. [Google Scholar] [CrossRef]
- Rauniyar, N.; Yates, J.R., III. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Elias, J.E. Relative Protein Quantification Using Tandem Mass Tag Mass Spectrometry. Methods Mol. Biol. 2017, 1550, 185–198. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A.; Alliance, A.L.; Lymphoma, G.; Eastern Cooperative Oncology, G.; et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J. Clin. Oncol. 2014, 32, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Masai, H. DNA binding and helicase actions of mouse MCM4/6/7 helicase. Nucleic Acids Res. 2005, 33, 3033–3047. [Google Scholar] [CrossRef]
- Rochman, M.; Postnikov, Y.; Correll, S.; Malicet, C.; Wincovitch, S.; Karpova, T.S.; McNally, J.G.; Wu, X.; Bubunenko, N.A.; Grigoryev, S.; et al. The interaction of NSBP1/HMGN5 with nucleosomes in euchromatin counteracts linker histone-mediated chromatin compaction and modulates transcription. Mol. Cell 2009, 35, 642–656. [Google Scholar] [CrossRef]
- Tessarz, P.; Santos-Rosa, H.; Robson, S.C.; Sylvestersen, K.B.; Nelson, C.J.; Nielsen, M.L.; Kouzarides, T. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 2014, 505, 564–568. [Google Scholar] [CrossRef]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, L.; Christopher, D.M.; Teng, Z.Q.; Fausett, S.R.; Liu, C.; George, O.L.; Klingensmith, J.; Jin, P.; Zhao, X. RNA-binding protein FXR2 regulates adult hippocampal neurogenesis by reducing Noggin expression. Neuron 2011, 70, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Agote-Aran, A.; Schmucker, S.; Jerabkova, K.; Jmel Boyer, I.; Berto, A.; Pacini, L.; Ronchi, P.; Kleiss, C.; Guerard, L.; Schwab, Y.; et al. Spatial control of nucleoporin condensation by fragile X-related proteins. EMBO J. 2020, 39, e104467. [Google Scholar] [CrossRef]
- Li, H.; Huo, Y.; He, X.; Yao, L.; Zhang, H.; Cui, Y.; Xiao, H.; Xie, W.; Zhang, D.; Wang, Y.; et al. A male germ-cell-specific ribosome controls male fertility. Nature 2022, 612, 725–731. [Google Scholar] [CrossRef]
- Khatter, H.; Myasnikov, A.G.; Natchiar, S.K.; Klaholz, B.P. Structure of the human 80S ribosome. Nature 2015, 520, 640–645. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Bruce, D.L.; Macartney, T.; Yong, W.; Shou, W.; Sapkota, G.P. Protein phosphatase 5 modulates SMAD3 function in the transforming growth factor-beta pathway. Cell Signal. 2012, 24, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Heliez, L.; Ricordel, C.; Becuwe, P.; Pedeux, R. Newly identified tumor suppressor functions of ING proteins. Curr. Opin. Pharmacol. 2023, 68, 102324. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Feng, X.; Qiao Shi, Z.; Ganapathy, A.; Kumar Mishra, M.; Atadja, P.; Morris, D.; Riabowol, K. ING1 and 5-azacytidine act synergistically to block breast cancer cell growth. PLoS ONE 2012, 7, e43671. [Google Scholar] [CrossRef] [PubMed]
- Rajarajacholan, U.K.; Thalappilly, S.; Riabowol, K. ING1 regulates rRNA levels by altering nucleolar chromatin structure and mTOR localization. Nucleic Acids Res. 2017, 45, 1776–1792. [Google Scholar] [CrossRef]
- Demir, S.; Wolff, G.; Wieder, A.; Maida, A.; Buhler, L.; Brune, M.; Hautzinger, O.; Feuchtinger, A.; Poth, T.; Szendroedi, J.; et al. TSC22D4 interacts with Akt1 to regulate glucose metabolism. Sci. Adv. 2022, 8, eabo5555. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Sakurai, M.; Mhamane, A.; Troullinaki, M.; Maida, A.; Deligiannis, I.K.; Yin, K.; Weber, P.; Morgenstern, J.; Wieder, A.; et al. Hepatocyte-specific activity of TSC22D4 triggers progressive NAFLD by impairing mitochondrial function. Mol. Metab. 2022, 60, 101487. [Google Scholar] [CrossRef] [PubMed]
- Corre, M.; Lebreton, A. Regulation of cold-inducible RNA-binding protein (CIRBP) in response to cellular stresses. Biochimie 2023, 217, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Vujicic, I.; Rusevski, A.; Stankov, O.; Popov, Z.; Dimovski, A.; Davalieva, K. Potential Role of Seven Proteomics Tissue Biomarkers for Diagnosis and Prognosis of Prostate Cancer in Urine. Diagnostics 2022, 12, 3184. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S. The prognostic value of immune-related genes AZGP1, SLCO5A1, and CTF1 in Uveal melanoma. Front. Oncol. 2022, 12, 918230. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhou, L.; Zhan, S.J.; Cheng, L.J.; Li, R.N.; Wang, H.; Liu, B.; Linghu, H. ALPL regulates the aggressive potential of high grade serous ovarian cancer cells via a non-canonical WNT pathway. Biochem. Biophys. Res. Commun. 2019, 513, 528–533. [Google Scholar] [CrossRef]
- Lou, Z.; Lin, W.; Zhao, H.; Jiao, X.; Wang, C.; Zhao, H.; Liu, L.; Liu, Y.; Xie, Q.; Huang, X.; et al. Alkaline phosphatase downregulation promotes lung adenocarcinoma metastasis via the c-Myc/RhoA axis. Cancer Cell Int. 2021, 21, 217. [Google Scholar] [CrossRef]
- Rao, S.R.; Snaith, A.E.; Marino, D.; Cheng, X.; Lwin, S.T.; Orriss, I.R.; Hamdy, F.C.; Edwards, C.M. Tumour-derived alkaline phosphatase regulates tumour growth, epithelial plasticity and disease-free survival in metastatic prostate cancer. Br. J. Cancer 2017, 116, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lv, D.; Cai, C.; Zhao, Z.; Wang, M.; Chen, W.; Liu, Y. A TP53-Associated Immune Prognostic Signature for the Prediction of Overall Survival and Therapeutic Responses in Muscle-Invasive Bladder Cancer. Front. Immunol. 2020, 11, 590618. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Narumi, R.; Nagayama, S.; Masuda, K.; Esaki, T.; Obama, K.; Tomonaga, T.; Sakai, Y.; Shimizu, Y.; Adachi, J. A large-scale targeted proteomics of plasma extracellular vesicles shows utility for prognosis prediction subtyping in colorectal cancer. Cancer Med. 2023, 12, 7616–7626. