
Nicotinamide Mononucleotide (NMN) Improves the Senescence of Mouse Vascular Smooth Muscle Cells Induced by Ang II Through Activating p-AMPK/KLF4 Pathway

 , , and
, , and
Abstract

1. Introduction
2. Results
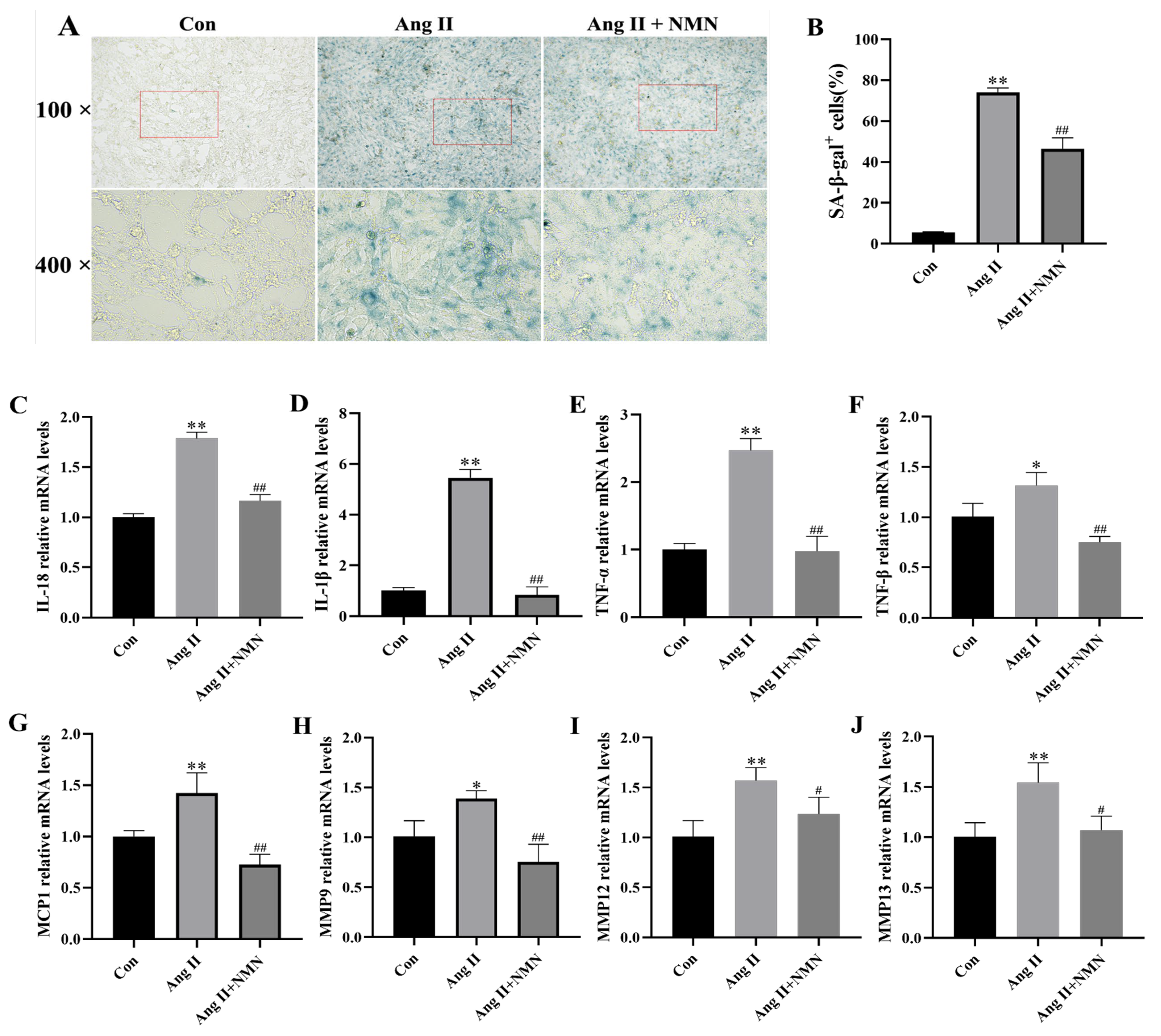
2.1. NMN Improved the Degree of Ang II-Induced Mouse VSMCs Cell Senescence and Reduced the Secretion of Key Senescence-Associated Secretory Phenotype (SASP) Factors
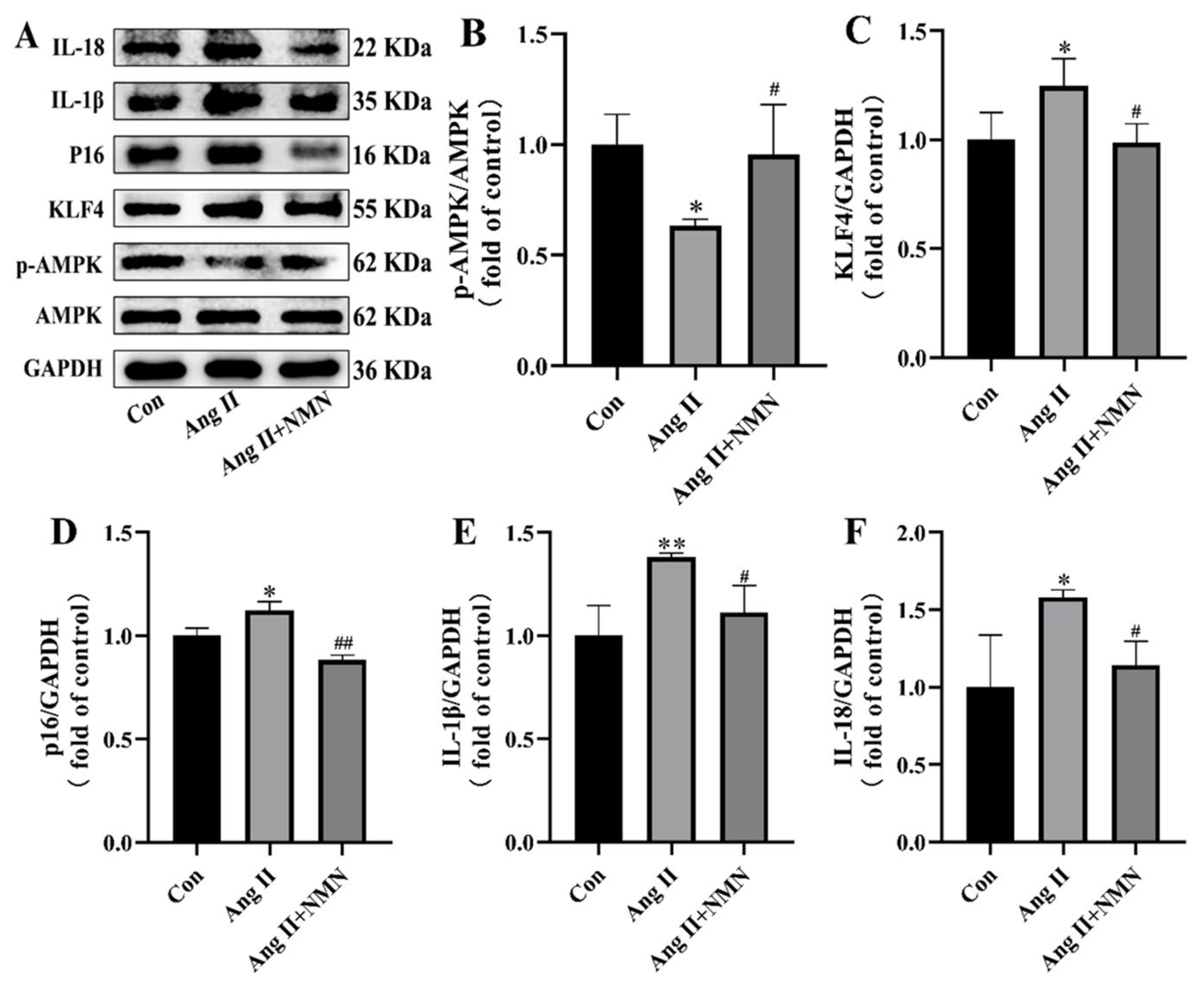
2.2. NMN Regulates the AMPK/KLF4/p16 Pathway Associated with Senescence in Mouse VSMCs
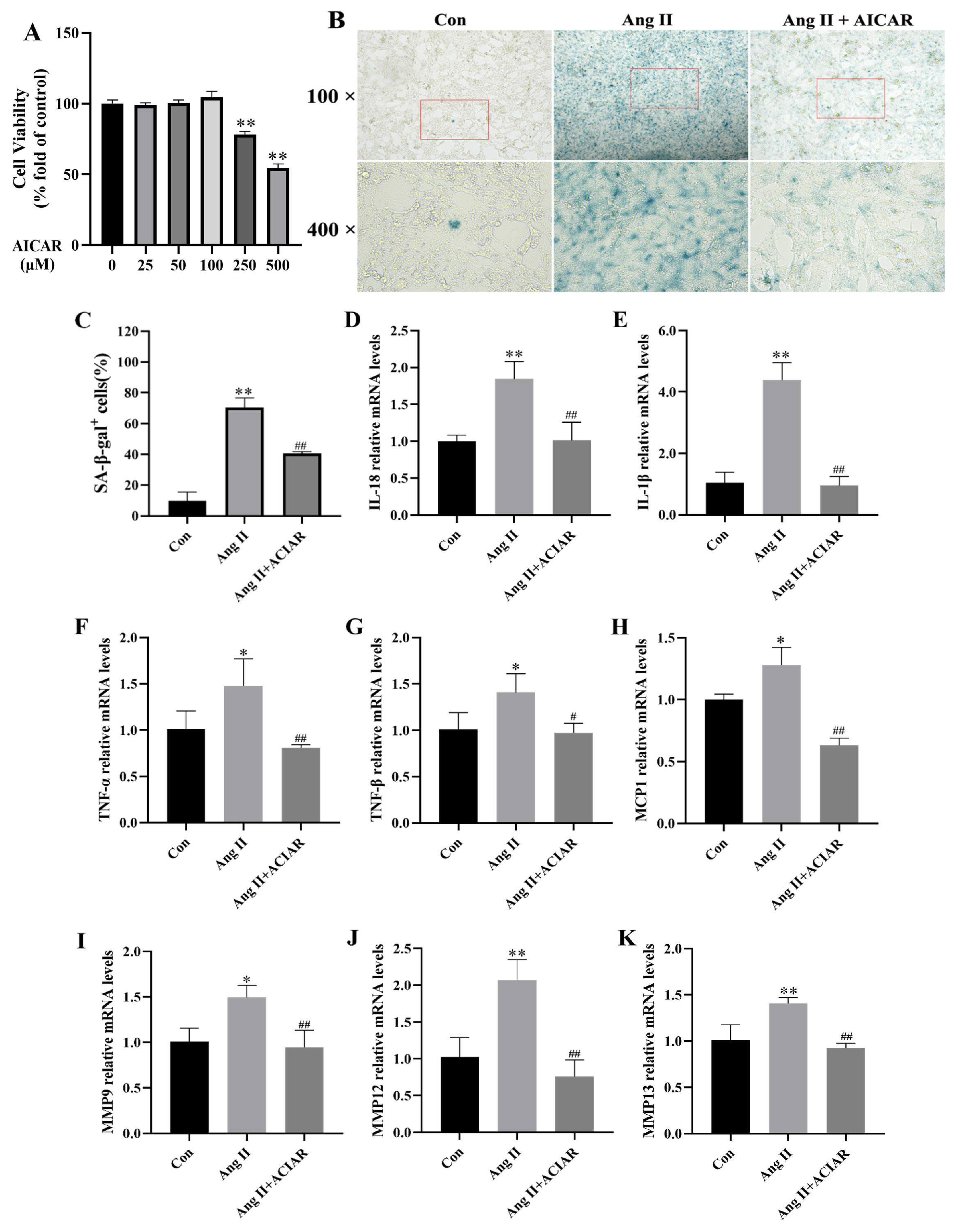
2.3. AICAR Can Improve the Senescence of Mouse VSMCs Induced by Ang II and Reduce the Secretion of Key SASP Factors
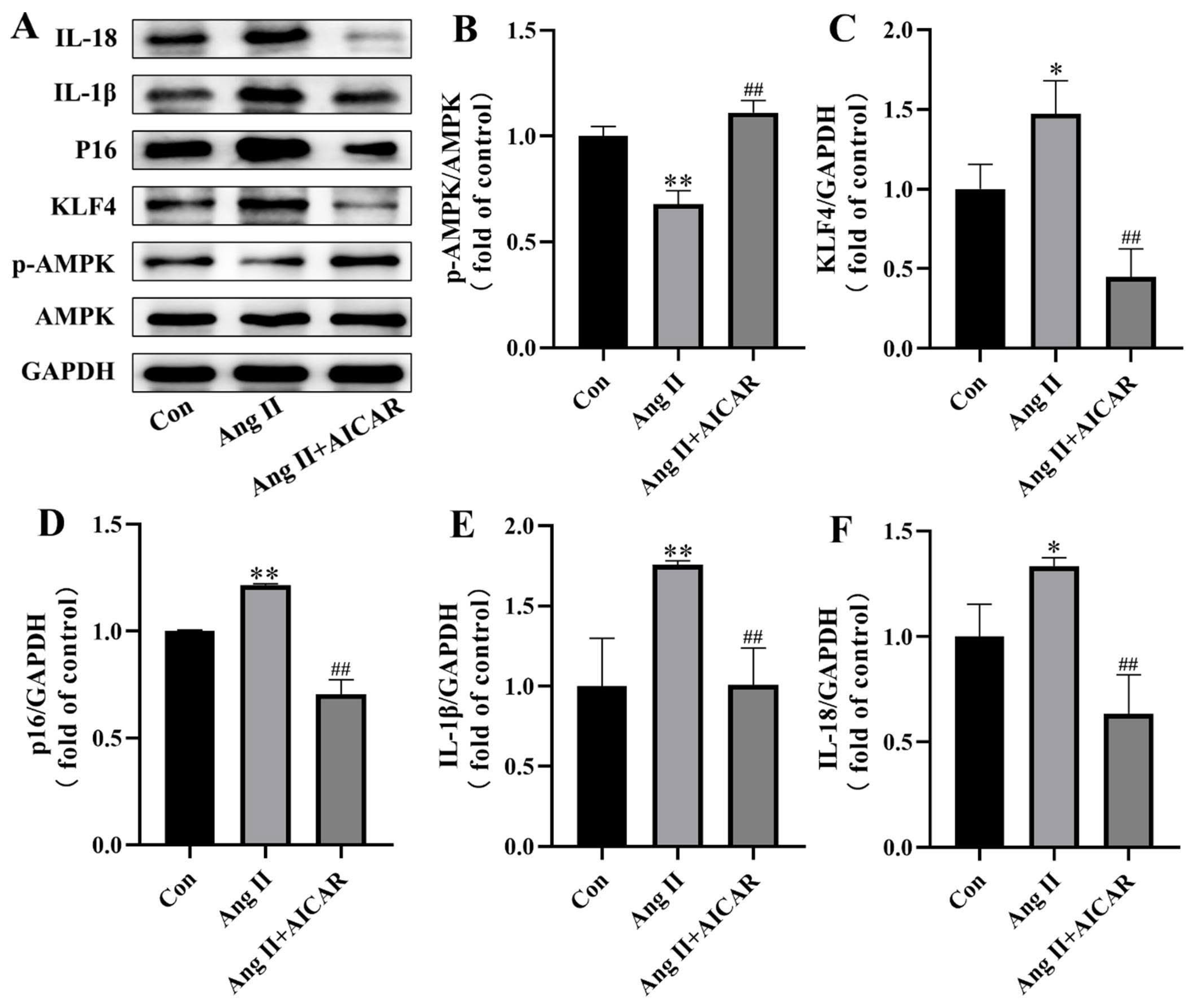
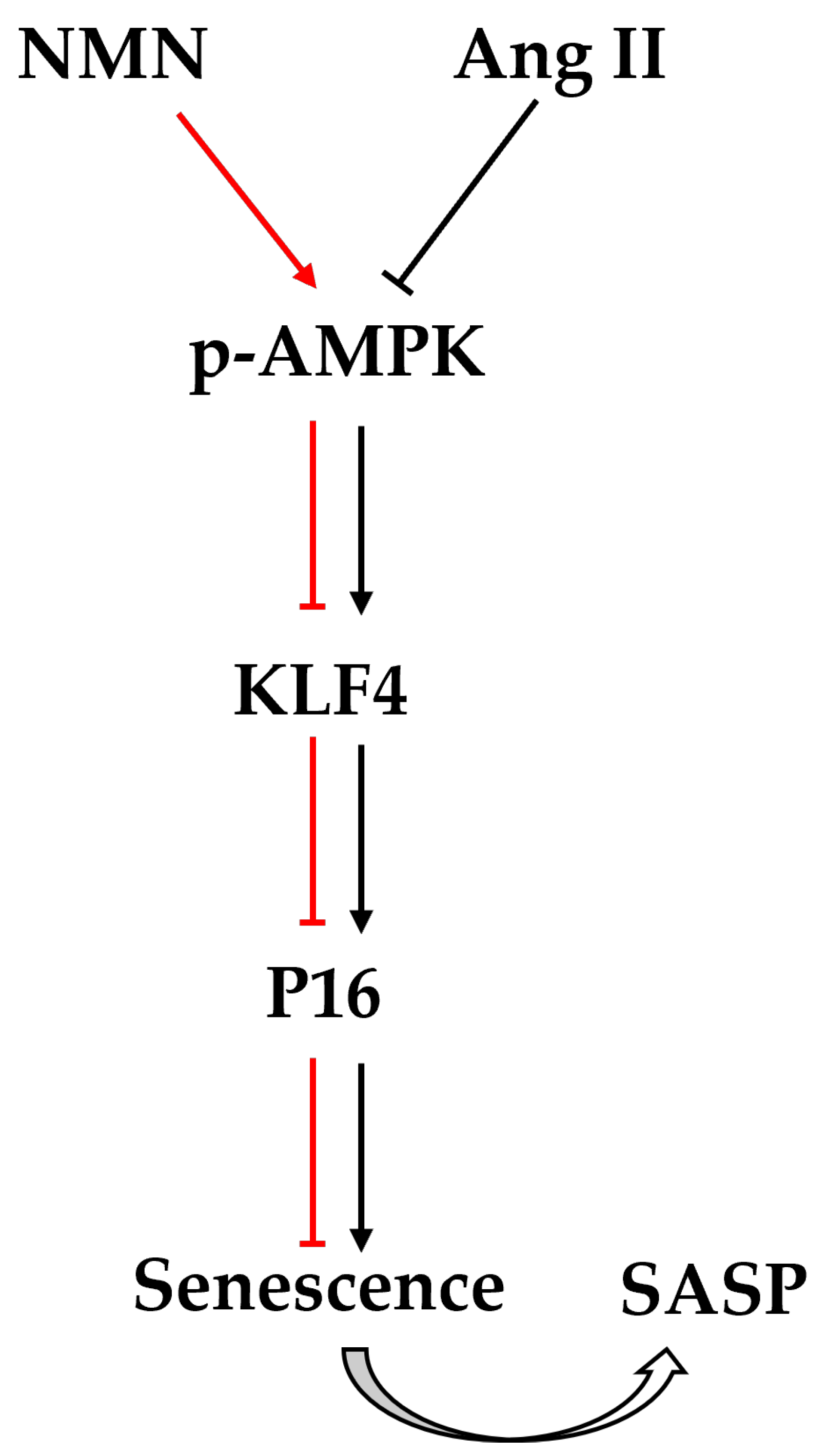
2.4. The Activated AMPK/KLF4/p16 Pathway Is Involved in Ang II-Induced Mouse VSMCs Cell Senescence
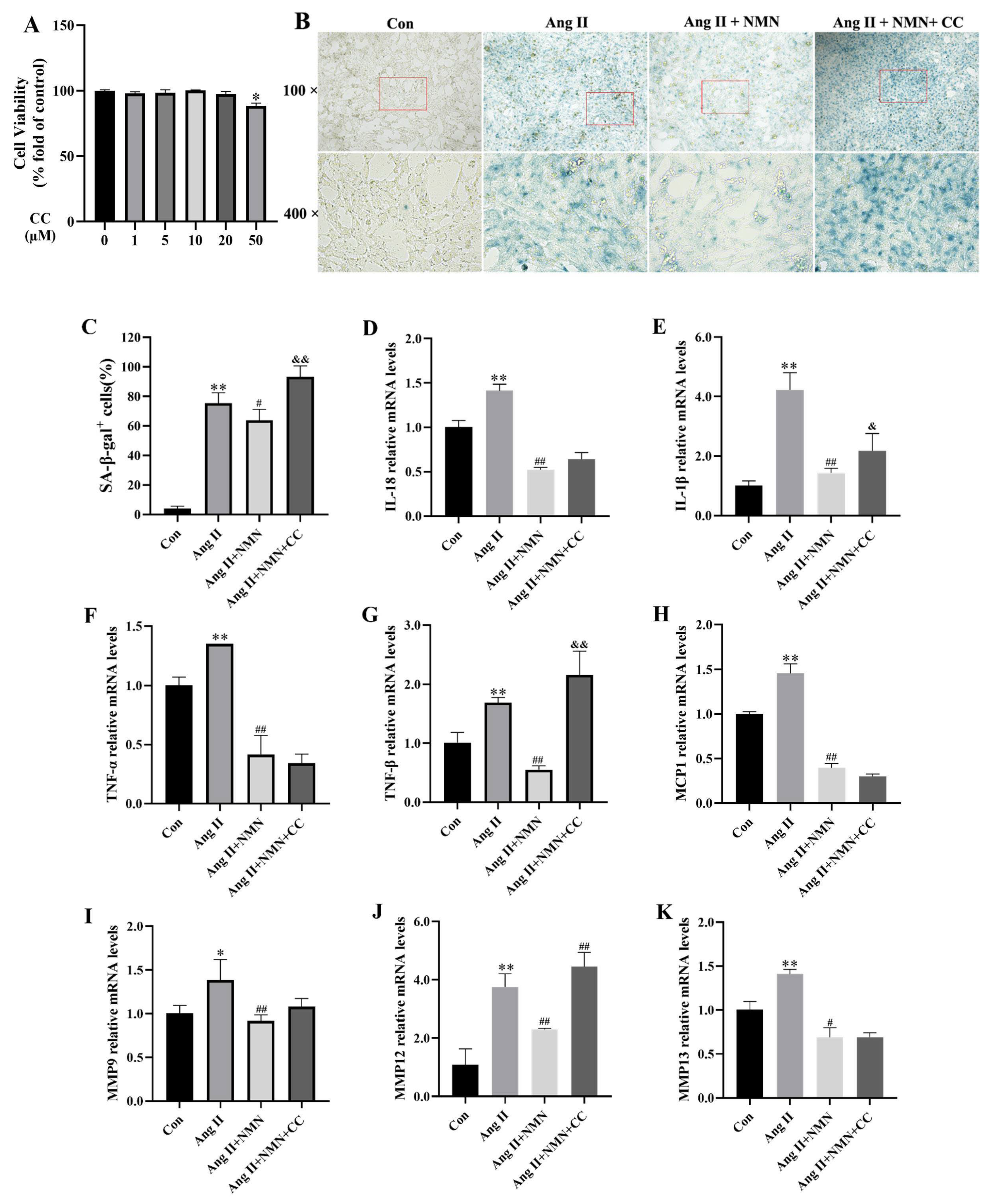
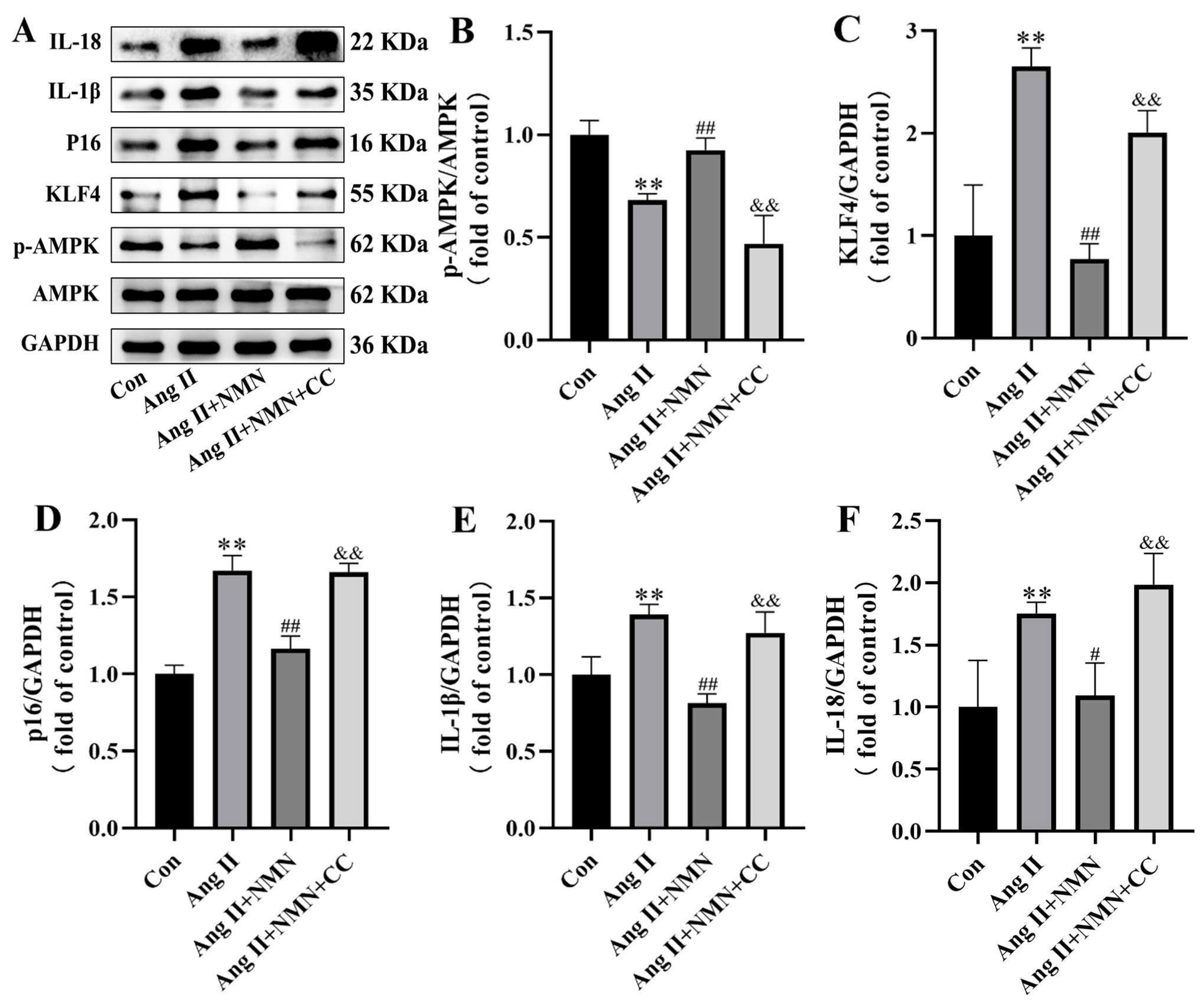
2.5. Compound C Could Mask the Effect of NMN on the Senescence of VSMCs Induced by Ang II
2.6. Inhibition of AMPK/KLF4/p16 Pathway Increased the Senescence of Mouse VSMCs Induced by Ang II
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Senescence-Galactosidase Staining (SA-β-Gal) Was Evaluated for Senescence
4.3. Total RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (qRT-PCR)
4.4. Cell Culture and Treatment
4.5. Cell Viability
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| VSMCs | Vascular smooth muscle cells |
