
Medicinal Phytocompounds as Potential Inhibitors of p300-HIF1α Interaction: A Structure-Based Screening and Molecular Dynamics Simulation Study


 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Screening of EANPDB to Shortlist Potential Inhibitors of p300-HIF-1 Interaction
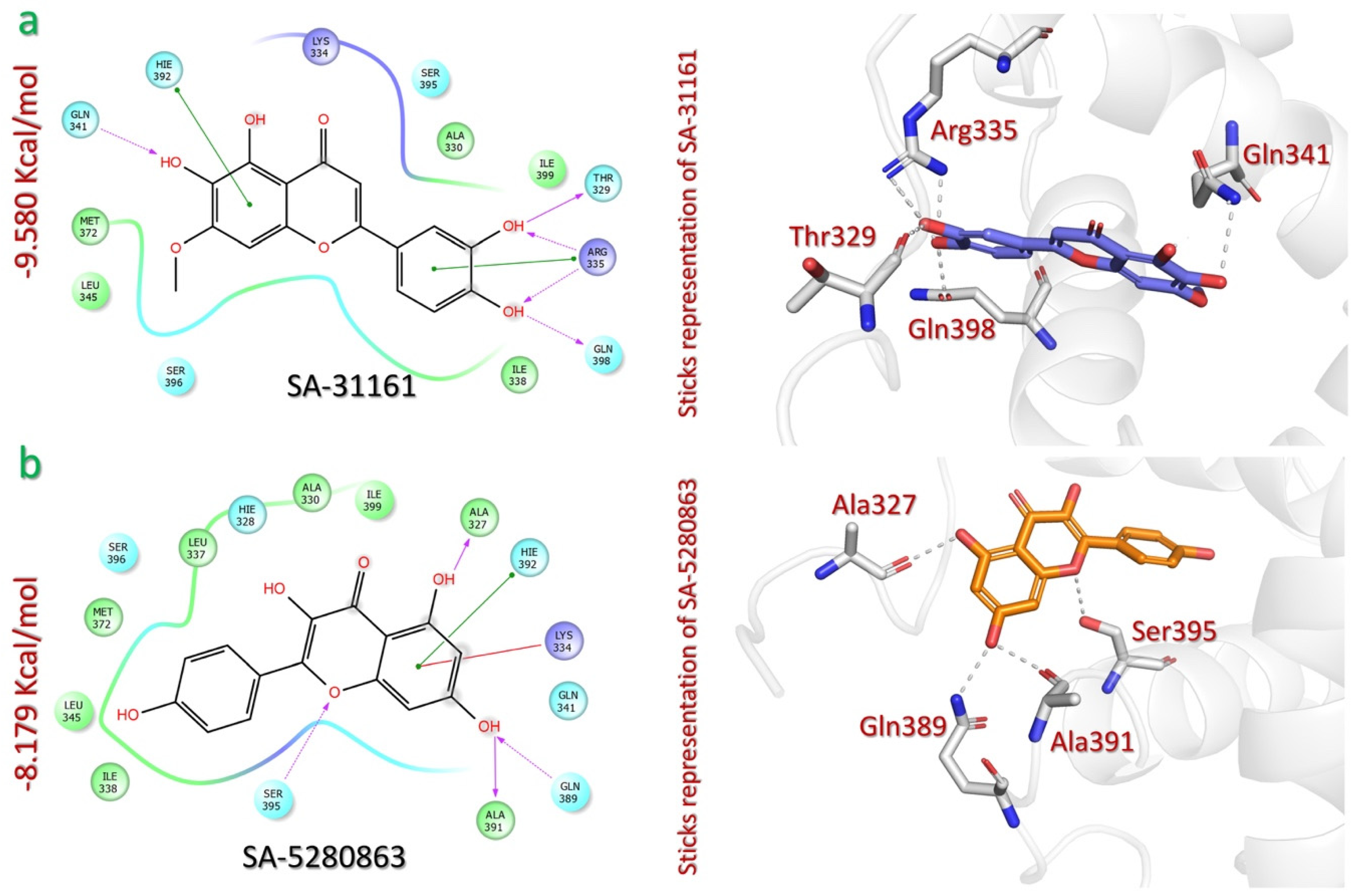
2.2. Screening of SANPDB to Shortlist Potential Inhibitors of p300-HIF-1 Interaction
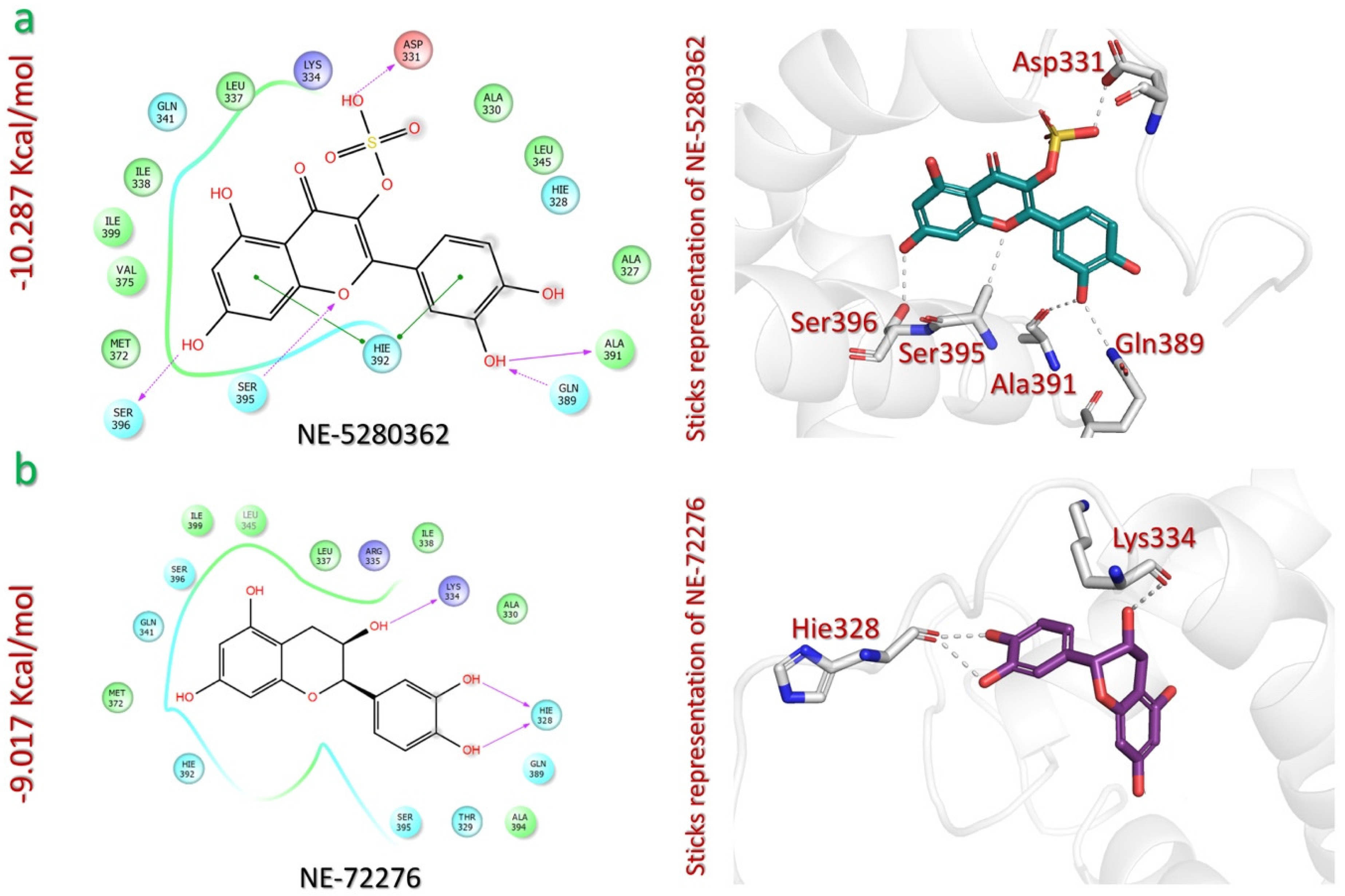
2.3. Screening of NEANPD to Shortlist Potential Inhibitors of p300-HIF-1 Interaction
2.4. Screening of NANPDB to Shortlist Potential Inhibitors of p300-HIF-1 Interaction
2.5. Screening of the TCM Database to Shortlist Potential Inhibitors of p300-HIF-1 Interaction
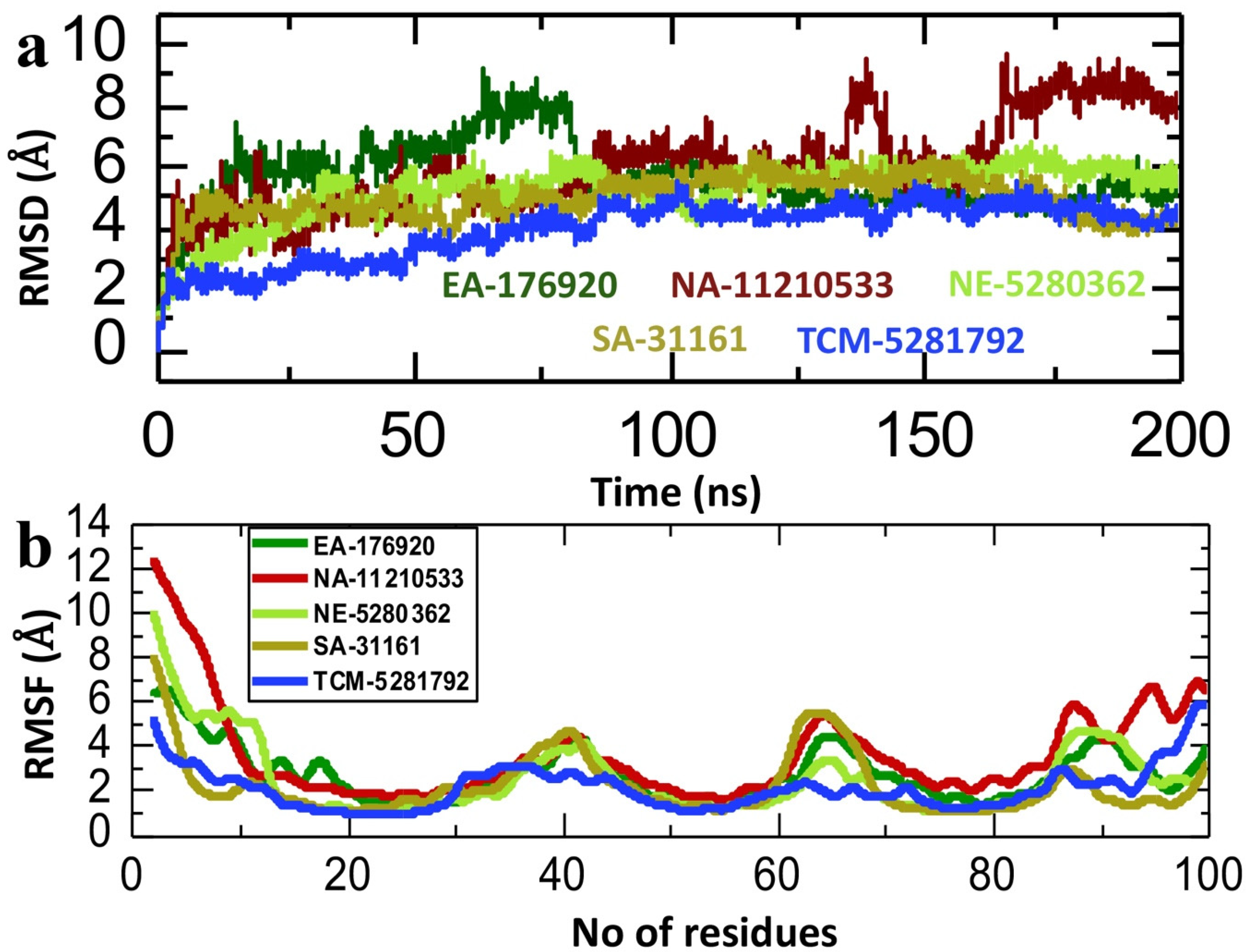
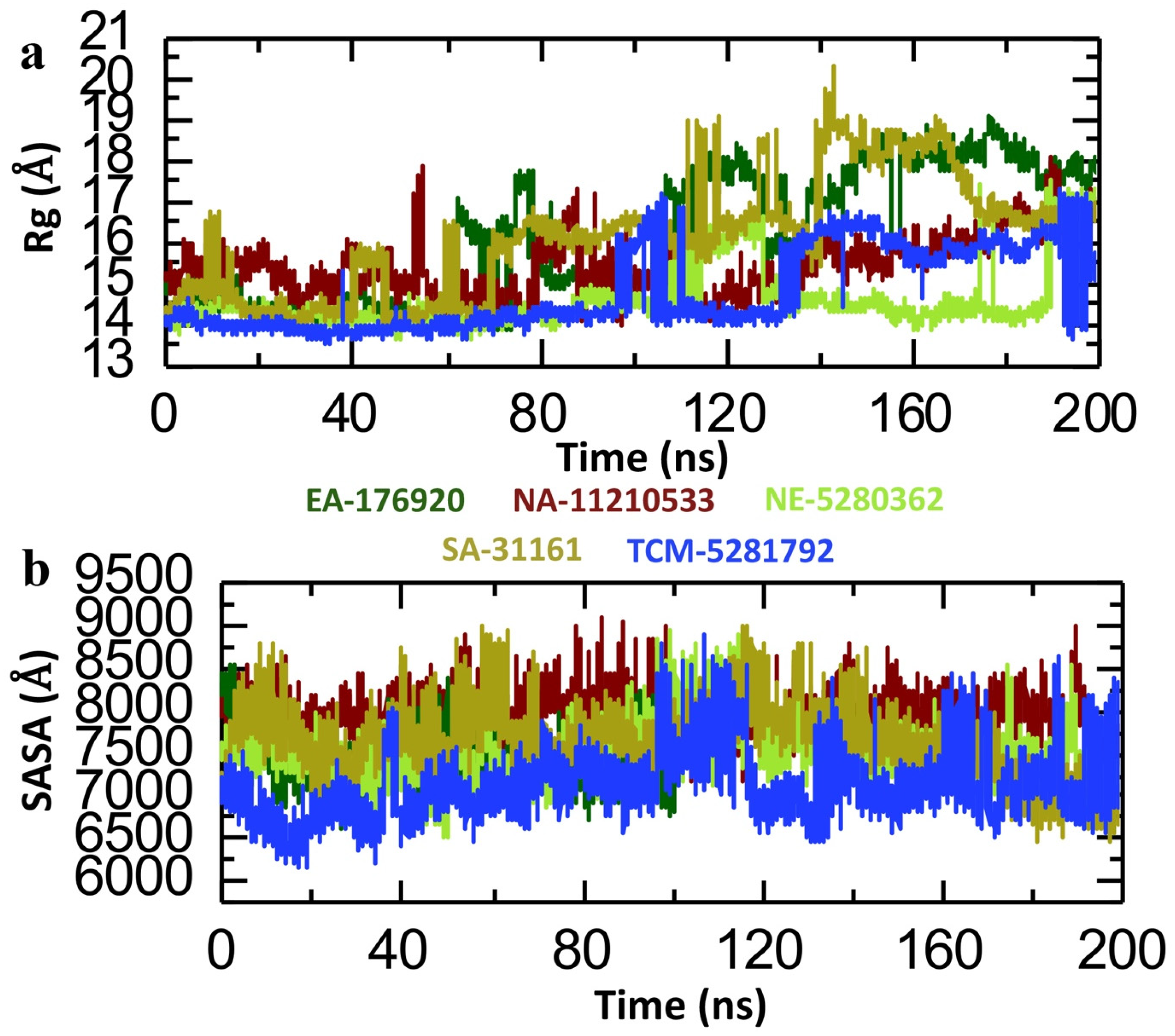
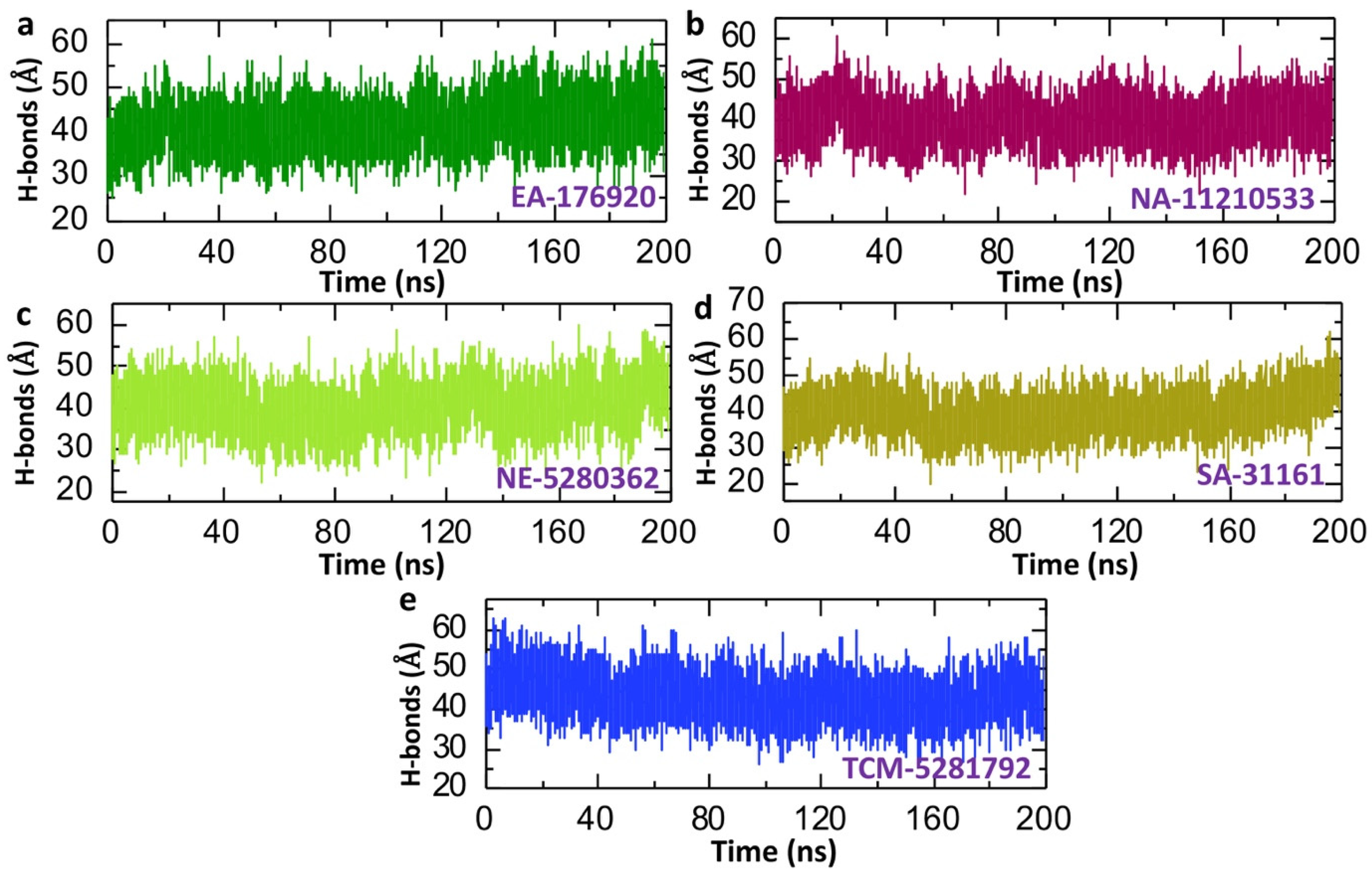
2.6. Dynamics Analysis of the Lead Compound from Each Database and p300
2.7. Binding Free Energy and Dissociation Constant (Kd) Analysis of the Five Lead Phytocompound–p300 Complexes
2.8. Oral Bioavailability and Drug Likeness Evaluation of the Five Lead Phytocompounds
2.9. ADMET Properties Evaluation of the Five Lead Phytocompounds
3. Materials and Methods
3.1. P300 Protein Structure Retrieval and Processing
3.2. Molecular Screening of Drug Databases Against p300
3.3. Molecular Dynamics Simulation
3.4. Dynamics Stability Analysis of Drug–p300 Complexes by Post-Simulation Trajectory
3.5. Kd (Dissociation Constant) Analysis of Shortlisted Compounds and p300 Complexes
3.6. Binding Free Energy Estimation Through MM/GBSA Analysis
3.7. Pharmacokinetics Analysis of Lead Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Kareem Azab, A. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Ohga, N.; Akiyama, K.; Hida, Y.; Maishi, N.; Towfik, A.M.; Inoue, N.; Shindoh, M.; Hida, K. Hypoxia-induced reactive oxygen species cause chromosomal abnormalities in endothelial cells in the tumor microenvironment. PLoS ONE 2013, 8, e80349. [Google Scholar] [CrossRef]
- Shimoda, L.A. KAT’s in the Cradle: p300/CBP and Regulation of HIF-1 and Blood Pressure during Intermittent Hypoxia. Am. Thorac. Soc. 2024, 70, 87–88. [Google Scholar] [CrossRef]
- Bayer, C.; Vaupel, P. Acute versus chronic hypoxia in tumors. Strahlenther. Onkol. 2012, 188, 616. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Milligan, J.; Huang, L.E. Molecular mechanism of Hypoxia-inducible Factor 1α-p300 interaction: A leucine-rich interface regulated by a single cysteine. J. Biol. Chem. 2001, 276, 3550–3554. [Google Scholar] [CrossRef]