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, J.; Sun, Y.; Liu, L.; Guo, L.; Zhao, C.; Zhang, J.; Qian, Q.; Cui, B.; Zhang, Y. The Plasma DIA-Based Quantitative Proteomics Reveals the Pathogenic Pathways and New Biomarkers in Cervical Cancer and High Grade Squamous Intraepithelial Lesion. J. Clin. Med. 2022, 11, 7155. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wen, Q.; Li, Q.; Li, P.; Liu, T.; Zhu, F.; Tan, Q.; Zhang, L. Identification of oxidative stress-related hub genes for predicting prognosis in diffuse large B-cell lymphoma. Gene 2024, 2024, 149077. [Google Scholar] [CrossRef] [PubMed]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Bianchi-Smiraglia, A.; Kunnev, D.; Limoge, M.; Lee, A.; Beckerle, M.C.; Bakin, A.V. Integrin-β5 and zyxin mediate formation of ventral stress fibers in response to transforming growth factor beta. Cell Cycle 2013, 12, 3377–3389. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Kostin, S.; Ngai, S.; Chan, K.; Lau, Y.; Lee, S.M.; Li, H.; Ng, E.K.; Schaper, J.; Tsui, S.K.; et al. Interaction of hCLIM1, an enigma family protein, with α-actinin 2. J. Cell. Biochem. 2000, 78, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Li, X.; Li, G.; Mizukami, T.; Liu, Y.; Wang, Y.; Xu, G.; Roder, H.; Zhang, L.; et al. PDLIM3 supports hedgehog signaling in medulloblastoma by facilitating cilia formation. Cell Death Differ. 2023, 30, 1198–1210. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Marimuthu, S.; Chaudhary, S.; Seshacharyulu, P.; Rachagani, S.; Muniyan, S.; Chirravuri-Venkata, R.; Atri, P.; Rauth, S.; Nimmakayala, R.K.; et al. Muc16 depletion diminishes KRAS-induced tumorigenesis and metastasis by altering tumor microenvironment factors in pancreatic ductal adenocarcinoma. Oncogene 2022, 41, 5147–5159. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, B.; Schooley, A.; Sachdev, R.; Eisenhardt, N.; Schneider, A.M.; Sieverding, C.; Madlung, J.; Gerken, U.; Macek, B.; Antonin, W. Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J. 2012, 31, 4072–4084. [Google Scholar] [CrossRef]
- Pulupa, J.; Prior, H.; Johnson, D.S.; Simon, S.M. Conformation of the nuclear pore in living cells is modulated by transport state. eLife 2020, 9, e60654. [Google Scholar] [CrossRef] [PubMed]
- Kalverda, B.; Pickersgill, H.; Shloma, V.V.; Fornerod, M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 2010, 140, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Bindra, D.; Samaiya, A.; Mishra, R.K. Overexpressed Nup88 stabilized through interaction with Nup62 promotes NF-κB dependent pathways in cancer. Front. Oncol. 2023, 13, 1095046. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lin, Y.; Jin, J.; Shen, H.; Dai, C. Nuclear Pore Complex 62 Promotes Metastasis of Gastric Cancer by Regulating Wnt/β-Catenin and TGF-β Signaling Pathways. J. Environ. Pathol. Toxicol. Oncol. 2021, 40, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Lu, Y.; Liu, B.; Ran, C.; Lei, X.; Wang, M.; Wu, S.; Yang, Y.; Wu, H. Glycolysis, a new mechanism of oleuropein against liver tumor. Phytomedicine 2023, 114, 154770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Tsai, C.C.; Su, Y.C.; Bamodu, O.A.; Chen, B.J.; Tsai, W.C.; Cheng, W.H.; Lee, C.H.; Hsieh, S.M.; Liu, M.L.; Fang, C.L.; et al. High-Grade B-Cell Lymphoma (HGBL) with MYC and BCL2 and/or BCL6 Rearrangements Is Predominantly BCL6-Rearranged and BCL6-Expressing in Taiwan. Cancers 2021, 13, 1620. [Google Scholar] [CrossRef]
- Alford, D.; Petkovic, I. Prognostic Significance of Bcl-2 Expression in Non-germinal Center B-cell-Like Diffuse Large B-cell Lymphoma. Cureus 2024, 16, e62031. [Google Scholar] [CrossRef]
- Pickering, M.C.; Cook, H.T. Translational mini-review series on complement factor H: Renal diseases associated with complement factor H: Novel insights from humans and animals. Clin. Exp. Immunol. 2008, 151, 210–230. [Google Scholar] [CrossRef]
- Xiang, M.; Zhang, H.; Kou, L.; Chen, J.; Xu, Z.; He, J. Low level of complement factor H increases the risk of cancer-related death in patients with small-cell lung cancer. Postgrad. Med. J. 2022, 98, 919–924. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Yan, J.; Wan, P.; Choksi, S.; Liu, Z.G. Necroptosis and tumor progression. Trends Cancer 2022, 8, 21–27. [Google Scholar] [CrossRef]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A., Jr.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef]
- Kuttanamkuzhi, A.; Panda, D.; Malaviya, R.; Gaidhani, G.; Lahiri, M. Altered expression of anti-apoptotic protein Api5 affects breast tumorigenesis. BMC Cancer 2023, 23, 374. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Nirmala, J.G.; Lopus, M. Cell death mechanisms in eukaryotes. Cell Biol. Toxicol. 2020, 36, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, I.A.; Abhari, B.A.; Fulda, S. Identification of a synergistic combination of Smac mimetic and Bortezomib to trigger cell death in B-cell non-Hodgkin lymphoma cells. Cancer Lett. 2017, 405, 63–72. [Google Scholar] [CrossRef]
- Koch, A.; Jeiler, B.; Roedig, J.; van Wijk, S.J.L.; Dolgikh, N.; Fulda, S. Smac mimetics and TRAIL cooperate to induce MLKL-dependent necroptosis in Burkitt’s lymphoma cell lines. Neoplasia 2021, 23, 539–550. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017-Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Arietta-Garcia, A.G.; Calzada, F.; Ramirez-Sanchez, I.; Bautista, E.; Garcia-Hernandez, N.; Ordonez-Razo, R.M. Advances in the Properties of Incomptine A: Cytotoxic Activity and Downregulation of Hexokinase II in Breast Cancer Cell Lines. Int. J. Mol. Sci. 2023, 24, 12406. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Liu, G.; Tang, Y. In silico ADMET prediction: Recent advances, current challenges and future trends. Curr. Top. Med. Chem. 2013, 13, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- UNECE. Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Available online: https://unece.org/about-ghs (accessed on 30 November 2024).