| NMN | Nicotinamide mononucleotide |
| Ang II | Angiotensin II |
| SASP | Senescence-associated secretory phenotype |
| SA-β-gal | Senescence-associated β-galactosidase |
| NAD+ | Nicotinamide adenine dinucleotide |
| AMPK | AMP-activated protein kinase |
| KLF4 | Kruppel-like factor 4 |
| IL-18 | Interleukin-18 |
| IL-1β | Interleukin-1β |
| TNF-α | Tumor necrosis factor α |
| TNF-β | Tumor necrosis factor β |
| MCP-1 | Monocyte chemotactic protein-1 |
| MMP9 | Matrix metalloproteinase-9 |
| MMP12 | Matrix metalloproteinase-12 |
| MMP13 | Matrix metalloproteinase-13 |
References
- Ding, Y.N. Epigenetic Regulation of Vascular Aging and Age-Related Vascular Diseases. Adv. Exp. Med. Biol. 2018, 1086, 55–75. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Yang, Y.; Cheng, A.; Xu, G.; He, F. Metabolism of vascular smooth muscle cells in vascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H613–H631. [Google Scholar] [CrossRef]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Jiang, G.; Cheng, C.; Lv, Z.; Liu, Y.; Wang, F. Inhibition of Platelets by Clopidogrel Suppressed Ang II-Induced Vascular Inflammation, Oxidative Stress, and Remodeling. J. Am. Heart Assoc. 2018, 7, e009600. [Google Scholar] [CrossRef]
- Chen, M.S.; Lee, R.T.; Garbern, J.C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2022, 118, 1173–1187. [Google Scholar] [CrossRef]