- Gou, P.; Zhang, W. Protein lysine acetyltransferase CBP/p300: A promising target for small molecules in cancer treatment. Biomed. Pharmacother. 2024, 171, 116130. [Google Scholar] [CrossRef]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Schölz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell 2018, 174, 231–244.e212. [Google Scholar] [CrossRef]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, B.; Liu, X.; Zhang, X.D.; Zhang, L.; Liu, T. Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics 2022, 12, 4935. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mouttet, B.; Warnatz, H.-J.; Risch, T.; Rietmann, F.; Frommelt, F.; Ngo, Q.A.; Dobay, M.P.; Marovca, B.; Jenni, S. The leukemogenic TCF3-HLF complex rewires enhancers driving cellular identity and self-renewal conferring EP300 vulnerability. Cancer Cell 2019, 36, 630–644.e639. [Google Scholar] [CrossRef]
- Kung, A.L.; Wang, S.; Klco, J.M.; Kaelin, W.G.; Livingston, D.M. Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat. Med. 2000, 6, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.J.; Sun, Z.-Y.J.; Poy, F.; Kung, A.L.; Livingston, D.M.; Wagner, G.; Eck, M.J. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1α. Proc. Natl. Acad. Sci. USA 2002, 99, 5367–5372. [Google Scholar] [CrossRef]
- Kushal, S.; Lao, B.B.; Henchey, L.K.; Dubey, R.; Mesallati, H.; Traaseth, N.J.; Olenyuk, B.Z.; Arora, P.S. Protein domain mimetics as in vivo modulators of hypoxia-inducible factor signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 15602–15607. [Google Scholar] [CrossRef]
- Semenza, G.L. Physiology meets biophysics: Visualizing the interaction of hypoxia-inducible factor 1α with p300 and CBP. Proc. Natl. Acad. Sci. USA 2002, 99, 11570–11572. [Google Scholar] [CrossRef]
- Freedman, S.J.; Sun, Z.-Y.J.; Kung, A.L.; France, D.S.; Wagner, G.; Eck, M.J. Structural basis for negative regulation of hypoxia-inducible factor-1α by CITED2. Nat. Struct. Mol. Biol. 2003, 10, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.-T.; Chu, X.; Wang, J. Investigations of the underlying mechanisms of HIF-1α and CITED2 binding to TAZ1. Proc. Natl. Acad. Sci. USA 2020, 117, 5595–5603. [Google Scholar] [CrossRef]
- Bai, L.; Merchant, J.L. A role for CITED2, a CBP/p300 interacting protein, in colon cancer cell invasion. FEBS Lett. 2007, 581, 5904–5910. [Google Scholar] [CrossRef]
- Chassagne, F.; Cabanac, G.; Hubert, G.; David, B.; Marti, G. The landscape of natural product diversity and their pharmacological relevance from a focus on the Dictionary of Natural Products®®. Phytochem. Rev. 2019, 18, 601–622. [Google Scholar] [CrossRef]
- Shahrajabian, M.H.; Sun, W.; Cheng, Q. The importance of flavonoids and phytochemicals of medicinal plants with antiviral activities. Mini-Rev. Org. Chem. 2022, 19, 293–318. [Google Scholar] [CrossRef]
- Mehrbod, P.; Safari, H.; Mollai, Z.; Fotouhi, F.; Mirfakhraei, Y.; Entezari, H.; Goodarzi, S.; Tofighi, Z. Potential antiviral effects of some native Iranian medicinal plants extracts and fractions against influenza A virus. BMC Complement. Med. Ther. 2021, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Hwang, S.J.; Park, J.-H.; Lee, H.-J. Chlorogenic acid inhibits hypoxia-induced angiogenesis via down-regulation of the HIF-1α/AKT pathway. Cell. Oncol. 2015, 38, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.; Wang, Y.; Gao, P.; Wang, L.; Wang, Q.; Zhang, Y.; Liu, K.; Xia, Q.; Tu, P. The natural compound sinometumine E derived from corydalis decumbens promotes angiogenesis by regulating HIF-1/VEGF pathway in vivo and in vitro. Biomed. Pharmacother. 2024, 178, 117113. [Google Scholar] [CrossRef]