- Raducka, A.; Swiatkowski, M.; Gobis, K.; Szymanski, P.; Czylkowska, A. In Silico ADME and Toxicity Prediction of Benzimidazole Derivatives and Its Cobalt Coordination Compounds. Synthesis, Characterization and Crystal Structure. Molecules 2022, 27, 8011. [Google Scholar] [CrossRef]
- Divya Rajaselvi, N.; Jida, M.D.; Nair, D.B.; Sujith, S.; Beegum, N.; Nisha, A.R. Toxicity prediction and analysis of flavonoid apigenin as a histone deacetylase inhibitor: An in-silico approach. In Silico Pharmacol. 2023, 11, 34. [Google Scholar] [CrossRef]
- Ansell, S.M. Non-Hodgkin Lymphoma: Diagnosis and Treatment. Mayo Clin. Proc. 2015, 90, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, K.; Dowling, P.; Bazou, D.; O’Gorman, P. Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers. Cancers 2021, 13, 1930. [Google Scholar] [CrossRef]
- Pagel, O.; Loroch, S.; Sickmann, A.; Zahedi, R.P. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev. Proteom. 2015, 12, 235–253. [Google Scholar] [CrossRef]
- Ralph, P.; Moore, M.A.; Nilsson, K. Lysozyme synthesis by established human and murine histiocytic lymphoma cell lines. J. Exp. Med. 1976, 143, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- NOM-062-ZOO-1999; Norma Oficial Mexicana. Especificaciones Técnicas para la Producción, Cuidado y Uso de Animales de Laboratorio. Diario Oficial de la Federación: Ciudad de México, Mexico, 2001.
- Douros, J.; Suffness, M. The National Cancer Institute’s Natural Products Antineoplastic Development Program. Recent Results Cancer Res. 1980, 70, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Zubair, A.C.; Ali, S.A.; Rees, R.C.; Goepel, J.R.; Winfield, D.A.; Goyns, M.H. Analysis of the colonization of unirradiated and irradiated SCID mice by human lymphoma and non-malignant lymphoid cells. Leuk. Lymphoma 1996, 22, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallah, R.; Abena, P.; Chetaille, B.; Aurran-Schleinitz, T.; Sainty, D.; Dubus, P.; Arnoulet, C.; Coso, D.; Xerri, L.; Gastaut, J.A. True histiocytic lymphoma following B-acute lymphoblastic leukaemia: Case report with evidence for a common clonal origin in both neoplasms. Br. J. Haematol. 2001, 113, 1047–1050. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, W.; Sun, H.; Zhou, H.; Xu, H. Expression of P120 catenin mRNA in Non-Hodgkin’s lymphoma cell lines. J. Huazhong Univ. Sci. Technolog Med. Sci. 2006, 26, 185–187. [Google Scholar] [CrossRef] [PubMed]