- Lin, M.J.; Hu, S.L.; Tian, Y.; Zhang, J.; Liang, N.; Sun, R.; Gong, S.X.; Wang, A.P. Targeting Vascular Smooth Muscle Cell Senescence: A Novel Strategy for Vascular Diseases. J. Cardiovasc. Transl. Res. 2023, 16, 1010–1020. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Kroemer, G. NAD(+) Metabolism in Cardiac Health, Aging, and Disease. Circulation 2021, 144, 1795–1817. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Cordeiro, H.S.; Tran, N.L.K.; Chini, E.N. NAD metabolism: Role in senescence regulation and aging. Aging Cell 2024, 23, e13920. [Google Scholar] [CrossRef]
- Hershberger, K.A.; Martin, A.S.; Hirschey, M.D. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 2017, 13, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Liang, B.; Liu, X.L.; Liu, W.J.; Huang, B.H.; Yang, S.B.; Gao, Y.Z.; Meng, J.S.; Li, M.J.; Ye, T.; et al. Targeting NAD+: Is it a common strategy to delay heart aging? Cell Death Discov. 2022, 8, 230. [Google Scholar] [CrossRef]
- Aman, Y.; Frank, J.; Lautrup, S.H.; Matysek, A.; Niu, Z.; Yang, G.; Shi, L.; Bergersen, L.H.; Storm-Mathisen, J.; Rasmussen, L.J.; et al. The NAD(+)-mitophagy axis in healthy longevity and in artificial intelligence-based clinical applications. Mech. Ageing Dev. 2020, 185, 111194. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Liang, Y.; Huang, S.; Zhang, J.; Mai, C.; He, B.; Shi, L.; Liu, B.; Li, W.; Huang, X.; et al. Ciprofloxacin Accelerates Angiotensin-II-Induced Vascular Smooth Muscle Cells Senescence Through Modulating AMPK/ROS pathway in Aortic Aneurysm and Dissection. Cardiovasc. Toxicol. 2024, 24, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, J.; Okon, I.S.; Zou, M.H.; Song, P. Absence of AMPKα2 accelerates cellular senescence via p16 induction in mouse embryonic fibroblasts. Int. J. Biochem. Cell Biol. 2016, 71, 72–80. [Google Scholar] [CrossRef]