- Torres, M.P.; Rachagani, S.; Purohit, V.; Pandey, P.; Joshi, S.; Moore, E.D.; Johansson, S.L.; Singh, P.K.; Ganti, A.K.; Batra, S.K. Graviola: A novel promising natural-derived drug that inhibits tumorigenicity and metastasis of pancreatic cancer cells in vitro and in vivo through altering cell metabolism. Cancer Lett. 2012, 323, 29–40. [Google Scholar] [CrossRef]
- Al, K. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 2004, 6, 33–43. [Google Scholar]
- Wu, D.; Zhang, R.; Zhao, R.; Chen, G.; Cai, Y.; Jin, J. A novel function of novobiocin: Disrupting the interaction of HIF 1α and p300/CBP through direct binding to the HIF1α C-terminal activation domain. PLoS ONE 2013, 8, e62014. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Jafari, M.; Tang, J. A critical assessment of traditional Chinese medicine databases as a source for drug discovery. Front. Pharmacol. 2024, 15, 1303693. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.A.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A Resource for Natural Products from Northern African Sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef]
- Simoben, C.V.; Qaseem, A.; Moumbock, A.F.; Telukunta, K.K.; Günther, S.; Sippl, W.; Ntie-Kang, F. Pharmacoinformatic investigation of medicinal plants from East Africa. Mol. Inform. 2020, 39, 2000163. [Google Scholar] [CrossRef]
- Suleman, M.; Sayaf, A.M.; Khan, A.; Khan, S.A.; Albekairi, N.A.; Alshammari, A.; Agouni, A.; Yassine, H.M.; Crovella, S. Molecular screening of phytocompounds targeting the interface between influenza A NS1 and TRIM25 to enhance host immune responses. J. Infect. Public Health 2024, 17, 102448. [Google Scholar] [CrossRef] [PubMed]
- Diallo, B.N.T.; Glenister, M.; Musyoka, T.M.; Lobb, K.; Tastan Bishop, Ö. SANCDB: An update on South African natural compounds and their readily available analogs. J. Cheminform. 2021, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminform. 2020, 12, 20. [Google Scholar] [CrossRef]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis e Sousa, C.; Cusack, S. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef] [PubMed]
- Maregesi, S.M.; Hermans, N.; Dhooghe, L.; Cimanga, K.; Ferreira, D.; Pannecouque, C.; Berghe, D.A.V.; Cos, P.; Maes, L.; Vlietinck, A.J. Phytochemical and biological investigations of Elaeodendron schlechteranum. J. Ethnopharmacol. 2010, 129, 319–326. [Google Scholar] [CrossRef]
- He, W. Study of Biological Active Principles from Kenyan Medicinal Plants. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2001. [Google Scholar]