- OECD. Guideline for Testing of Chemicals. Acute Oral Toxicity-Acute Toxic Class Method; Organización para la Cooperación y el Desarrollo Económicos, OECD/OCDE: Paris, France, 2001; pp. 1–14. [Google Scholar]
- Calzada, F.; Solares-Pascasio, J.I.; Valdes, M.; Garcia-Hernandez, N.; Velázquez, C.; Ordoñez-Razo, R.M.; Barbosa, E. Antilymphoma potential of the ethanol extract and rutin obtained of the leaves from schinus molle linn. Pharmacognosy Res. 2018, 10, 119–123. [Google Scholar] [CrossRef]
- Yu, K.; Wang, Z.; Wu, Z.; Tan, H.; Mishra, A.; Peng, J. High-Throughput Profiling of Proteome and Posttranslational Modifications by 16-Plex TMT Labeling and Mass Spectrometry. Methods Mol. Biol. 2021, 2228, 205–224. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Blanco, D.B.; Xie, B.; Li, Y.; Wu, Z.; Chi, H.; Peng, J. Quantifying Proteome and Protein Modifications in Activated T Cells by Multiplexed Isobaric Labeling Mass Spectrometry. Methods Mol. Biol. 2021, 2285, 297–317. [Google Scholar] [CrossRef]
- Pagel, O.; Kollipara, L.; Sickmann, A. Quantitative Proteome Data Analysis of Tandem Mass Tags Labeled Samples. Methods Mol. Biol. 2021, 2228, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, Y.; Zhang, T.; Shu, L.; Roepstorff, P.; Yang, F. Quantitative Proteomics Using Isobaric Labeling: A Practical Guide. Genom. Proteom. Bioinform. 2021, 19, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Sivanich, M.K.; Gu, T.J.; Tabang, D.N.; Li, L. Recent advances in isobaric labeling and applications in quantitative proteomics. Proteomics 2022, 22, e2100256. [Google Scholar] [CrossRef] [PubMed]
- Gautam, P.; Nair, S.C.; Gupta, M.K.; Sharma, R.; Polisetty, R.V.; Uppin, M.S.; Sundaram, C.; Puligopu, A.K.; Ankathi, P.; Purohit, A.K.; et al. Proteins with altered levels in plasma from glioblastoma patients as revealed by iTRAQ-based quantitative proteomic analysis. PLoS ONE 2012, 7, e46153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Li, S.; Zhang, X.; Li, W.; Luo, S.; Sun, Z.; Nie, R. ITRAQ-based quantitative proteomic analysis of processed Euphorbia lathyris L. for reducing the intestinal toxicity. Proteome Sci. 2018, 16, 8. [Google Scholar] [CrossRef]