- Yang, C.; Xiao, X.; Huang, L.; Zhou, F.; Chen, L.H.; Zhao, Y.Y.; Qu, S.L.; Zhang, C. Role of Kruppel-like factor 4 in atherosclerosis. Clin. Chim. Acta 2021, 512, 135–141. [Google Scholar] [CrossRef]
- Sunaga, H.; Matsui, H.; Anjo, S.; Syamsunarno, M.R.; Koitabashi, N.; Iso, T.; Matsuzaka, T.; Shimano, H.; Yokoyama, T.; Kurabayashi, M. Elongation of Long-Chain Fatty Acid Family Member 6 (Elovl6)-Driven Fatty Acid Metabolism Regulates Vascular Smooth Muscle Cell Phenotype Through AMP-Activated Protein Kinase/Krüppel-Like Factor 4 (AMPK/KLF4) Signaling. J. Am. Heart Assoc. 2016, 5, e004014. [Google Scholar] [CrossRef]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef]
- Li, H.R.; Liu, Q.; Zhu, C.L.; Sun, X.Y.; Sun, C.Y.; Yu, C.M.; Li, P.; Deng, X.M.; Wang, J.F. β-Nicotinamide mononucleotide activates NAD+/SIRT1 pathway and attenuates inflammatory and oxidative responses in the hippocampus regions of septic mice. Redox Biol. 2023, 63, 102745. [Google Scholar] [CrossRef] [PubMed]
- Okabe, K.; Yaku, K.; Uchida, Y.; Fukamizu, Y.; Sato, T.; Sakurai, T.; Tobe, K.; Nakagawa, T. Oral Administration of Nicotinamide Mononucleotide Is Safe and Efficiently Increases Blood Nicotinamide Adenine Dinucleotide Levels in Healthy Subjects. Front. Nutr. 2022, 9, 868640. [Google Scholar] [CrossRef]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an Endothelial NAD(+)-H(2)S Signaling Network Is a Reversible Cause of Vascular Aging. Cell 2018, 173, 74–89.e20. [Google Scholar] [CrossRef]