- Zaidi, F.; Voirin, B.; Jay, M.; Viricel, M.R. Free flavonoid aglycones from leaves of Mentha pulegium and Mentha suaveolens (Labiatae). Phytochemistry 1998, 48, 991–994. [Google Scholar] [CrossRef]
- Eldahshan, O.A.; Ayoub, N.A.; Singab, A.-N.B.; Al-Azizi, M.M. Potential superoxide anion radical scavenging activity of doum palm (Hyphaene thebaica L.) leaves extract. Rec. Nat. Prod. 2008, 2, 83. [Google Scholar]
- Abou-El-Hamd, E.; Mohamed-Elamir, F.; Moustafa, M.F. Euphorbia helioscopia: Chemical constituents and biological activities. Int. J. Phytopharm. 2012, 3, 78–90. [Google Scholar]
- Benabdelaziz, I.; Haba, H.; Lavaud, C.; Harakat, D.; Benkhaled, M. Lignans and other constituents from Helianthemum sessiliflorum Pers. Rec. Nat. Prod. 2015, 9, 342. [Google Scholar]
- El Amrani, M.; Lai, D.; Debbab, A.; Aly, A.H.; Siems, K.; Seidel, C.; Schnekenburger, M.; Gaigneaux, A.; Diederich, M.; Feger, D. Protein kinase and HDAC inhibitors from the endophytic fungus Epicoccum nigrum. J. Nat. Prod. 2014, 77, 49–56. [Google Scholar] [CrossRef]
- Hammerschmidt, L.; Aly, A.H.; Abdel-Aziz, M.; Müller, W.E.; Lin, W.; Daletos, G.; Proksch, P. Cytotoxic acyl amides from the soil fungus Gymnascella dankaliensis. Bioorg. Med. Chem. 2015, 23, 712–719. [Google Scholar] [CrossRef]
- Hawas, U.W. A new 8-hydroxy flavone O-xyloside sulfate and antibacterial activity from the Egyptian seagrass Thalassia hemprichii. Chem. Nat. Compd. 2014, 50, 629–632. [Google Scholar] [CrossRef]
- Hammoda, H.M.; Ghazy, N.M.; Harraz, F.M.; Radwan, M.M.; ElSohly, M.A.; Abdallah, I.I. Chemical constituents from Tribulus terrestris and screening of their antioxidant activity. Phytochemistry 2013, 92, 153–159. [Google Scholar] [CrossRef]
- Fatriansyah, J.F.; Boanerges, A.G.; Kurnianto, S.R.; Pradana, A.F.; Surip, S.N. Molecular Dynamics Simulation of Ligands from Anredera cordifolia (Binahong) to the Main Protease (M pro) of SARS-CoV-2. J. Trop. Med. 2022, 2022, 1178228. [Google Scholar] [CrossRef]
- Ghufran, M.; Khan, H.A.; Ullah, M.; Ghufran, S.; Ayaz, M.; Siddiq, M.; Bungau, S. In silico strategies for designing of peptide inhibitors of oncogenic K-ras G12V mutant: Inhibiting cancer growth and proliferation. Cancers 2022, 14, 4884. [Google Scholar] [CrossRef]
- Rashid, F.; Suleman, M.; Shah, A.; Dzakah, E.E.; Chen, S.; Wang, H.; Tang, S. Structural analysis on the severe acute respiratory syndrome coronavirus 2 non-structural protein 13 mutants revealed altered bonding network with TANK binding kinase 1 to evade host immune system. Front. Microbiol. 2021, 12, 789062. [Google Scholar] [CrossRef]
- Shah, A.; Rehmat, S.; Aslam, I.; Suleman, M.; Batool, F.; Aziz, A.; Rashid, F.; Nawaz, M.A.; Ali, S.S.; Junaid, M. Comparative mutational analysis of SARS-CoV-2 isolates from Pakistan and structural-functional implications using computational modelling and simulation approaches. Comput. Biol. Med. 2022, 141, 105170. [Google Scholar] [CrossRef]