- Li, S.; Su, X.; Jin, Q.; Li, G.; Sun, Y.; Abdullah, M.; Cai, Y.; Lin, Y. iTRAQ-Based Identification of Proteins Related to Lignin Synthesis in the Pear Pollinated with Pollen from Different Varieties. Molecules 2018, 23, 548. [Google Scholar] [CrossRef]
- Bailey, A. Chapter 5 Transforming and Visualising Proteomics Data. Available online: https://ab604.github.io/docs/bspr_workshop_2018/transform.html (accessed on 26 November 2024).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Calzada, F.; Valdes, M.; Martinez-Solis, J.; Velazquez, C.; Barbosa, E. Annona cherimola Miller and Its Flavonoids, an Important Source of Products for the Treatment of Diabetes Mellitus: In Vivo and In Silico Evaluations. Pharmaceuticals 2023, 16, 724. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Santos, J.; Calzada, F.; Mendieta-Wejebe, J.E.; Ordonez-Razo, R.M.; Martinez-Casares, R.M.; Valdes, M. Understanding the Antilymphoma Activity of Annona macroprophyllata Donn and Its Acyclic Terpenoids: In Vivo, In Vitro, and In Silico Studies. Molecules 2022, 27, 7123. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Dong, S.; Ge, Y.; Fonseca, J.P.; Robinson, Z.T.; Mysore, K.S.; Mehta, P. DiVenn: An Interactive and Integrated Web-Based Visualization Tool for Comparing Gene Lists. Front. Genet. 2019, 10, 421. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 20 June 2023).
- Perez-Riverol, Y.; Bandla, C.; Kundu, D.J.; Kamatchinathan, S.; Bai, J.; Hewapathirana, S.; John, N.S.; Prakash, A.; Walzer, M.; Wang, S.; et al. The PRIDE database at 20 years: 2025 Update. Nucleic Acids Res. 2025, 53, D543–D553. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Perez-Riverol, Y.; Sharma, V.; Carver, J.J.; Mendoza, L.; Kundu, D.J.; Wang, S.; Bandla, C.; Kamatchinathan, S.; et al. The ProteomeXchange consortium at 10 years: 2023 Update. Nucleic Acids Res. 2023, 51, D1539–D1548. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C− (DMSO) vs. | Down Regulated: FC < 0.67 (<1/1.5) | Up Regulated: FC > 1.5 |
|---|---|---|
| MTX | 111 | 63 |
| 5LANM | 76 | 69 |
| 5RINM | 117 | 72 |
| 10LANM | 78 | 80 |
| 10RINM | 83 | 132 |
| Protein | Ligands | |||||||
|---|---|---|---|---|---|---|---|---|
| Incomptine A | Methotrexate (MTX) | |||||||
| ΔG | Ki | H-BR | NPI | ΔG | Ki | H-BR | NPI | |
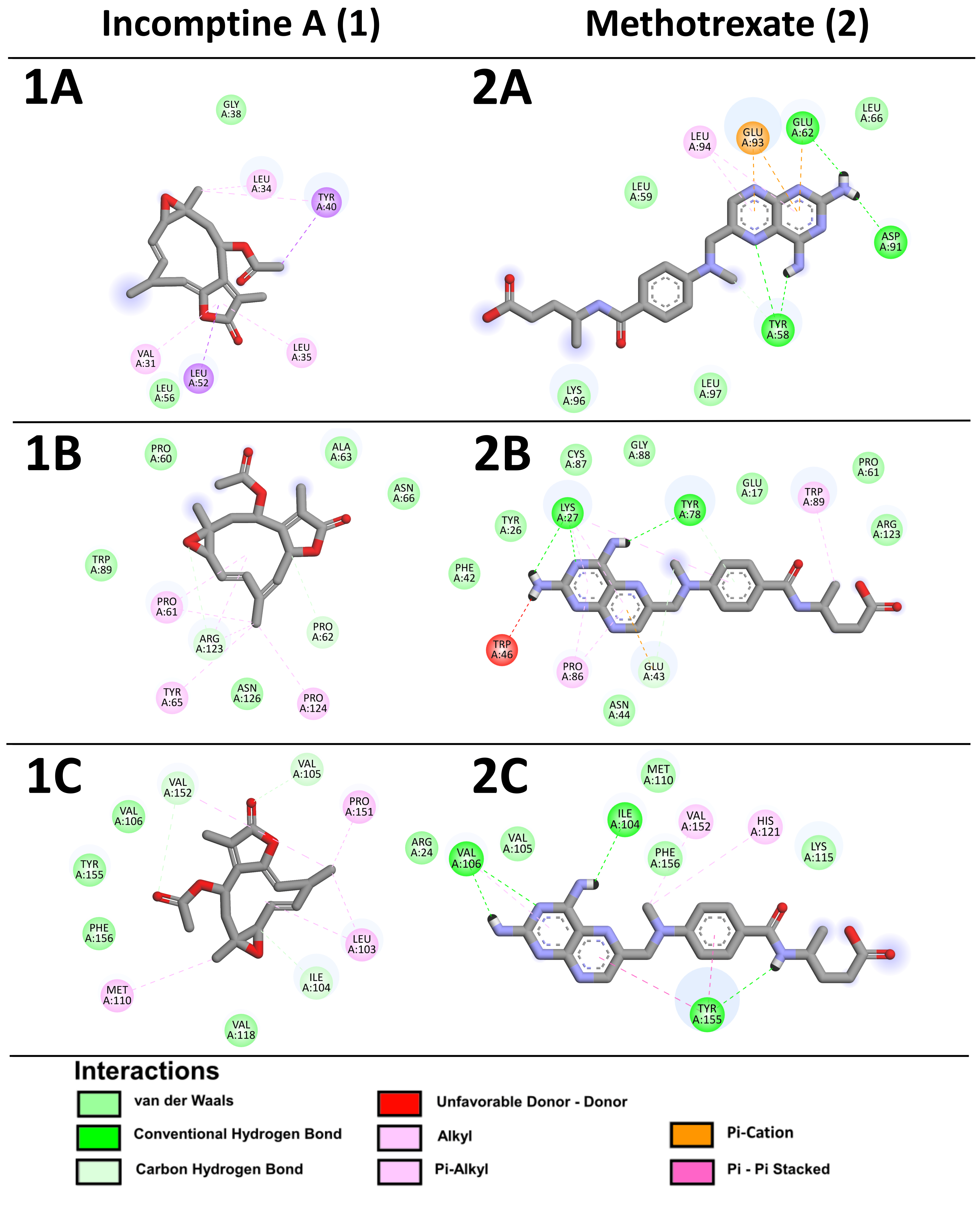
| Histone H2A type 1-F | −4.08 | 1.03 mM | Gly38, Leu56 | Val31, Leu34, Leu35, Tyr40, Leu52 | −4.24 | 778.4 µM | Tyr58, Leu 59, Glu62, Leu66, Asp91, Lys96, Leu97 | Glu93, Leu94 |