- Wang, H.; Sun, Y.; Pi, C.; Yu, X.; Gao, X.; Zhang, C.; Sun, H.; Zhang, H.; Shi, Y.; He, X. Nicotinamide Mononucleotide Supplementation Improves Mitochondrial Dysfunction and Rescues Cellular Senescence by NAD(+)/Sirt3 Pathway in Mesenchymal Stem Cells. Int. J. Mol. Sci. 2022, 23, 14739. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Abudupataer, M.; Ming, Y.; Xiang, B.; Lai, H.; Wang, C.; Li, J.; Zhu, K. Nicotinamide Mononucleotide Alleviates Angiotensin II-Induced Human Aortic Smooth Muscle Cell Senescence in a Microphysiological Model. J. Cardiovasc. Pharmacol. 2023, 81, 280–291. [Google Scholar] [CrossRef]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun. Signal 2022, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yu, M.; Yu, Y.; Li, Y.; Yang, F.; Liu, Y.; Han, L.; Xu, Z.; Wang, G. KLF4 prevented angiotensin II-induced smooth muscle cell senescence by enhancing autophagic activity. Eur. J. Clin. Investig. 2022, 52, e13804. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef]
- Vellasamy, D.M.; Lee, S.J.; Goh, K.W.; Goh, B.H.; Tang, Y.Q.; Ming, L.C.; Yap, W.H. Targeting Immune Senescence in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 13059. [Google Scholar] [CrossRef]