- MIu, L.; Bogatyreva, N.; Galzitskaia, O. Radius of gyration is indicator of compactness of protein structure. Mol. Biol. 2008, 42, 701–706. [Google Scholar]
- Marsh, J.A.; Teichmann, S.A. Relative solvent accessible surface area predicts protein conformational changes upon binding. Structure 2011, 19, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Suleman, M.; Yousafi, Q.; Ali, J.; Ali, S.S.; Hussain, Z.; Ali, S.; Waseem, M.; Iqbal, A.; Ahmad, S.; Khan, A. Bioinformatics analysis of the differences in the binding profile of the wild-type and mutants of the SARS-CoV-2 spike protein variants with the ACE2 receptor. Comput. Biol. Med. 2021, 138, 104936. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Khan, A.; Adil, S.; Qudsia, H.A.; Waheed, Y.; Alshabrmi, F.M.; Wei, D.-Q. Structure-based design of promising natural products to inhibit thymidylate kinase from Monkeypox virus and validation using free energy calculations. Comput. Biol. Med. 2023, 158, 106797. [Google Scholar] [CrossRef]
- Suleman, M.; Murtaza, A.; Khan, H.; Rashid, F.; Alshammari, A.; Ali, L.; Khan, A.; Wei, D.-Q. The XBB. 1.5 slightly increase the binding affinity for host receptor ACE2 and exhibit strongest immune escaping features: Molecular modeling and free energy calculation. Front. Mol. Biosci. 2023, 10, 1153046. [Google Scholar] [CrossRef]
- Kralj, S.; Jukič, M.; Bren, U. Molecular Filters in Medicinal Chemistry. Encyclopedia 2023, 3, 501–511. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Wang, X.; Lyu, Y.; Chen, X.; Liu, K. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef]
- Reichel, A.; Lienau, P. Pharmacokinetics in drug discovery: An exposure-centred approach to optimising and predicting drug efficacy and safety. New Approaches Drug Discov. 2016, 232, 235–260. [Google Scholar]
- Xu, W.; Ling, P.; Zhang, T. Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. J. Drug Deliv. 2013, 2013, 340315. [Google Scholar] [CrossRef]
- Tsaioun, K.; Bottlaender, M.; Mabondzo, A.; Alzheimer’s Drug Discovery Foundation not@ valid. com. ADDME–Avoiding Drug Development Mistakes Early: Central nervous system drug discovery perspective. BMC Neurol. 2009, 9, S1. [Google Scholar] [CrossRef] [PubMed]
- Lea, T. Caco-2 cell line. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Springer: Berlin/Heidelberg, Germany, 2015; pp. 103–111. [Google Scholar]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Agnihotry, S.; Pathak, R.K.; Srivastav, A.; Shukla, P.K.; Gautam, B. Molecular docking and structure-based drug design. In Computer-Aided Drug Design; Springer: Berlin/Heidelberg, Germany, 2020; pp. 115–131. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sayaf, A.M.; Khalid, S.U.; Hameed, J.A.; Alshammari, A.; Khan, A.; Mohammad, A.; Alghamdi, S.; Wei, D.-Q.; Yeoh, K. Exploring the natural products chemical space through a molecular search to discover potential inhibitors that target the hypoxia-inducible factor (HIF) prolyl hydroxylase domain (PHD). Front. Pharmacol. 2023, 14, 1202128. [Google Scholar] [CrossRef]