| Fragile X mental retardation syndrome-related protein 2 | −6.83 | 9.9 µM | Pro60, Pro62, Ala63, Asn66, Trp89, Arg123, Asn126 | Pro61, Tyr65, Pro124 | −4.99 | 220.3 µM | Glu17, Tyr26, Lys27, Phe42, Glu43, Asn44, Pro61, Tyr78, Cys87, Gly88, Arg123 | Trp46, Pro86, Trp89 |
| DNA-directed RNA polymerase II subunit E | −5.18 | 160.5 µM | Arg24, Ile104, Val105, Val106, Val118, Val152, Tyr155, Phe156 | Leu103, Met110, Pro151 | −4.15 | 904.1 µM | Ile104, Val105, Val106, Met110, Lys115, Tyr155, Phe156 | His121, Val152 |
| 40S ribosomal protein S2 | −5.17 | 162.6 µM | Ile81, Ser85, Leu86, Pro87, Ile88, Ser161, Ile162, Pro164 | Tyr82, Val163 | −7.02 | 1.81 µM | Phe84, Ser85, Leu86, Ile162, Ser245, Lys246, Tyr248, Ser249 | Tyr82, Pro87, Ile88, Val163, Pro164 |
| Signal transducer and activator of transcription 1 | −4.43 | 565.7 µM | Val362, Leu363, Phe364, Asn381, Leu383, Gly 384 | Lys361, Ile382, His386 | −5.12 | 175.6 µM | Asn233, Val237, Trp239, Lys240, Arg241, Gln243, Gln244, Val318, Val319, Gln322, Ala479, Glu480 | Leu453, Pro481 |
| Transforming growth factor beta-1 proprotein | −4.34 | 653.7 µM | Ala89, Tyr317, His318, Glu362, Pro363, Pro365 | Tyr81, Arg85, Val88, Pro314, Leu364, Ile383 | −4.72 | 347.2 µM | Val88, Ala89, Ser92, Tyr299, Gly316, Tyr317, His318, Leu364, Pro365 | Pro314, Glu362, Ile383 |
| TSC22 domain family protein 4 | −4.37 | 629.8 µM | Glu101, His103, Ser104 | Leu100, Pro102, Phe105 | −2.08 | 29.86 µM | Leu152, Arg153, Pro154, Asn355, Ala356, Ala357, Glu359, Gln360 | Pro155 |
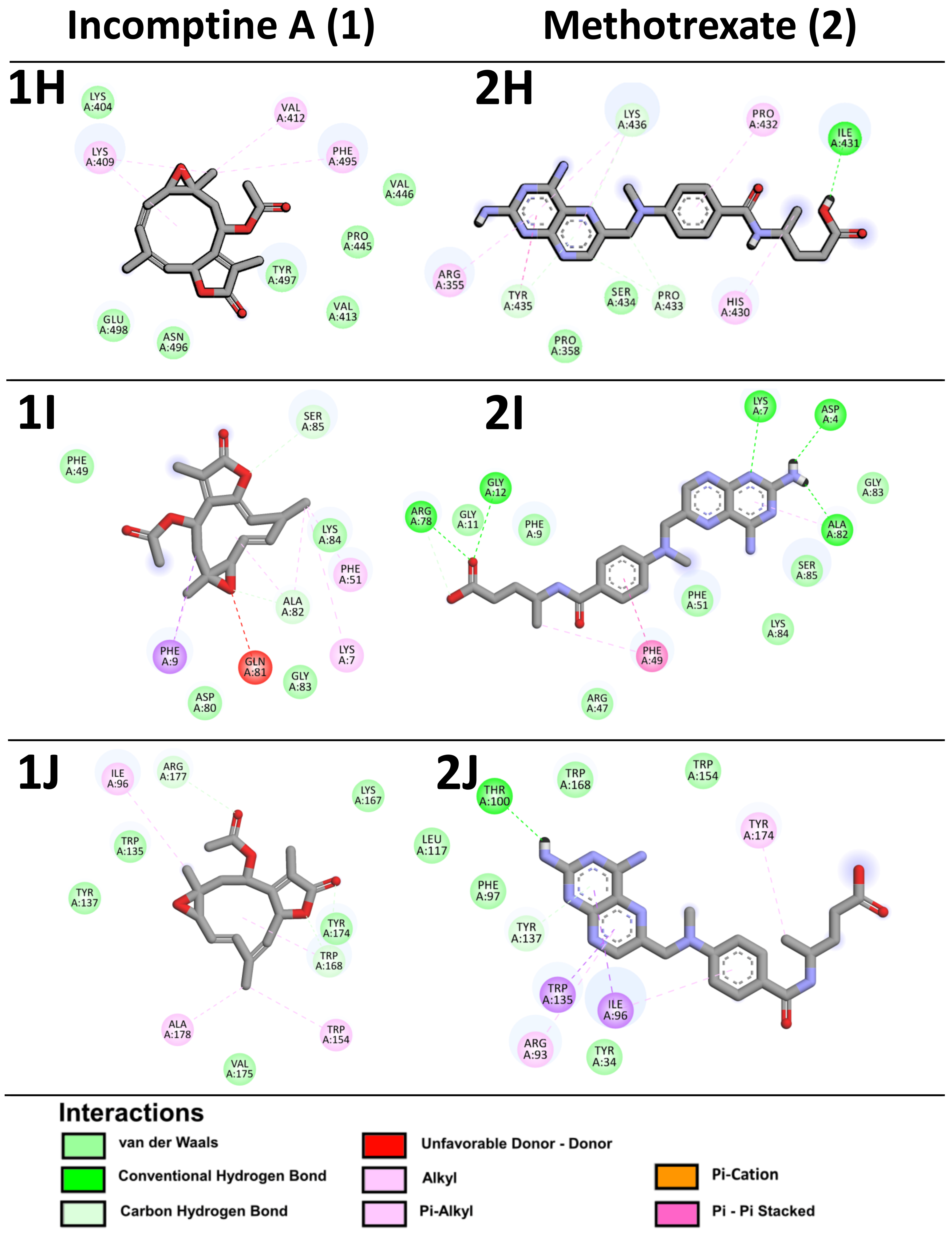
| Apoptosis inhibitor 5 | −5.19 | 158.2 µM | Lys404, Val413, Pro445, Val446, Asn496, Tyr497, Glu498 | Lys409, Val412, Phe495 | −5.1 | 182.68 µM | Pro358, Ile431, Pro433, Ser434, Tyr435, Lys436 | Arg335, His430, Pro432 |
| Cold-inducible RNA-binding protein | −5.92 | 45.5 µM | Phe49, Asp80, Ala82, Gly83, Lys84, Ser85 | Lys7, Phe9, Phe51, Gln81 | −4.58 | 436.7 µM | Asp4, Lys7, Phe9, Gly11, Gly12, Arg47, Phe51, Arg78, Ala82, Gly83, Lys84, Ser85 | Phe49 |
| Zinc-alpha-2-glycoprotein | −6.08 | 35.0 µM | Trp135, Tyr137, Lys167, Trp168, Tyr174, Val175, Arg177 | Ile96, Trp154, Ala178 | −4.0 | 436.7 µM | Tyr34, Phe97, Thr100, Leu117, Tyr137, Trp154, Trp168 | Arg93, Ile96, Trp135, Tyr174 |