- Ma, D.; Zheng, B.; Liu, H.L.; Zhao, Y.B.; Liu, X.; Zhang, X.H.; Li, Q.; Shi, W.B.; Suzuki, T.; Wen, J.K. Klf5 down-regulation induces vascular senescence through eIF5a depletion and mitochondrial fission. PLoS Biol. 2020, 18, e3000808. [Google Scholar] [CrossRef]
- Gan, L.; Liu, D.; Liu, J.; Chen, E.; Chen, C.; Liu, L.; Hu, H.; Guan, X.; Ma, W.; Zhang, Y.; et al. CD38 deficiency alleviates Ang II-induced vascular remodeling by inhibiting small extracellular vesicle-mediated vascular smooth muscle cell senescence in mice. Signal Transduct. Target. Ther. 2021, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Lan, C.; Fan, C.; Gong, X.; Chen, C.; Yu, C.; Wang, J.; Luo, X.; Hu, C.; Jose, P.A.; et al. Down-regulation of AMPK/PPARδ signalling promotes endoplasmic reticulum stress-induced endothelial dysfunction in adult rat offspring exposed to maternal diabetes. Cardiovasc. Res. 2022, 118, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Hu, Y.; Zhang, Y.; Yu, F. Ghrelin improves endothelial function and reduces blood pressure in Ang II-induced hypertensive mice: Role of AMPK. Clin. Exp. Hypertens. 2023, 45, 2208774. [Google Scholar] [CrossRef]
- Zhan, J.K.; Wang, Y.J.; Li, S.; Wang, Y.; Tan, P.; He, J.Y.; Chen, Y.Y.; Deng, H.Q.; Huang, W.; Lin, X.; et al. AMPK/TSC2/mTOR pathway regulates replicative senescence of human vascular smooth muscle cells. Exp. Ther. Med. 2018, 16, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef]
- Chi, C.; Li, D.J.; Jiang, Y.J.; Tong, J.; Fu, H.; Wu, Y.H.; Shen, F.M. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1810–1821. [Google Scholar] [CrossRef]