- Aziz, M.; Ejaz, S.A.; Zargar, S.; Akhtar, N.; Aborode, A.T.; Wani, T.A.; Batiha, G.E.-S.; Siddique, F.; Alqarni, M.; Akintola, A.A. Deep learning and structure-based virtual screening for drug discovery against NEK7: A novel target for the treatment of cancer. Molecules 2022, 27, 4098. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Roe, D.R.; Brooks, B.R. A protocol for preparing explicitly solvated systems for stable molecular dynamics simulations. J. Chem. Phys. 2020, 153, 054123. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz Jr, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Luty, B.A.; Davis, M.E.; Tironi, I.G.; Van Gunsteren, W.F. A comparison of particle-particle, particle-mesh and Ewald methods for calculating electrostatic interactions in periodic molecular systems. Mol. Simul. 1994, 14, 11–20. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A. Thermodynamic fluctuations in protein molecules. Proc. Natl. Acad. Sci. USA 1976, 73, 2740–2741. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Ma, W.; Yang, L.; He, L. Overview of the detection methods for equilibrium dissociation constant KD of drug-receptor interaction. J. Pharm. Anal. 2018, 8, 147–152. [Google Scholar] [CrossRef]
- Suleman, M.; Luqman, M.; Wei, D.-Q.; Ali, S.; Ali, S.S.; Khan, A.; Khan, H.; Ali, Z.; Khan, W.; Rizwan, M. Structural insights into the effect of mutations in the spike protein of SARS-CoV-2 on the binding with human furin protein. Heliyon 2023, 9, e15083. [Google Scholar] [CrossRef]
- Tonge, P.J. Drug–target kinetics in drug discovery. ACS Chem. Neurosci. 2018, 9, 29–39. [Google Scholar] [CrossRef]
- Vangone, A.; Schaarschmidt, J.; Koukos, P.; Geng, C.; Citro, N.; Trellet, M.E.; Xue, L.C.; Bonvin, A.M. Large-scale prediction of binding affinity in protein–small ligand complexes: The PRODIGY-LIG web server. Bioinformatics 2019, 35, 1585–1587. [Google Scholar] [CrossRef]
- Pollastri, M.P. Overview on the Rule of Five. Curr. Protoc. Pharmacol. 2010, 49, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Blundell, T.; Ascher pkCSM, D. Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Compound Name, PubChem ID, and Source | Docking Score | Ligand Atom/Functional Group | P300 (1P4Q) | Interaction | Distance (Å) |
|---|---|---|---|---|---|---|
| EANPDB | 4′-O-methylepigallocatechin (EA-176920) Elaeodendron schlechteranum | −8.719 | OH(resorcinol) | Lys334 | HB | 2.16 |
| O(oxanol) | Ser395 | HB | 2.50 | |||
| OH(Methoxyresorcinol) | Ala327 | HB | 2.40 | |||
| OH(Methoxyresorcinol) | Ala391 | HB | 1.86 | |||
| OH(Methoxyresorcinol) | Gln389 | HB | 1.89 | |||
| Methoxyresorcinol ring | Hie392 | π-π | 5.16 | |||
| (+)Sesamin(EA-46881231) Zanthoxylum usambarense | −8.642 | OH(catechol) | Gln398 | HB | 1.74 | |