| Alpha-1-acid glycoprotein 1 | −5.15 | 167.8 µM | Val59, Thr65, Glu82, Gln84, Val110, Leu130, Ser143, Tyr145 | Tyr45, Phe50, Ile62, Arg108, Phe132 | −4.92 | 246.1 µM | Val27, Pro30, Ile31, Thr32, Thr35, Gln38, Leu119, Ile121, Arg123 | Ile20, Ala24, Asn25, Val29, Leu120, Leu122, Met129 |
| Nuclear pore glycoprotein p62 | −4.07 | 1.04 nM | Ile225, Ala226, Thr227, Pro229, Asp394, Ile396, Gln400 | Ala228, Leu393, Leu397 | −2.35 | 18.8 mM | Ser238, Leu239, Thr241, Asn459, Lys476, Ile477, Asn479, Ala480, Asp483, Gln486 | Cys240, Met482 |
| Beta-enolase | −4.81 | 298.8 µM | Lys228, Thr229, Gln232, Val240, Asn286, Tyr287 | Ile231, Pro237, Pro280 | −5.83 | 53.5 µM | Ser37, Gly38, Glu45, Ala46, Leu47, Glu48, Lys54, Leu58, Gly59, Asn345, Gln346, Arg372 | Thr41, Arg50, Lys60 |
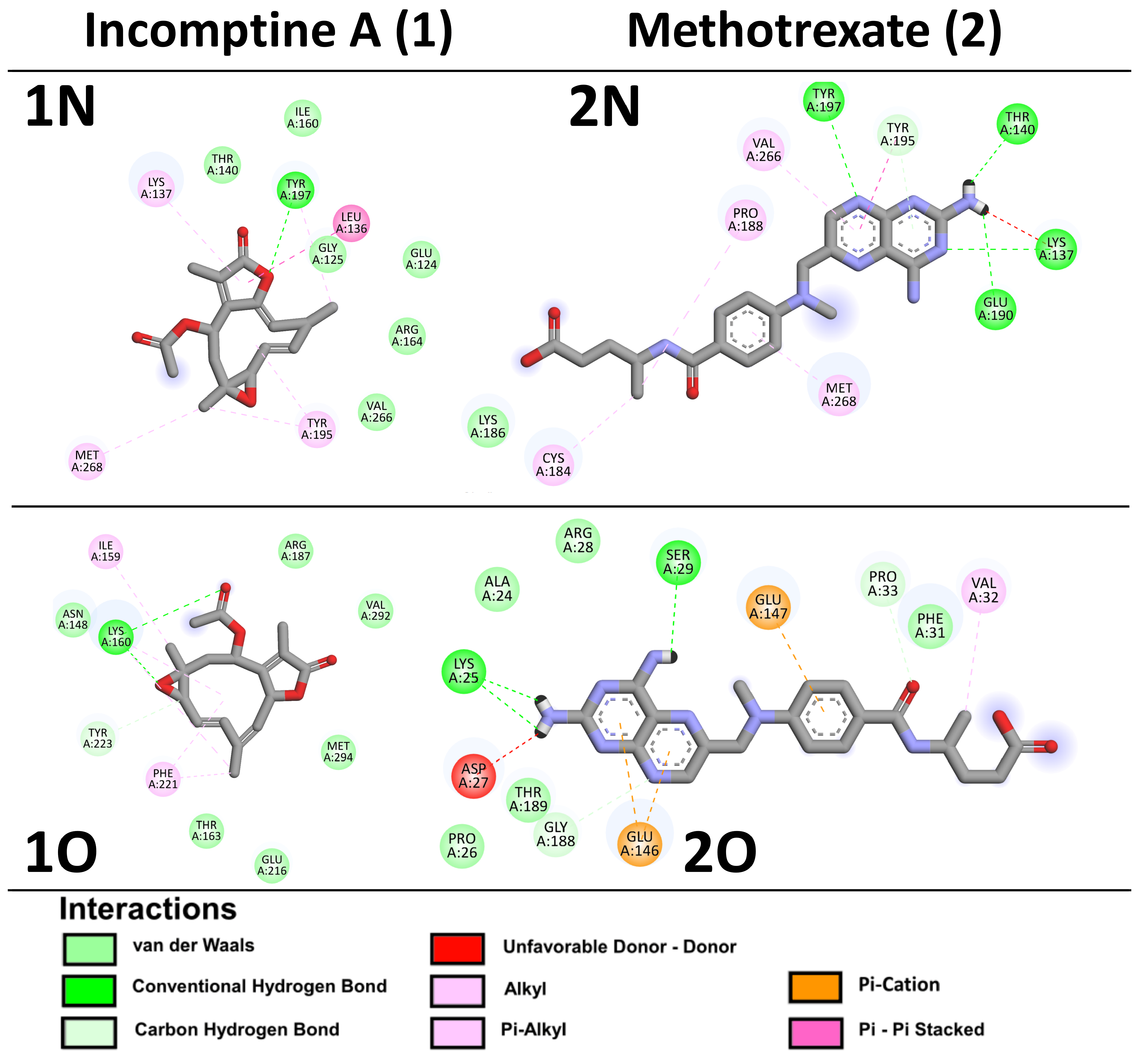
| Caspase-3 | −5.29 | 137.7 µM | Glu124, Gly125, Thr140, Ile160, Arg164, Tyr197, Val266 | Leu136, Lys137, Tyr195, Met268 | −3.38 | 3.34 mM | Lys137, Ther140, Lys186, Glu190, Tyr195, Tyr197 | Cys184, Pro188, Val266, Met268 |
| Caspase-7 | −6.12 | 89.2 µM | Asn148, Lys160, Thr163, Arg187, Glu216, Tyr223, Val292, Met294 | Ile159, Phe221 | −4.1 | 995.2 µM | Ala24, Lys25, Pro26, Arg28, Ser29, Phe31, Pro33, Gly188, Thr189 | Asp27, Val32, Glu146, Glu147 |
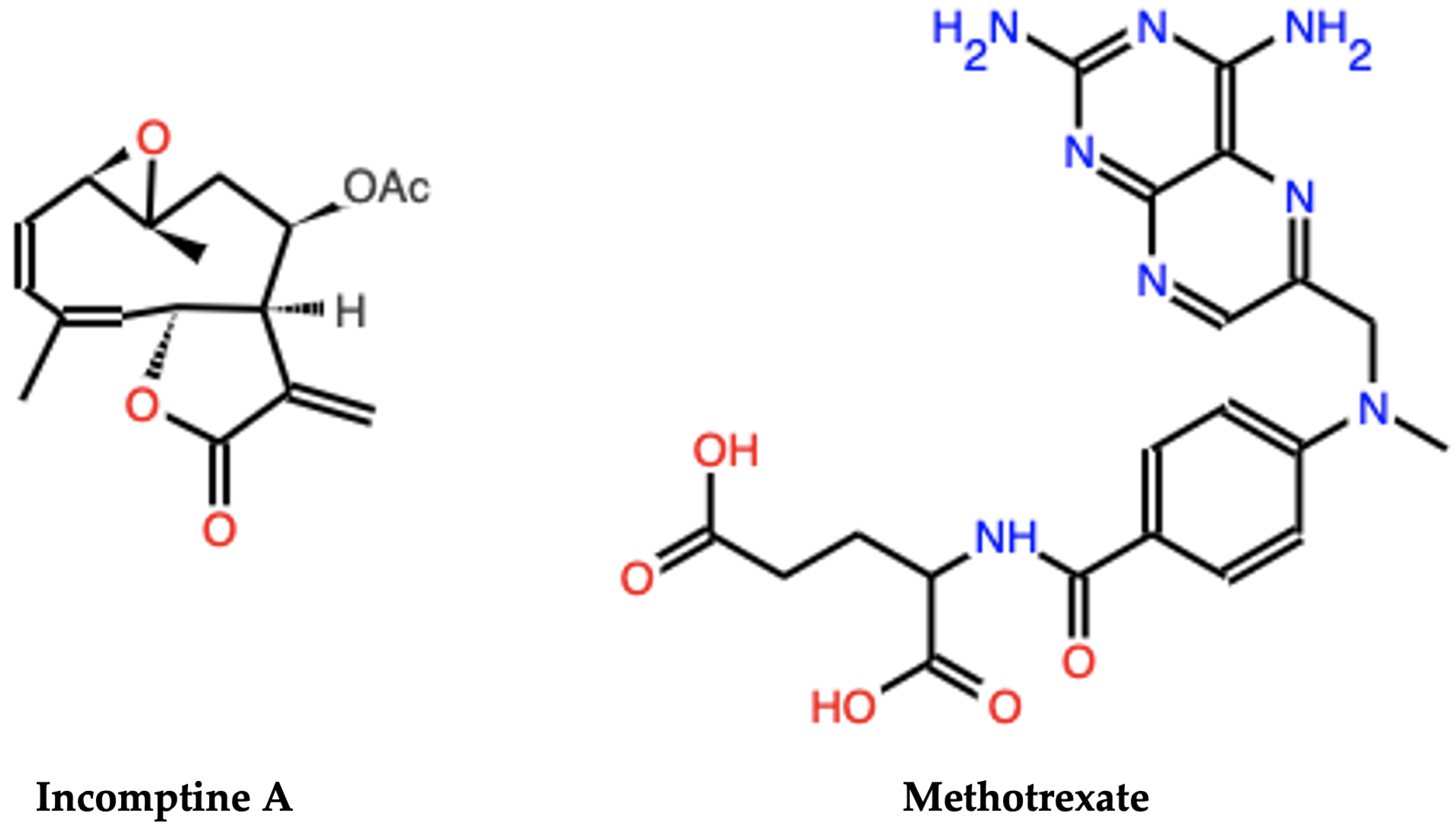
| Smiles | IA | C=C1C(=O)O[C@@H]2/C=C(/C)CC[C@H]3O[C@@]3(C)C[C@@H](OC(C)=O)[C@H]12 | ||||
| MTX | CN(Cc1cnc2nc(N)nc(N)c2n1)c1ccc(C(=O)N[C@@H](CCC(=O)O)C(=O)O)cc1 | |||||
| Physicochemical Properties | ||||||
| IA | MTX | Druglikeness | ||||
| Molecular formula | C17H22O5 | C20H22N8O5 | ||||
| Molecular weight | 306.35 g/mol | 454.44 g/mol | IA | MTX | ||
| TPSA | 65.13 Å2 | 210.54 Å2 | Lipinsky | Yes | Yes | |
| Lipophilicity (LogP) | 2.33 | −0.32 | Ghose | Yes | Yes | |
| Water solubility (LogS) | −2.71 | −2.41 | Veber | Yes | No, 1 volation | |
| Solubility class | Soluble | Very soluble | Egan | Yes | No, 1 volation | |
| Number of rotating links | 2 | 10 | Muegge | Yes | No, 1 volation | |
| Number of H-bond donors | 0 | 5 | PAINS | 0 | 0 | |
| Number of H-bond acceptors | 5 | 9 | ||||
| Pharmacokinetics Properties | ||||||
| Absorption | Metabolism | |||||
| IA | MTX | IA | MTX | |||
| Gastrointestinal absorption | High | Low | CYP2C9 substrate | No | No | |
| Hematoencephalic barrier | Yes | No | CYP2D6 substrate | No | No | |
| Caco-2 permeability | High | Low | CYP3A4 substrate | Yes | Yes | |
| p-glycoprotein substrate | Yes | Yes | CYP2C9 inhibitor | No | No | |