- Niu, K.M.; Bao, T.; Gao, L.; Ru, M.; Li, Y.; Jiang, L.; Ye, C.; Wang, S.; Wu, X. The Impacts of Short-Term NMN Supplementation on Serum Metabolism, Fecal Microbiota, and Telomere Length in Pre-Aging Phase. Front. Nutr. 2021, 8, 756243. [Google Scholar] [CrossRef]
- Nadeeshani, H.; Li, J.; Ying, T.; Zhang, B.; Lu, J. Nicotinamide mononucleotide (NMN) as an anti-aging health product—Promises and safety concerns. J. Adv. Res. 2022, 37, 267–278. [Google Scholar] [CrossRef]
- Tarantini, S.; Valcarcel-Ares, M.N.; Toth, P.; Yabluchanskiy, A.; Tucsek, Z.; Kiss, T.; Hertelendy, P.; Kinter, M.; Ballabh, P.; Süle, Z.; et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019, 24, 101192. [Google Scholar] [CrossRef]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, Q.; Gao, W.; Sehgal, S.A.; Wu, H. The multifaceted regulation of mitophagy by endogenous metabolites. Autophagy 2022, 18, 1216–1239. [Google Scholar] [CrossRef]
- Shi, T.; Fan, G.Q.; Xiao, S.D. SIRT3 reduces lipid accumulation via AMPK activation in human hepatic cells. J. Dig. Dis. 2010, 11, 55–62. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer 5′-3′ | Reverse Primer 5′-3′ |
|---|---|---|
| IL-18 | ATAAATGACCAAGTTCTCTTCGTTGAC | CACAGCCAGTCCTCTTACTTCAC |
| IL-1β | TCGCAGCAGCACATCAACAAG | TCCACGGGAAAGACACAGGTAG |
| TNF-α | CACGCTCTTCTGTCTACTGAACTTC | CTTGGTGGTTTGTGAGTGTGAGG |
| TNF-β | TCTGGAGAGCAAGCACGGATC | ACCACCTGGGAGTAGACAAAGTAG |
| MCP-1 | CACTCACCTGCTGCTACTCATTC | GCTTCTTTGGGACACCTGCTG |
| MMP9 | AGTATCTGTATGGTCGTGGCTCTAAG | GGAGGTGCTGTCGGCTGTG |