| OH(Catechol) | Gln398 | HB | 1.88 | |||
| Catechol ring | Lys334 | π-cation | 4.59 | |||
| SANPDB | Pedalitin (SA-31161) Arctotis arctotoides | −9.580 | OH(catechol) | Thr329 | HB | 1.67 |
| OH(Catechol) | Gln398 | HB | 1.90 | |||
| OH(catechol) | Arg335 | HB | 2.68 | |||
| OH(Catechol) | Arg335 | HB | 2.17 | |||
| OH(Methoxyresorcinol) | Gln341 | HB | 2.31 | |||
| Methoxyresorcinol ring | Hie392 | π-π | 3.58 | |||
| Catechol ring | Arg335 | π-π | 4.74 | |||
| Kaempferol (SA5280863) Combretum apiculatum | −8.179 | OH(resorcinol) | Ala327 | HB | 1.78 | |
| OH(resorcinol) | Ala391 | HB | 1.82 | |||
| OH(resorcinol) | Gln389 | HB | 1.92 | |||
| O(pyranone) | Ser395 | HB | 2.10 | |||
| Resorcinol ring | Lys334 | π-Cation | 5.68 | |||
| Resorcinol ring | Hie392 | π-π | 5.18 | |||
| NEANPDB | Quercetin 3-sulfate (NE-5280362) Euphorbia helioscopia | −10.287 | OH(HSO4) | Asp331 | HB | 1.33 |
| OH(catechol) | Ala391 | HB | 1.88 | |||
| OH(catechol) | Gln389 | HB | 1.95 | |||
| OH(resorcinol) | Ser396 | HB | 1.95 | |||
| O(pyranone) | Ser396 | HB | 2.39 | |||
| Catechol ring | Hie392 | π-π | 5.48 | |||
| Resorcinol ring | Hie392 | π-π | 5.06 | |||
| (−)Epicatechin(NE-72276) Helianthemum sessiliflorum | −9.017 | O(oxanol) | Lys334 | HB | 1.92 | |
| OH(catechol) | Hie328 | HB | 1.69 | |||
| OH(catechol) | Hie328 | HB | 2.03 | |||
| NANPDB | Epicoccolide B (NA-11210533) Epicoccum nigrum | −10.366 | OH(Trihydroxybenzaldehyde) | Gln398 | HB | 1.71 |
| OH(Trihydroxybenzaldehyde) | Gln398 | HB | 1.69 | |||
| OH(Trihydroxybenzaldehyde) | Gln389 | HB | 1.95 | |||
| OH(Trihydroxybenzaldehyde) | Hie328 | HB | 2.24 | |||
| Dihydroxybenzaldehyde ring | Hie392 | π-π | 4.38 | |||
| Farinosone C (NA-11336960) Gymnascella dankaliensis | −7.818 | OH(methanol) | Hie328 | HB | 1.77 | |
| OH(phenol) | Ala394 | HB | 2.36 | |||
| OH(phenol) | Ala391 | HB | 1.85 | |||
| CO(COOH) | Ser395 | HB | 2.20 | |||
| TCM | Rosmarinic acid (TCM-5281792) Thalassia hemprichii | −12.648 | OH(catechol) | Ala391 | HB | 1.91 |
| OH(catechol) | Ser395 | HB | 2.26 | |||
| OH(catechol) | Gln389 | HB | 1.94 | |||
| OH(catechol) | Hie392 | HB | 2.27 | |||
| OH(catechol) | Gln341 | HB | 2.08 | |||
| CO(COOH) | Arg335 | HB | 1.89 | |||
| Catechol ring | Hie392 | π-π | 4.74 | |||
| O(COOH) | Arg335 | SB | 4.96 | |||
| 5-p-trans-coumaroylquinic acid (TCM-6441280) Tribulus terrestris | −9.470 | OH(catechol) | His368 | HB | 2.17 | |
| Co(easter) | Lys334 | HB | 2.01 | |||
| OH(cyclohexane) | Ala327 | HB | 2.06 | |||
| OH(cyclohexane) | Gln389 | HB | 2.33 | |||
| OH(cyclohexane) | Ala391 | HB | 2.13 | |||
| OH(COOH) | Ala394 | HB | 2.36 | |||
| CO(COOH) | Ser395 | HB | 1.79 |
| MM/GBSA | |||||
|---|---|---|---|---|---|
| Parameter | EA-176920 | NA-11210533 | NE-5280362 | SA-31161 | TCM-5281792 |
| ΔEvdw | −31.8393 | −3.7149 | −45.9966 | −41.6263 | −47.3801 |