| p-glycoprotein inhibitor | No | No | CYP2D6 Inhibitor | No | No | |
| Log Kp (skin permeation) | −6.89 cm/s | −10.39 cm/s | CYP3A4 Inhibitor | No | No | |
| CYP1A2 Inhibitor | No | No | ||||
| CYP2C19 Inhibitor | No | No | ||||
| Distribution | Excretion | |||||
| Mitochondrial | Yes | Yes | CL | 4.64 mL/min/Kg | 2.41 mL/min/Kg | |
| Protein plasma binding | 75.9% | 63.4% | T 1/2 | 0.78 h | 0.39 h | |
| Volume Distribution | 1.38 L/Kg | 0.32 L/Kg | ||||
| Toxicity | ||||||
| IA | MTX | IA | MTX | |||
| Hepatotoxicity | Inactive | Active | Carcinogenicity | Inactive | Inactive | |
| Neurotoxicity | Inactive | Active | Immunotoxicity | Inactive | Inactive | |
| Nephrotoxicity | Inactive | Inactive | Mutagenicity | Inactive | Inactive | |
| Respiratory toxicity | Active | Active | Cytotoxicity | Inactive | Inactive | |
| Cardiotoxicity | Inactive | Inactive | ||||
| Predicted rats LD50 | 2.68 mol/Kg | 3.49 mol/Kg | ||||
| Predicted human LD50 | 1330 mg/Kg | 3 mg/Kg | ||||
| Expected toxicity class * | IV | I | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Hernández, N.; Calzada, F.; Bautista, E.; Sánchez-López, J.M.; Valdes, M.; Hernández-Caballero, M.E.; Ordoñez-Razo, R.M. Quantitative Proteomics and Molecular Mechanisms of Non-Hodgkin Lymphoma Mice Treated with Incomptine A, Part II. Pharmaceuticals 2025, 18, 242. https://doi.org/10.3390/ph18020242
García-Hernández N, Calzada F, Bautista E, Sánchez-López JM, Valdes M, Hernández-Caballero ME, Ordoñez-Razo RM. Quantitative Proteomics and Molecular Mechanisms of Non-Hodgkin Lymphoma Mice Treated with Incomptine A, Part II. Pharmaceuticals. 2025; 18(2):242. https://doi.org/10.3390/ph18020242
Chicago/Turabian StyleGarcía-Hernández, Normand, Fernando Calzada, Elihú Bautista, José Manuel Sánchez-López, Miguel Valdes, Marta Elena Hernández-Caballero, and Rosa María Ordoñez-Razo. 2025. "Quantitative Proteomics and Molecular Mechanisms of Non-Hodgkin Lymphoma Mice Treated with Incomptine A, Part II" Pharmaceuticals 18, no. 2: 242. https://doi.org/10.3390/ph18020242
APA StyleGarcía-Hernández, N., Calzada, F., Bautista, E., Sánchez-López, J. M., Valdes, M., Hernández-Caballero, M. E., & Ordoñez-Razo, R. M. (2025). Quantitative Proteomics and Molecular Mechanisms of Non-Hodgkin Lymphoma Mice Treated with Incomptine A, Part II. Pharmaceuticals, 18(2), 242. https://doi.org/10.3390/ph18020242