| MMP12 | GGCAACTGGACAACTCAACTCTG | CGCTTCATCCATCTTGACCTCTG |
| MMP13 | CAGTTGACAGGCTCCGAGAAATG | CACATCAGGCACTCCACATCTTG |
| GAPDH | GCAAATTCAACGGCACAGTCAAG | TCGCTCCTGGAAGATGGTGATG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, N.; Liu, S.; Wang, Y.; Ying, L.; Zhang, K.; Li, H.; Xiao, L.; Hu, Y.; Luo, G. Nicotinamide Mononucleotide (NMN) Improves the Senescence of Mouse Vascular Smooth Muscle Cells Induced by Ang II Through Activating p-AMPK/KLF4 Pathway. Pharmaceuticals 2025, 18, 553. https://doi.org/10.3390/ph18040553
Liang N, Liu S, Wang Y, Ying L, Zhang K, Li H, Xiao L, Hu Y, Luo G. Nicotinamide Mononucleotide (NMN) Improves the Senescence of Mouse Vascular Smooth Muscle Cells Induced by Ang II Through Activating p-AMPK/KLF4 Pathway. Pharmaceuticals. 2025; 18(4):553. https://doi.org/10.3390/ph18040553
Chicago/Turabian StyleLiang, Na, Si Liu, Yan Wang, Linyao Ying, Keyi Zhang, Hao Li, Lin Xiao, Yuming Hu, and Gang Luo. 2025. "Nicotinamide Mononucleotide (NMN) Improves the Senescence of Mouse Vascular Smooth Muscle Cells Induced by Ang II Through Activating p-AMPK/KLF4 Pathway" Pharmaceuticals 18, no. 4: 553. https://doi.org/10.3390/ph18040553
APA StyleLiang, N., Liu, S., Wang, Y., Ying, L., Zhang, K., Li, H., Xiao, L., Hu, Y., & Luo, G. (2025). Nicotinamide Mononucleotide (NMN) Improves the Senescence of Mouse Vascular Smooth Muscle Cells Induced by Ang II Through Activating p-AMPK/KLF4 Pathway. Pharmaceuticals, 18(4), 553. https://doi.org/10.3390/ph18040553