| ΔEele | −3.7244 | −1.5064 | −23.7432 | −12.2433 | −288.4422 |
| EGB | 17.7219 | −23.5780 | 42.7170 | 24.9116 | 284.4164 |
| ESURF | −4.1602 | 3.3495 | −5.4301 | −5.0337 | −6.3696 |
| Delta G Gas | −35.5638 | −5.2213 | −69.7399 | −53.8696 | −335.8223 |
| Delta G Solv | 13.5618 | −20.2286 | 37.2869 | 19.8778 | 278.0468 |
| ∆G Total | −22.0020 | −25.4499 | −32.4530 | −33.9918 | −57.7755 |
| Drugs ID | Molecular Weight | Hydrogen Acceptors | Hydrogen Donors | Consensus Log P | Lipinski’s Rule | Bioavailability | |
|---|---|---|---|---|---|---|---|
| Results | Violation | ||||||
| EA-176920 | 320.297 | 7 | 5 | 1.55 | Yes | 0 | 0.55 |
| NA-11210533 | 358.302 | 8 | 5 | 2.86 | Yes | 0 | 0.55 |
| NE-5280362 | 382.302 | 9 | 5 | 1.46 | Yes | 0 | 0.11 |
| SA-31161 | 316.265 | 7 | 4 | 2.29 | Yes | 0 | 0.55 |
| TCM-5281792 | 360.318 | 7 | 5 | 1.76 | Yes | 0 | 0.56 |
| Drugs ID | Water Solubility Log S | Caco-2 Permeability ×10−6 | Human Intestinal Absorption (%) | VDss (Human) | BBB Permeability | AMES Toxicity | Skin Sensitization | Hepatotoxicity |
|---|---|---|---|---|---|---|---|---|
| EA-176920 | −3.085 | −0.119 | 60.725 | 1.635 | No | No | No | No |
| NA-11210533 | −3.271 | −0.137 | 62.577 | 0.482 | No | No | No | No |
| NE-5280362 | −2.892 | −1.143 | 30.235 | −0.067 | No | No | No | No |
| SA-31161 | −3.168 | −0.364 | 79.877 | 0.661 | Yes | No | No | No |
| TCM-5281792 | −3.059 | −0.937 | 32.516 | 0.393 | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suleman, M.; Sayaf, A.M.; Aftab, S.; Alissa, M.; Alghamdi, A.; Alghamdi, S.A.; Alshehri, M.A.; Yeoh, K.K.; Crovella, S.; Shaito, A.A. Medicinal Phytocompounds as Potential Inhibitors of p300-HIF1α Interaction: A Structure-Based Screening and Molecular Dynamics Simulation Study. Pharmaceuticals 2025, 18, 602. https://doi.org/10.3390/ph18040602
Suleman M, Sayaf AM, Aftab S, Alissa M, Alghamdi A, Alghamdi SA, Alshehri MA, Yeoh KK, Crovella S, Shaito AA. Medicinal Phytocompounds as Potential Inhibitors of p300-HIF1α Interaction: A Structure-Based Screening and Molecular Dynamics Simulation Study. Pharmaceuticals. 2025; 18(4):602. https://doi.org/10.3390/ph18040602
Chicago/Turabian StyleSuleman, Muhammad, Abrar Mohammad Sayaf, Sohail Aftab, Mohammed Alissa, Abdullah Alghamdi, Suad A. Alghamdi, Mohammed A. Alshehri, Kar Kheng Yeoh, Sergio Crovella, and Abdullah A. Shaito. 2025. "Medicinal Phytocompounds as Potential Inhibitors of p300-HIF1α Interaction: A Structure-Based Screening and Molecular Dynamics Simulation Study" Pharmaceuticals 18, no. 4: 602. https://doi.org/10.3390/ph18040602
APA StyleSuleman, M., Sayaf, A. M., Aftab, S., Alissa, M., Alghamdi, A., Alghamdi, S. A., Alshehri, M. A., Yeoh, K. K., Crovella, S., & Shaito, A. A. (2025). Medicinal Phytocompounds as Potential Inhibitors of p300-HIF1α Interaction: A Structure-Based Screening and Molecular Dynamics Simulation Study. Pharmaceuticals, 18(4), 602. https://doi.org/10.3390/ph18040602